Abstract
Marek’s disease (MD) is characterized as a T cell lymphoma induced by a cell-associated α-herpesvirus, Marek’s disease virus type 1 (MDV1). As with many viral infectious diseases, DNA methylation variations were observed in the progression of MD; these variations are thought to play an important role in host-virus interactions. We observed that DNA methyltransferase 3a (DNMT3a) and 3b (DNMT3b) were differentially expressed in chicken MD-resistant line 63 and MD-susceptible line 72 at 21 d after MDV infection. To better understand the role of methylation variation induced by MDV infection in both chicken lines, we mapped the genome-wide DNA methylation profiles in each line using Methyl-MAPS (methylation mapping analysis by paired-end sequencing). Collectively, the data sets collected in this study provide a more comprehensive picture of the chicken methylome. Overall, methylation levels were reduced in chickens from the resistant line 63 after MDV infection. We identified 11,512 infection-induced differential methylation regions (iDMRs). The number of iDMRs was larger in line 72 than in line 63, and most of iDMRs found in line 63 were overlapped with the iDMRs found in line 72. We further showed that in vitro methylation levels were associated with MDV replication, and found that MDV propagation in the infected cells was restricted by pharmacological inhibition of DNA methylation. Our results suggest that DNA methylation in the host may be associated with disease resistance or susceptibility. The methylation variations induced by viral infection may consequentially change the host transcriptome and result in diverse disease outcomes.
Introduction
Marek’s disease (MD) induced by Marek’s disease virus (MDV) is a T cell lymphoma in chickens and other birds. The disease is characterized by infiltration of proliferating lymphoid cells into many tissuesCitation1 and is also a unique natural model for lymphomas that overexpress Hodgkin’s antigen (CD30) in humans.Citation2 Although vaccines against MD have been developed, vaccination efforts have driven the virus to greater virulence. MDV infection is divided into four different phases. The entry of virus to target cells initiates early cytolytic infection from 3–7 days post infection (DPI). Latent infection starts at 7–8 DPI and primarily occurs at activated CD4+ T cells. Late lytic infection is reactivated from latency around 2–3 weeks post infection in susceptible chickens, and then switches to the transformation stage with observed lymphoid tumors in chickens.Citation3 Thus, the late cytolytic stage is a critical step for MD progression and disease outcome. As with many viruses, MDV infection induces changes in DNA methylation patterns in the hosts and virus itself that contribute to MD progression by activating or silencing genes crucial for MD immunity.Citation4-Citation6
In somatic cells, DNA methylation usually occurs at CG dinucleotides (CpG sites), and is catalyzed by the three DNA methyltransferases, including DNMT1 for maintaining methylation and the de novo methyltransferases DNMT3a and DNMT3b. Inhibition of DNMTs contributes to DNA demethylation through a passive mechanism.Citation7 Methylation is associated with the repression of transcription and is essential for key biological processes, including development, X-chromosome inactivation, imprinting and tissue specific gene expression.Citation8 Tissue-specific variations in DNA methylation patterns have been observed. In addition, abnormal DNA methylation contributes to disease, including cancer and infectious disease.Citation5,Citation7,Citation9 In a normal vertebrate genome, the genome is primarily heavily methylated except for CpG dense promoters. Methylome analysis in a variety of cancer cells has shown that DNA methylation levels decrease genome-wide, including hypomethylation in repetitive regions, while some tumor suppressor genes are observed to become hypermethylated.Citation10 DNA methylation alterations have been observed in host-virus interactions, such as those identified in Epstein-Barr virus (EBV) transformed lymphoblastoid cell lines.Citation11 Proper maintenance of DNA methylation likely plays a vital role to prevent tumorigenesis and disease progression.
To explore the role of DNA methylation in MD immunology, two inbred chicken lines were used. Most birds in line 72 develop tumors after MDV infection, while line 63 is resistant to the disease. Both lines have the same major histocompatibility complex (MHC) haplotype but show significantly different MD incidences after MDV infection, which allows us to examine mechanisms of MD resistance. DNMTs were found to be differentially regulated in line 63 and 72 after MDV infection, suggesting that promoter methylation alterations are triggered by the infection.Citation6 DNA methylation was lost at active promoters in MDV carrying T cell lines and MD tumors, and was present in some CpG regions in MDV genome.Citation6 However, conclusions from these studies were limited since they focused on methylation variations at specific loci. Genome-wide alterations have not yet been proved.
