Abstract
5-methylcytosine (5-mC) can be oxidized to 5-hydroxymethylcytosine (5-hmC). Genome-wide profiling of 5-hmC thus far indicates 5-hmC may not only be an intermediate form of DNA demethylation but could also constitute an epigenetic mark per se. Here we describe a cost-effective and selective method to detect both the hydroxymethylation and methylation status of cytosines in a subset of cytosines in the human genome. This method involves the selective glucosylation of 5-hmC residues, short-Sequence tag generation and high-throughput sequencing. We tested this method by screening H9 human embryonic stem cells and their differentiated embroid body cells, and found that differential hydroxymethylation preferentially occurs in bivalent genes during cellular differentiation. Especially, our results support hydroxymethylation can regulate key transcription regulators with bivalent marks through demethylation and affect cellular decision on choosing active or inactive state of these genes upon cellular differentiation. Future application of this technology would enable us to uncover the status of methylation and hydroxymethylation in dynamic biological processes and disease development in multiple biological samples.
Introduction
5-methylcytosine (5-mC) in mammalian genomic DNA is essential for normal development and diverse biological functions. 5-mC can be oxidized to 5-hydroxymethylcytosine (5-hmC) by the ten-11 translocation (TET) enzyme family proteins.Citation1-Citation3 5-hmC was found to be widespread in many tissues and cell types, playing important roles in gene transcription regulation, and can be further oxidized to 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC) by TET proteins.Citation4-Citation7 A full appreciation of the biological importance of 5-hmC will require the development of tools that allow 5-hmC and 5-mC to be distinguished unequivocally. Previously, strategies based on DNA immunoprecipitation (hydroxymethylated DNA immunoprecipitation sequencing, hMeDIP-Seq),Citation8 chemical labeling of 5-hmC coupled with affinity enrichment (glucosylation, periodate oxidation, biotinylation, GLIB),Citation4 and improved immunohistochemical visualization to distinguish 5-mC and 5-hmC were applied.Citation4,Citation8-Citation10 Among these methods, both hMeDIP and GLIB can be used to generate a genome-wide hydroxymethylome, but suffer from low resolution and might be biased by regional CpG density and the amount of hydroxymethylated cytosines. Most recently, oxBS-SeqCitation11 and TAB-Seq,Citation12 which can map 5-hmC at single-base resolution at the genome-wide level, have been developed as well, enabling most comprehensive investigation of the genomic distribution of DNA hydroxymethylation in a genome. However, a massive amount of data was required to determine 5-mC and 5-hmC in parallel for the human genome. (In theory, 180Gb clean data are required to achieve 30X sequencing depth.) Furthermore, minimal amount of incomplete conversion may lead to large bias for these two methods; regardless, they are not readily available yet. Thus, a cost-effective and high-resolution approach is urgently required to facilitate the genome-wide screen studies for multiple samples.
Here, we present a hydroxymethylation and methylation sensitive tag sequencing (HMST-Seq) strategy, which involves selective glucosylation of 5-hmC residues, short-Sequence tag generation and high-throughput sequencing and provides a method for single-base resolution detection of both 5-hmC and 5-mC in MspI sites in the human genome. We tested this method by screening H9 human embryonic stem cells (hESCs) and their differentiated embroid bodies (EBs) and found broad distribution with variable abundance of 5-hmC as well as 5-mC. High levels of 5-hmC and, reciprocally, low levels of 5-mC can be found within regions around transcription binding sites, distal-regulatory elements and binding sites of transcription factors. Differential hydroxymethylation preferentially occurs in bivalent genes during cellular differentiation. Finally, HMST-Seq generated base-resolution maps of 5-hmC and 5-mC in MspI sites in the human genome. Our results support that hydroxymethylation can regulate key transcription regulators with bivalent marks through demethylation and affect cellular decisions on choosing active or inactive states of these genes upon cellular differentiation.
Results
Design and assessment of HMST-Seq
Previously, we developed a high-throughput tagging technology to detect genome-wide DNA methylation patterns by combining methylation-sensitive enzyme enrichment of unmethylated DNA tags and massively parallel tag-Sequencing technology.Citation13 We here took advantage of the differential enzymatic sensitivities of the isoschizomers MspI and HpaII and adapted our method to parallel 5-hmC/5-mC detection. HpaII cleaves only a completely unmodified site, any modification at either cytosine blocks the cleavage, while MspI recognizes and cleaves both 5-mC and 5-hmC, but not the newly discovered 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC).Citation7 Furthermore, β-glucosyltransferase (β-GT) can transfer a glucose to the hydroxyl group of 5-hmC and generate β-glucosyl-5-hydroxymethylcytosine (5-ghmC), which blocks MspI digestion.Citation14 Thus, either by combining β-GT treatment with MspI digestion or simply applying MspI/HpaII digestion, short sequence tags generated can be used for inferring hydroxymethylation or methylation status in around 1.8 million cytosine sites in the human genome (). As the MspI/HpaII recognition sites are distributed extensively in the human genome and 5-hmC preferentially exist in CG contextCitation12 in ESCs, a representative profiling of 5-hmC/5-mC can thus be achieved by our method, hereafter termed as “Hydroxymethylation and Methylation Sensitive Tag sequencing” (HMST-Seq). In theory, this method only requires 5.67 GB sequencing data (162 million single-end 35 bp-length reads) for parallel 5-hmC/5-mC detection of around 1.8 million CCGG sites to be sequenced in 30X sequencing depth in the human genome, thus massively decreasing the sequencing cost.
Figure 1. Schematic presentation of the HMST-Seq method. For (A) “C + mC” library, the genomic DNA was first glucosylated, and then digested with MspI. For (B) “C” library and (C) “C + mC + hmC” library, genomic DNA was directly digested with MspI or HpaII, respectively. After digestion, the DNA fragments from each of the three libraries were ligated to biotinylated linkers, fragmented by NlaIII cleavage and captured by streptavidin-conjugated beads. Then, the captured DNA fragments were ligated with linkers (N) containing a MmeI recognition site, and digested with MmeI that generates short sequence tags. Finally, the tag sequences were ligated with P7 adapters, amplified by PCR and sequenced.
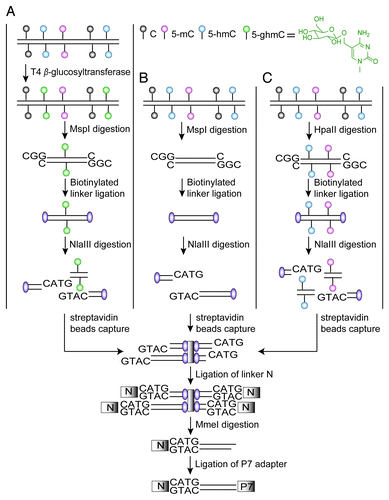
As the efficacy of glucosylation by T4 β-glucosyltransferase (β-GT) is a key step for identifying 5-hmC, we first examined the ability of glucosylation of 5-hmC residues by in vitro digestion of synthesized double-stranded oligonucleotides containing a single 5-hmC residue prior to Q-PCR assay. Meanwhile, we tested the digestion efficiency of MspI on CCGG, ChmCpGG and CmCpGG sites. We performed three replicates of experiments and confirmed the consistency of protecting effects exerted on 5-hmC sites by glucosylation as well as high efficiency of MspI digestion on ChmCpGG and CmCpGG sites (Fig. S1; Tables S1 and S2), which is in agreement with a report from Kinney SM et al.Citation14 In regard to a previous report on inhibition of MspI cleavage activity by hydroxymethylation of the CpG site,Citation15 our results probably benefited from high dose of MspI enzyme and prolonged time of digestion.
Then, we performed HMST-Seq using H9 hESCs and their differentiated EBs. Three different libraries were constructed for each cell line. Tags in the first library generated from MspI digestion were comprised of unmodified, methylated and hydroxymethylated cytosines, except for 5-fC and 5-caC (termed as “C + mC + hmC” library). If genomic DNA is first glucosylated, MspI digestion on 5-hmC will be blocked by 5-ghmC and leads to the second library with tags referring to only unmodified and methylated cytosines (termed as “C + mC” library). In addition, we applied HpaII digestion and generated the third library containing tags referring to only unmodified cytosines (termed as “C” library). Experimental replicates were performed to check for technical repeatability, which was well supported by high correlation coefficients (Pearson’s R2 = 0.87 on average) (Table S3).
We then used the average tag counts from two replicate libraries for further data normalization. Theoretically, the cytosine without any modification should stand for an invariant level within each library. However, random and systematic variation might occur during the library construction and sequencing, creating measurement bias for the tags. Thus, we adapted a previously published method global rank-invariant set normalization (GRSN)Citation16 to our data normalization among libraries. A rank-invariant set of sites were selected in an iterative manner as unmodified “C” tags and used to generate a robust average reference, and lowest algorithm was used to normalize tag counts among all libraries. The hydroxymethylation abundance of specific CCGG site can be determined as the ratio between tag counts of “C + mC + hmC” and “C + mC” libraries. In a similar way, ratio between tag counts of “C + mC” and “C” libraries can represent the methylation abundance. Furthermore, a statistics test based on Poisson distribution was performed between two libraries based on the normalized tag counts to identify significantly modified sites. The CCGG sites possessing significantly different tag counts between two libraries (sequencing depth > 10X, FDR < 0.05), meanwhile ratio of tag counts larger than 1, were determined as significantly modified sites with 5-hmC or 5-mC. As a result, 35,906 (3.28%) and 311,661 (28.47%) of the total CCGG sites in H9 cells were identified as significant 5-hmC and 5-mC, respectively. In EB cells, 21,913 (1.95%) and 353,159 (31.37%) of CCGG sites were significantly hydroxymethylated or methylated, respectively.
To evaluate accuracy of HMST-Seq, we generated profiles of DNA modification by hMeDIP-Seq and MeDIP-Seq technologies, which can be comparable to our profiles of modified cytosines (). The results showed generally similar enriched or depleted patterns of 5-hmC or 5-mC in genic regions generated by the two types of methods, despite of the existence of non-CG methylation and influence of GC content on antibody-based methods (Fig. S2). We also made a pair-wise comparison between methylation levels detected by whole genome bisulfate sequencing (WGBS) (downloaded from GEO with accession number GSM706059) and the methylation abundance deduced by HMST-Seq (Pearson’s R = 0.57) (Fig. S3A). As the WGBS approach cannot distinguish methylated cytosines with other modified cytosines (5-hmC, 5-fC and 5-caC), it is reasonable to compare the whole genome methylome with tag counts of “C” library, which inversely represents the total counts of modified cytosines. A better correlation therefore (Pearson’s R = -0.79) was obtained (Fig. S3B). Considering the culturing condition or passage number of the H9 cells might differ from ours, these results indicated reasonably acceptable correlation between our HMST-Seq and the WGBS data.