The emergence of next generation sequencing (NGS) provides a unique opportunity to understand global methylation variation following MDV infection in MD resistant and susceptible chickens. Since DNA methylation cannot be reliably directly detected, several approaches are available for genome-wide methylation analysis, including antibody affinity enrichment of methylated DNA, sodium bisulfite conversion and enzymatic digestion.Citation12 To study MDV induced methylation fluctuation in MD resistant and susceptible chickens, we adopted a recently developed method called Methyl-MAPS (methylation mapping analysis by paired-end sequencing).Citation13 This method yields high-resolution information with no bias toward the CpG rich regions: both single-copy and repetitive regions are directly probed. Our previous studies revealed that DNMTs were differentially expressed in the line 63 and line 72 chickens,Citation14 especially at 21 DPI, when the virus was reactivated from the latent stage.Citation6,Citation15,Citation16 The results point to a profound distinction between epigenetic profiles and expression of selected genes in line 63 and 72 chickens. The interesting finding that many of these differences, particularly in response to MDV challenge, affect genes of the immune system or tumor progression and related pathways provides a strong rationale for pursuit of our hypothesis that epigenetic signatures are associated with resistance or susceptibility of MDV induced tumorigenesis. In this study, global methylation levels were found to differ between non-infected 63 and 72 lines, and were reduced after MDV infection in line 63. Analysis of differential methylation regions induced by MDV infection (iDMRs) suggests that DNA methylation likely regulates distinct pathways in resistant and susceptible chickens. We further demonstrate that drugs that inhibit DNA methylation repressed the spread of MDV in vitro. Collectively, DNA methylation is an important regulator of MD resistance and susceptibility, and these findings improve our understanding of the role of DNA methylation in infectious diseases and viral-infection induced tumors.
Results
Expression of DNMTs in MDV-infected and non-infected chickens
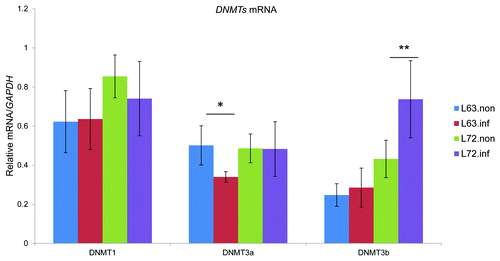
To test the influence of MDV infection on DNMT expression, the mRNA levels of each of the three DNA methyltransferases were examined by qPCR in the thymus of lines 63 and 72 at 21 DPI as shown in . While transcriptional levels of DNMT1 were ~20% higher in line 72 compared with line 63 (p > 0.05), DNMT1 expression was not significantly altered in either line after MDV exposure. DNMT3a mRNA was similar in non-infected line 63 and line 72. However, its expression was repressed by 30% in infected line 63 chickens (p < 0.05). The expression of DNMT3b was 2 fold higher in line 72 than in line 63 before MDV infection (p > 0.05), and its transcription was further activated in line 72, by a ~50% increase after MDV infection (p > 0.01).
Figure 1. Quantification of chicken DNMT expression. Expression of DNMT1, DNMT3a and DNMT3b was measured by qRT-PCR in thymus samples from non-infected and infected line 63 and line 72 chickens, and normalized to GAPDH. The quantitative results are represented as mean ± SEM (n = 4). A single asterisk (p values < 0.05) and double asterisks (p values < 0.01) indicate the transcription level in the specific group was significantly different when compared with the adjoined group. L72.inf: infected line 72; L72.non.inf: non-infected line 72; L63.non.inf: non-infected line 63; L63.inf: infected line 63.
DNA methylation in MD-resistant and -susceptible chickens
To understand the biological consequence of DNMT3a and DNMT3b expression variations, DNA methylation global levels were measured using 5-mC dot blots and methylation was mapped genome-wide using Methyl-MAPS in MDV-infected and non-infected line 63 and line 72. Prior to infection, the average methylation level was higher in line 72 (0.44) than in line 63 (0.39) (). In agreement, the distribution of methylation levels in the sequencing data were shifted downwards in line 63. To explore how methylation was altered among each of the four samples, we categorized the CpG sites as methylated (methylation level greater than 75%), intermediately methylated (25–75%), and unmethylated (< 25%). Based on this criterion, the initial methylation differences were attributed to more methylated CpGs in line 72 and more moderately methylated CpGs in line 63 (). We also analyzed the methylation difference according to their distribution in the genome. Generally, methylation levels in CpG islands (CGIs) were similar in both lines, 0.16 in line 63 and 0.13 in line 72, before infection. Meta-gene analysis of the methylation patterns across gene regions is plotted in .
Figure 2. (A) Boxplots of methylation levels for the detected CpGs. The y-axis was the methylation levels for all CpG sites calculated as the number of reads from methylated (RE) library divided by the number of reads from both methylated and unmethylated (McrBC) libraries. L72.inf, infected line 72; L72.non, non-infected line 72; L63.non, non-infected line 63; L63.inf, infected line 63. p values (B) Quantification of 5-mC content by anti-5-mC dot blot. The 5-mC content was measured in DNA from thymus using dot blot, shown as (mean ± STD, n = 4). A single asterisk (p values < 0.05) indicated the 5-mC contents in the specific group was significantly different when compared with other groups. L72.inf, infected line 72; L72.non, non-infected line 72; L63.non, non-infected line 63; L63.inf, infected line 63.