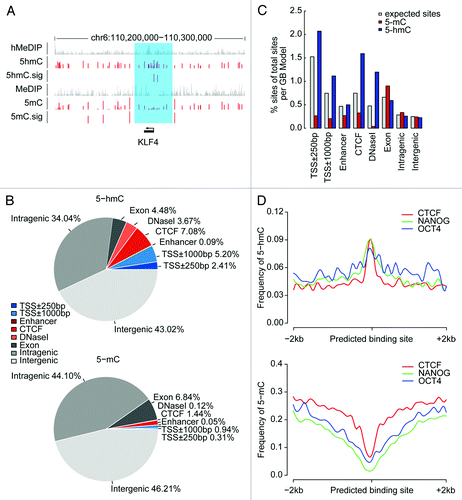
Figure 2. Genomic distribution of 5-hmC and 5-mC sites in H9 hESCs. (A) Snapshot of 5-hmC and 5-mC maps (red) compared with affinity-based 5-mC and 5-hmC maps (gray) near the KLF4 gene. For HMST-Seq, the vertical axis shows the abundance of methylation or hydroxymenthylation and limits are 0 to 3. 5-hmC.sig and 5-mC.sig sites represent significantly hydroxymethylated of methylated sites respectively. For hMeDIP and MeDIP sequencing, the vertical axis limits are 0 to 15. The sapphirine box shows the region of KLF4 and its promoter. (B) Overlap of 5-hmC and 5-mC with genomic regions. Genic features were extracted from the UCSC hg19 database. Regulatory elements (CTCF and DNase I) were download from previously published data generated by ChIP-Seq and DNase-Seq experiments and enhancer were download from VISTA Enhancer Browser database. Each 5-hmC or 5-mC site is counted once: the cytosines located in previous regions are excluded counterclockwise from TSS ± 250bp. Blue, promoter-proximal elements; Red, distal-regulatory elements; Gray, genic regions and intergenic regions. (C) Relative enrichment of modified sites within several genomic elements. The observed percentage of 5-mC (red) or 5-hmC (blue) counts within these genomic elements out of all modified CCGG sites in human genome and the expected percentage of CCGG counts (gray) within these genomic elements out of all CCGG counts in human genome are presented. All values of counts were normalized to the length of each region (per Gb). (D) Frequency of 5-hmC and 5-mC around protein-DNA interaction sites. The frequency of 5-hmC and 5-mC in each 100bp windows are displayed throughout regions of 2 kilobases (kb) upstream and downstream from predicted sites of protein-DNA interaction (CTCF, NANOG and OCT4), where 5-hmC is enriched but 5-mC is depleted.
Genomic distribution of 5-hmC or 5-mC in pluripotent cells
We next focused our analysis on the profile of H9 hESCs. A broad distribution of 5-hmC as well as 5-mC was observed, not only in gene bodies and gene proximal regions, but also in distal-regulatory elements including predicted enhancers, CTCF-binding sites, and DNase I hypersensitive sites (). In agreement with previous studies,Citation12 both 5-hmC and 5-mC tended to be biased to low observed/expected CpG (CpGo/e) context (Fig. S4). Furthermore, by comparing with genomic coverage of MspI sites, 5-hmC was relatively more enriched within regions around TSS and distal-regulatory elements than other genic regions. In contrast, the frequency of 5-mC was low in these regions (). Such inverse tendency with enrichment of 5-hmC and depletion of 5-mC was also clearly observed around the binding sites of CTCF protein and pluripotency factors NANOG and OCT4 (), which were observed previously.Citation17 Considering that 5-hmC can inhibit the binding of DNA methyltransferase DNMT1Citation18 and methyl-CpG-binding protein (MBP) MeCP2 that normally recognize 5-mC,Citation19 these results might support the notion that 5-hmC may regulate 5-mC levels at certain protein-DNA interaction sites.
As 5-hmC is derived from 5-mC,Citation20 we also looked at the correlation between modification levels of hydroxymethylation and methylation. However, we did not observe a linearly inverse relation between 5-hmC and 5-mC (Fig. S5), which might be due to the fact that 5-hmC can be further oxidized to 5-fC and 5-caC by TET enzymes.Citation6,Citation7
The overall bias of 5-hmC distribution in regions around transcription start sites (TSSs) might indicate a possible regulatory role of 5-hmC in gene expression, we therefore compared the 5-hmC or 5-mC level of sets of genes with varying expression levels measured by digital gene expression (DGE) (Fig. S6). The results indicated that lowly expressed genes exhibited both higher abundance and frequency of hydroxymethylation around TSSs than genes with intermediate or high expression. Similarly, both abundance and frequency of methylation around TSSs increased along with decreased gene expression, supporting the negative regulatory role of 5-mC within promoter regions.
Changes of 5-hmC or 5-mC of H9 ESCs upon cellular differentiation to EBs
We further examined the changes of the two types of DNA modifications during cellular differentiation. As indicated in the above results, we identified slightly more 5-mCs (31.37% vs. 28.47%) but less 5-hmCs (1.95% vs. 3.28%) in a global scale in EBs in comparison with H9s. To confirm this result, we further performed ultra-performance liquid chromatography/tandem mass spectrometry (UPLC–MS/MS). We obtained a similar trend of global 5-hmC changes (2.00% vs. 3.03% 5-hmCG in total CG sites). Meanwhile, as 5-hmCs preferentially occur at CpG sites, the percentage of total 5-hmC detected by HMST-Seq and UPLC–MS/MS can be largely comparable; the similar results we obtained from the two methods thus suggested for the feasibility of HMST-Seq. As the “CCGG” sites are preferentially located at CpG islands that are normally protected from methylation, the total number of 5-mCs detected by the two methods cannot be compared directly, but we also confirmed by UPLC–MS/MS that the total number of 5-mCs was slightly increased (3.07% vs. 2.77%) upon differentiation to EBs (Table S4). We also examined the digital gene expression data (DGE) for the DNA methyltransferase (DNMT) and Tet gene families. We found only DNMT3A and TET2 genes showed slightly varied expression levels with similar tendency corresponding to the small changes in 5-mC and 5-hmC (Table S5). However, whether DNMT3A and TET2 were the main modifiers responsible for the dynamic changes of two types of DNA modifications during cellular differentiation is currently not clear. On the other hand, general patterns of 5-hmC and 5-mC distribution in different genomic elements were similar between H9 and EB cells (Fig. S7).
We then performed pair-wise comparison for hydroxymethylation and methylation of CCGG sites. By sliding through the genome, differentially modified regions containing at least five CCGG sites were identified by a criterion of p value of Wilcoxon rank sum test less than 0.05, which resulted in 1,033 differentially methylated regions (DMRs) and 4,007 differentially hydroxymethylated regions (DhMRs), both distributed extensively in the genome (). In comparison with hESCs, a majority of the DMRs (889, 86.06%) were hypomethylated, while more than half of the DhMRs (2,196, 54.80%) were hyper-hydroxymethylated in EBs. Interestingly, only a small portion (129) of the differential regions was overlapped, among which 103 (79.84%) were hypomethylated but hyper-hydroxymethylated in EBs, supporting the role of 5-hmC as an intermediate in DNA demethylation. To sum up, the above results indicated decreased methylation in dynamic genomic regions in EBs that underwent demethylation through TET oxidation mechanism, despite the overall similarity of DNA modification patterns.
Figure 3. Changes of DNA hydroxymethylation and methylation upon cellular differentiation. (A) Distribution of differentially hydroxymethylated regions (DhMRs) and differentially methylated regions (DMRs) within genomic elements. The DhMR or DMR located in two different elements are attributed to the element that overlaps with a bigger proportion of the DhMR or DMR and counted only once. Blue, promoter-proximal elements; red, distal-regulatory elements; gray, genic regions and intergenic regions. (B). Changes of DNA hydroxymethylation upon cellular differentiation in relation with gene expression. The hyper- (blue) or hypo-hydroxymethylated (pink) genes upon cellular differentiation were cross-matched with genes enriched with histone H3 trimethylation (categorized as “K4 only,” “K27 only” and bivalently “K4 + K27”) or targeted by PRC components of EZH2 (PRC2) and RING1B (PRC1) at their promoters in H9 ESCs. The fraction of the matched genes in all DhMR-associated genes and the fraction of all genes (gray) with these promoter markers are presented. Number of genes in each category is indicated and * represents Chi-square test p value < 0.05. (C) Boxplots of methylation, hydroxymethylation and gene expression levels of genes containing promoter bivalent marks in the top four Gene Ontology terms enriched with DhMR-associated genes. The vertical axis shows the abundance of methylation (left) and hydroxymethylation (middle) in the promoters and TPM/100 of gene expression levels (right).
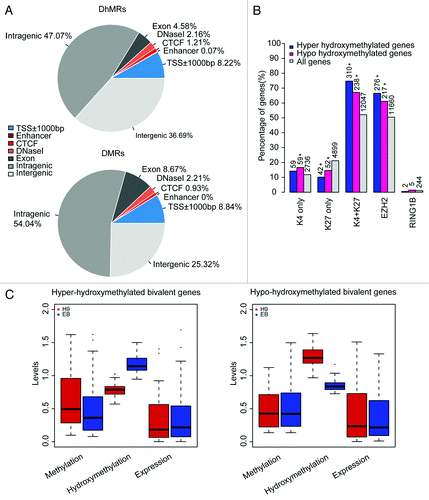
Furthermore, we found 771 genes containing DhMRs and 116 genes containing DMRs in their promoters (Table S6). As our results agreed with a previous study indicating that 5-hmC are enriched at bivalent genes with both H3K4me3 and H3K27me3 marks (Fig. S8),Citation9 and TET1 depletion impairs PRC2 binding to these targets,Citation5,Citation21 we then cross-matched the DhMR- or DMR-associated genes with bivalent genes and EZH2 target genes in ESCs. We found that the proportion of DhMRs-associated genes containing bivalent marks between hESCs and EBs out of all DhMRs-associated genes was significantly higher than the proportion of bivalent genes in all genes (Chi-square test p value < 0.05). DhMRs were also preferentially found in promoters of EZH2 target genes, but the proportion of H3K27me3-only genes containing DhMRs out of all H3K27me3-only genes was significantly lower than the proportion of H3K27me3-only genes out of all genes (). These results indicated hydroxymethylation changes preferentially occurred in bivalent genes during cellular differentiation.
In order to further explore the role of differential hydroxymethylation on cellular differentiation, Gene Ontology (GO) analyses were performed on the 648 DhMR-associated genes (Table S7). Top four terms in relation with transcription regulation (GO:0030528, GO:0043565, GO:0006350 and GO:0045449) were most significantly enriched, which were comprised of 126 genes in total. Out of these 126 DhMR-associated genes, 79 ones were bivalently marked in H9 cells, also suggesting a significantly (Chi-square test p value = 0.023) high frequency (62.70%) of hydroxymethylation alterations in bivalent genes than the odds of bivalent genes in all genes (52.16%). Furthermore, we observed that increased hydroxymethylation generally endued decreased methylation in these DhMRs and thus activated gene expression of these bivalent genes in EBs and vice versa (). These results supported that hydroxymethylation might play an important role in regulating key transcription regulators during cellular differentiation, especially in affecting methylation status of bivalent genes; thus, they may affect cellular decisions on choosing active or inactive state of these genes upon differentiation to EBs.
Discussion
Recent studies show that 5-hmC is widespread in the mammalian genome and has been implicated in the process of DNA demethylation and binding inhibition of certain methyl-CpG-binding proteins.Citation6,Citation19 Genome-wide single-base resolution map of 5-hmC will enable the comprehensive appreciation of the biological importance of hydroxymethylation. Most recently, oxBS-SeqCitation11 and TAB-SeqCitation12 could locate 5-hmC in the genome and determine the relative abundance at each modified site; however, generating a massive amount of data in the process. Here we described a cost-effective and selective method to detect both 5-hmC and 5-mC in MspI sites in the human genome. Using digestion of in vitro synthesized double-stranded oligonucleotides, we demonstrated the efficacy of glucosylation of 5-hmC residues by T4 β-GT: after glucosylation, 5-hmC can be protected from MspI digestion. The high correlation coefficients between experimental replicates also well supported technical repeatability. In this method, we constructed three different libraries (“C,” “C + mC” and “C + mC + hmC”) for each sample. Theoretically, cytosine without any modification (“C” library) should stand for an invariant level within each library, and we adapted a previously published method, global rank-invariant set normalization (GRSN),Citation16 to normalize our data among libraries. After normalization, hydroxymethylation abundance of specific CCGG sites can be determined as the ratio between tag counts of “C + mC + hmC” and “C + mC” libraries. In a similar way, ratio between tag counts of “C + mC” and “C” libraries can represent the methylation abundance. Besides, hydroxymethylated or methylated sites were determined based on significantly different tag counts between two libraries. All together, we can also locate 5-hmC and 5-mC, and determine the relative abundance of hydroxymethylation and methylation.
We applied this method to H9 cells to generate representative single-base resolution maps of 5-hmC and 5-mC. We showed that, generally, similar patterns of these maps compared with the methylation and hydroxymethylation maps generated by MeDIP-Seq and hMeDIP-Seq technologies. A widespread distribution of 5-hmC as well as 5-mC was observed, not only in gene bodies and gene proximal regions, but also in distal-regulatory elements, including predicted enhancers, CTCF-binding sites, and DNase I hypersensitive sites. Both 5-hmC and 5-mC tended to be biased to low observed/expected CpG (CpGo/e) context, which was consistent with a previous study.Citation12 Importantly, enriched 5-hmC and reciprocally lacking 5-mC regions can be found within regions around transcription binding sites, distal-regulatory elements and binding sites of transcription factors. Previous studies suggested that 5-hmC can inhibit the binding of DNA methyltransferase DNMT1Citation18 and methyl-CpG-binding protein (MBP) MeCP2 that normally recognize 5-mC.Citation19 These results might support the notion that 5-hmC is derived from 5-mCCitation20 and regulates 5-mC levels at certain protein-DNA interaction sites.
In order to explore possible functional roles of 5-hmC or 5-mC upon cellular differentiation, we also examined these two types of DNA modifications on EBs. Though globally similar modifications were observed between these two cell lines, we identified 1,033 DMRs and 4,007 DhMRs, both of which distributed extensively in the genome. A majority (79.84%) of the intersection of DMRs and DhMRs were hypomethylated but hyper-hydroxymethylated in EBs, supporting the role of 5-hmC as an intermediate in DNA demethylation. We observed that 5-hmC is enriched at bivalent genes with both H3K4me3 and H3K27me3 marks. This observation is consistent with a previous report.Citation9 More interestingly, we also found that hydroxymethylation changes preferentially occurred in bivalent genes during cellular differentiation. Gene ontology (GO) analyses based on DhMR-associated genes showed that the top four terms in relation with transcription regulation were most significantly enriched, and genes in the top four enriched terms preferentially contained bivalent markers. Furthermore, we observed that increased hydroxymethylation generally endued decreased methylation in these DhMRs and thus activated gene expression of these bivalent genes in EBs and vice versa. These results supported that hydroxymethylation might play an important role in regulating key transcription regulators during cellular differentiation, especially in affecting methylation status of bivalent genes, and thus may affect cellular decisions on choosing active or inactive state of these genes upon differentiation to EBs.
In summary, we have developed a cost-effective and selective method to examine both hydroxymethylation and methylation at base-resolution, thus making it possible to study the genomic loci that are targeted for demethylation during cellular differentiation. Future application of this technology would enable us to uncover the status of methylation and hydroxymethylation in dynamic biological process and disease development in multiple biological samples.
Materials and Methods
Culturing of H9 human embryonic stem cells (hESCs) and in vitro differentiation into embryoid bodies (EBs)
For maintenance of H9 hESCs, MEF feeder layer was obtained from CF-1 mouse embryos and cultured in MEF medium, which is composed of 90% DMEM (GIBCO 12430) supplemented with 10% FBS (Biochrom S0615). Before freezing cells, MEFs were cultured for 3h at 37°C in mitomycin C (Sigma M0503) medium that is composed of MEF medium supplemented with 10 ug/ml mitomycin C. For culturing H9 ES cells, MEF-conditioned medium was produced by conditioning MEFs for at least 24 h in the medium composed of DMEM/F12 (GIBCO 11330) supplemented with 20% knockout serum replacement (GIBCO 10828), 2 mM L-glutamine (GIBCO 25030), 2 mM nonessential amino acids (GIBCO 11140), 0.1 mM 2-mercaptoethanol (GIBCO 21985–023), and 4 ng/ml recombinant human fibroblast growth factor-basic (bFGF; GIBCO 13256–029).
Prior to differentiation, hESCs were cultured on MEFs that were prepared 24 h in advance, transferred from MEFs onto Matrigel (Invitrogen, 354234), and cultured in MEF-conditioned medium. Clumps of hESCs (~5 million cells per plate) were then plated in suspension onto ultra-low adhesion dishes (Corning) in DMEM (Gibco 12430) containing 20% FBS (Biochrom S0615), which promoted the formation of embryoid bodies (EBs).
In vitro analysis of MspI and HpaII sensitivity to glucosylation of 5-hmC
The efficiency of glucosylation for hydoxymethylcytosines and enzyme digestion was confirmed using EpiMark 5-hmC and 5-mC Analysis Kit (NEB). Double stranded oligonucleotides containing either a single 5-hmC, 5-mC or C residue within the MspI recognition site on both strands and all primers are included in the kit. Briefly, 0.5 ng of each type of oligonucleotides were mixed with 600 ng genomic DNA by addition of 12.4 μl of UDP-Glucose, 31 μl of NEBuffer 4 and nuclease-free water to a final volume 305.5 μl. Each reaction mixture was then split into three tubes (98.5 μl each), in which one was supplemented with 1.5 μl T4-glucosyltransferase (β-GT) and incubated for 18 h at 37°C. Negative control for β-GT treatment was performed by the same treatment without any oligonucleotides supplemented. The other two tubes were supplemented with 1.5 μl of water instead of β-GT, in which one was used as “input” tube without further digestion. Digestion with 1 μl (100 units) of MspI enzyme was performed for 16 h at 37°C. All incubation was performed on a thermal cycler with the lid heated at 47°C. Finally, Q-PCR assay was performed to test the efficiency of glucosylation and MspI digestion using primers complemented with control DNA.
Hydroxymethylation and methylation sensitive tag sequencing (HMST-Seq)
Genomic DNA was extracted from H9 hESCs and EBs using QIAamp DNA Blood Mini Kit (QIAGEN). HSMT-Seq library construction followed the same protocol we reported previously,Citation13 except that three independent libraries with initially different enzyme digestion were constructed for each sample, which were termed “C” library, “C + mC” library and “C + mC + hmC,” respectively. For “C + mC” library construction, an aliquot of genomic DNA from each sample was first glucosylated by incubating 1 µg of DNA substrates with 3 μl (30 units) of T4 β-glucosyltransferase (β-GT) (NEB) for 16 h at 37°C in a total 100 μl reaction containing 1X NEBuffer 4 supplemented with 80 µM UDP-Glc. After glucosylation, the aliquot of glucosylated DNA was then digested with 300 units of MspI (NEB). For the “C” or “C + mC + hmC” libraries, the aliquots of DNA without glucosylation were directly digested with 300 units of HpaII or MspI (NEB), respectively. The enzyme digestion reactions were performed at 37°C for 16 h in a 50 μl of either NEBuffer 1 (HpaII) or NEBuffer 4 (MspI). After digestion, the digested aliquots of DNA from all three libraries were ligated with biotinylated linker, fragmented by NlaIII, captured by streptavidin-conjugated beads, digested with MmeI to generate short sequence tags (16–17bp), and ligated with sequencing linkers and amplified by PCR. To avoid the bias from PCR, amplification was stopped after 12 cycles. Then, the purified tags were sequenced using Illumina HiSeq analyzer according to the manufacturer’s instructions.
Data analysis for HMST-Seq
In silico analysis indicated that tags can be extracted for 1.79 million MspI/HpaII sites out of 3.2 million total sites in the human genome. We applied our previous strategy of sequence matching based on virtual libraryCitation13 for high confidence genome wide mapping of the 16–17 bp tags generated from sequencing reads after base calling, adaptor removal and low-quality reads filtering. The virtual library was constructed as follows: The human genome sequence (hg19) was in silico digested by MspI or NlaIII, we defined the DNA sequences between the nearest NlaIII sites around each MspI site in both directions as the virtual library reference for mapping. Using the open-source programming language Perl, all the tags were mapped to the virtual library with no more than one mismatch, and the unambiguous mapped tags were used for further analysis.
To normalize the data from three libraries, we adapted a previously published method (global rank-invariant set normalization, GRSN)Citation16 and took a rank-invariant set of sites selected in an iterative manner as “C” tags, which were further used to generate a robust average reference that is used to normalize tag counts among all libraries. Based on the normalized tag counts, the modification abundance of specific CCGG sites were determined as the ratio between tag counts of two libraries. For instance, hydroxymethylation level can be defined as ratio of “C + mC + hmC” and “C + mC” tags. Furthermore, a statistics test based on Poisson distribution was performed.Citation22 The CCGG sites with significantly different tag counts (sequencing depth > 10X, FDR < 0.05), meanwhile ratio of tags between two libraries larger than 1, were determined as significantly modified sites. For a given genomic interval, modification frequency of CCGG sites was defined as the ratio of significant modified sites within all CCGG sites. Differentially hydroxymethylated or methylated regions (DhMR or DMR) were defined in the following steps: (A) the first 5 CCGG sites that contain at least 4 CCGG sites with the same changing trend and Wilcoxon rank sum test p value < 0.05 were taken as the seed sites of a candidate DMR; then, (B) a 3′ downstream adjacent CCGG with the same changing trend was incorporated with this candidate DMR. Up to 2,000 bp inter-distance was allowed between the two adjacent CCGGs, Wilcoxon rank sum test was performed in the incorporated region, (C) these steps were repeated until Wilcoxon rank sum test p value > = 0.05 and (D) the incorporated region were defined as a DhMR or DMR.
Library construction and data analysis of MeDIP-Seq and hMeDIP-Seq
Prior to immunoprecipitation, 5 µg of genomic DNA was sonicated to a mean size of 200 bp fragments, followed with end repair, < A > base addition and adaptor ligation steps according to Illumina Paired-End protocol.
MeDIP-Seq and hMeDIP-Seq libraries were constructed on 500 ng of adaptor ligated DNA using the Magnetic Methylated DNA Immunoprecipitation kit (Diagenode) and hMeDIP 10rxns kit (Active Motif) following the manufacturer’s instructions, respectively. After immunoprecipitation, the captured products were then eluted and purified on a single ZYMO DNA Clean and Concentrator-5 column following the manufacturer’s instructions. Finally, PCR amplification was performed using platinum pfx DNA polymerase (Invitrogen) with a thermal cycling program of 94°C 2 min, 12 cycles of 94°C 15s, 60°C 30s, 72°C 30 then prolonging with 5min at 72°C and products could be hold at 12°C. Amplification quality and quantity were evaluated by 2100 Analyzer and QPCR. The libraries were sequenced using Illumina HiSeq analyzer according to the manufacturer’s instructions. After base calling, low-quality reads were omitted and the clean reads were aligned to the UCSC human reference genome (hg19) using SOAP2 (Version 2.21).Citation23 Mismatches of no more than two bases were allowed in the alignment.
UPLC–MS/MS analysis of global DNA methylation and hydroxymethylation
Genomic DNA (0.2 μg) extracted from H9 and EB was digested with 1U DNase I, 2U Alkaline Phosphatase, Calf Intestinal (CIP) and 0.005U snake venom phosphodiesterase I at 37°C for 24 h. A microcon centrifugal filter device with a 3,000 D cutoff membrane was used to remove protein from the digested DNA samples by centrifuging at 12,000 rpm for 60 min. The mobile phase consisted of 0.1% formic acid (solvent A) and methanol containing 0.1% formic acid (solvent B). The ñow rate was set to 500 μl/min. Enzymatically digested DNA samples (5 μl each) were injected for UFLC-MS/MS analysis with the LC gradient as follows: 0.0–4.0 min, 0 to 50% of solvent B; 4.0–6.0 min, 50% solvent B; 6.0–6.1 min, 50% to 5% of solvent B and 6.1–15 min, 5% of solvent B. The separated analytes were detected using a 5500 Qtrap linear ion trap quadruple mass spectrometer with Analyst software (Version 1.5) equipped with a Turbo V ion source operated in the ESI mode (AB Sciex). The Source and Gas were set as follows: gas 1, nitrogen (45 psi); gas 2 nitrogen (40 psi); ion spray voltage, 5500 V; ion source temperature, 400°C and curtain gas, nitrogen (30 psi). The mass spectrometer was operated in the multi-reaction monitoring mode (MRM).
Library construction and data analysis of digital gene expression (DGE)
Four μg of the total RNA isolated from each sample was used for DGE library construction as previously described.Citation24 After base calling, low-quality reads were omitted. The clean reads were aligned to the UCSC human reference genome (hg19) using SOAP2 (Version 2.21).Citation23 Mismatches of no more than two bases were allowed in the alignment. Differentially expressed genes were identified according to a previously published method,Citation22 p values were adjusted by false discovery rate (FDR) for multiple tests. A threshold of FDR < 0.05 and fold change > 2 was applied.
Public data used
We downloaded the human reference genome (hg19) annotation data from UCSC database to identify the genomic elements. The whole genome methylome data of H9 was downloaded from GEO (GEO accession number GSM706059). The regulatory elements DNase I were downloaded from NIH Roadmap Epigenomics Project Data generated by DNase-Seq experiments (GEO accession number GSM878612). The enhancers (hg19) were downloaded from VISTA Enhancer Browser database. Previously published data on the protein-DNA interaction sites for CTCF, NANOG and OCT4 were downloaded.Citation25 ChIP-Seq data of histone H3 trimethylation (GEO accession number GSM616128 and GSM706066) and target regions of EZH2 (PRC2) and RING1B (PRC1) (GEO accession number GSM327665 and GSM327666)Citation26 were re-analyzed using RSEG with default parameters.Citation27
Accession number
Sequencing data have been deposited to GEO with accession number GSE41071.
| Abbreviations: | ||
| 5-mC | = | 5-methylcytosine |
| 5-hmC | = | 5-hydroxymethylcytosine |
| HMST-Seq | = | hydroxymethylation and methylation sensitive Tag sequencing |
| 5-ghmC | = | β-glucosyl-5-hydroxymethylcytosine |
| CpGo/e | = | Observed/Expected CpG |
| DMRs | = | differentially methylated regions |
| DhMRs | = | differentially hydroxymethylated regions |
Additional material
Download Zip (636.8 KB)Acknowledgments
We would like to thank Laurie Goodman and Songgang Li for helpful discussion and critical reading of the manuscript. This work was supported by Science and Technology Department, BGI-Shenzhen through grants from the Chinese 863 program (NO. 2012AA02A201) and Guangdong Innovative Research Team Program (NO. 2009010016).
Author Contributions
F. Gao conceived the project and designed the experiments with help from J.W. Wang and Y.D. Xia; J.W. Wang, H.J. Luo, Z.W. Gao, X. Han, J.Y. Zhang, X.J. Huang and Y. Yao performed the experiments; Y.D. Xia performed data analysis with help from H.L. Lu and N. Yi; B.J. Zhou, Z.L. Lin and B. Wen performed the UPLC–MS/MS analysis; F. Gao interpreted data with help from X.Q. Zhang, H.M. Yang and J. Wang; F. Gao wrote the manuscript help from Y.D. Xia.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009; 8:1698 - 710; http://dx.doi.org/10.4161/cc.8.11.8580; PMID: 19411852
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324:930 - 5; http://dx.doi.org/10.1126/science.1170116; PMID: 19372391
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009; 324:929 - 30; http://dx.doi.org/10.1126/science.1169786; PMID: 19372393
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol 2011; 29:68 - 72; http://dx.doi.org/10.1038/nbt.1732; PMID: 21151123
- Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, Zhao K, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev 2011; 25:679 - 84; http://dx.doi.org/10.1101/gad.2036011; PMID: 21460036
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011; 333:1303 - 7; http://dx.doi.org/10.1126/science.1210944; PMID: 21817016
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011; 333:1300 - 3; http://dx.doi.org/10.1126/science.1210597; PMID: 21778364
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011; 473:398 - 402; http://dx.doi.org/10.1038/nature10008; PMID: 21460836
- Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 2011; 473:394 - 7; http://dx.doi.org/10.1038/nature10102; PMID: 21552279
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, et al. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun 2011; 2:241; http://dx.doi.org/10.1038/ncomms1240; PMID: 21407207
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012; 336:934 - 7; http://dx.doi.org/10.1126/science.1220671; PMID: 22539555
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012; 149:1368 - 80; http://dx.doi.org/10.1016/j.cell.2012.04.027; PMID: 22608086
- Li J, Gao F, Li N, Li S, Yin G, Tian G, et al. An improved method for genome wide DNA methylation profiling correlated to transcription and genomic instability in two breast cancer cell lines. BMC Genomics 2009; 10:223; http://dx.doi.org/10.1186/1471-2164-10-223; PMID: 19439076
- Kinney SM, Chin HG, Vaisvila R, Bitinaite J, Zheng Y, Estève PO, et al. Tissue-specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genomes. J Biol Chem 2011; 286:24685 - 93; http://dx.doi.org/10.1074/jbc.M110.217083; PMID: 21610077
- Ichiyanagi K. Inhibition of MspI cleavage activity by hydroxymethylation of the CpG site: a concern for DNA modification studies using restriction endonucleases. Epigenetics 2012; 7:131 - 6; http://dx.doi.org/10.4161/epi.7.2.18909; PMID: 22395461
- Pelz CR, Kulesz-Martin M, Bagby G, Sears RC. Global rank-invariant set normalization (GRSN) to reduce systematic distortions in microarray data. BMC Bioinformatics 2008; 9:520; http://dx.doi.org/10.1186/1471-2105-9-520; PMID: 19055840
- Kunarso G, Chia NY, Jeyakani J, Hwang C, Lu X, Chan YS, et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 2010; 42:631 - 4; http://dx.doi.org/10.1038/ng.600; PMID: 20526341
- Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res 2007; 67:946 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-06-3123; PMID: 17283125
- Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res 2004; 32:4100 - 8; http://dx.doi.org/10.1093/nar/gkh739; PMID: 15302911
- Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell 2011; 146:866 - 72; http://dx.doi.org/10.1016/j.cell.2011.08.042; PMID: 21925312
- Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011; 473:389 - 93; http://dx.doi.org/10.1038/nature09934; PMID: 21451524
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res 1997; 7:986 - 95; PMID: 9331369
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 2009; 25:1966 - 7; http://dx.doi.org/10.1093/bioinformatics/btp336; PMID: 19497933
- Zhou L, Chen J, Li Z, Li X, Hu X, Huang Y, et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS One 2010; 5:e15224; http://dx.doi.org/10.1371/journal.pone.0015224; PMID: 21253009
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005; 122:947 - 56; http://dx.doi.org/10.1016/j.cell.2005.08.020; PMID: 16153702
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 2008; 4:e1000242; http://dx.doi.org/10.1371/journal.pgen.1000242; PMID: 18974828
- Song Q, Smith AD. Identifying dispersed epigenomic domains from ChIP-Seq data. Bioinformatics 2011; 27:870 - 1; http://dx.doi.org/10.1093/bioinformatics/btr030; PMID: 21325299