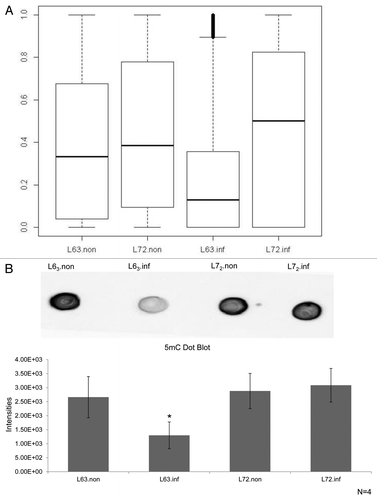
Figure 3. The classification of CpG based on the methylation levels. The CpGs were divided into 3 categories based on the methylation levels greater than 75% (Methy), 25–75% (Inter) and < 25% (Unmethyl). The number on the top of each bar represented the number of analyzed CpGs, and the percentage of CpGs in each category was labeled in the bar. L72.inf: infected line 72; L72.non: non-infected line 72; L63.non: non-infected line 63; L63.inf: infected line 63.
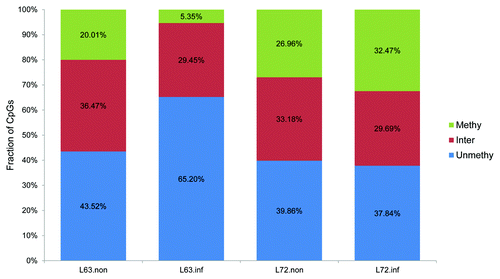
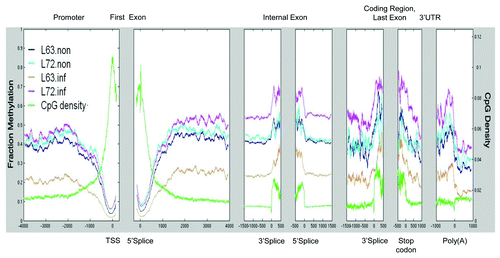
Figure 4. CpG distribution and DNA methylation pattern in chicken genes. Average CpG density and methylated CpG density were plotted across all genes from the promoter to the end of annotated 3′ UTRs.
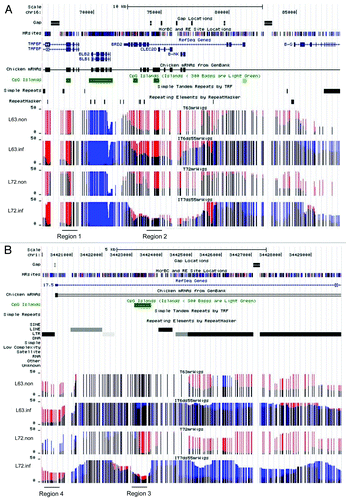
In addition to global changes, we found methylation differences at a variety of loci in the two lines. For example, differential methylation was identified in MHCII (major histocompatibility complex II) (). MHCII is one of the determinants for MD resistance, and its haplotype is the same between line 63 and line 72. Prior to MDV infection, region 1 was less methylated in line 72 than line 63, and methylation levels were similar in region 2. In repetitive DNA regions, the average methylation levels were 0.42 in both lines; however, we found methylation variation at individual loci. For example, the long-terminal repeat (LTR) (region 3, ) were highly methylated in uninfected line 63, and unmethylated in uninfected line 72. The CGI (region 4, ) near the LTR was highly methylated in line 72, and showed intermediate methylation in line 63.
Figure 5. Genome-wide DNA methylation profiling. Screen shot from UCSC genome browser tracks of methylation data. The top includes the genomic location and annotation information. MR site track indicated potential McrBC and RE cut sites. The bars indicate the absolute read counts mapped to the CpG site. The red bars represent CpGs cleaved by McrBC and thus methylated, while the blue indicates CpGs cleaved by RE and thus unmethylated. The black denotes CpGs cleaved by both methyl-dependent and methyl-sensitive enzymes that have moderate methylation levels. The height indicates the number of observances. (A) Genome browser view of the methylation status of the MHCII locus in lines 63 and 72 before and after MDV treatment. Regions 1 and 2 showed methylation variations in two lines after infection. (B) The methylation status of a region of repetitive DNA sequences on chromosome 1. Region 3 and 4 are two examples showing methylation variations induced by MDV infection. It was clear that the LTR repeat (region 4) upstream of chicken transcript was less methylated in line 72 than line 63 before MDV infection, whereas its methylation was raised in line 72 after the virus challenge.
DNA methylation change induced by MDV infection
Methylation alterations were triggered by MDV infection in line 63 and line 72. Methylation levels decreased ~38% in infected line 63 (p < 0.05), and increased ~6% in infected line 72 chickens (p > 0.05) (). In addition, we found sweeping decreases in the methylation levels of individual CpG sites (). This difference was interpreted by a lost of methylation in line 63 and a slight gain of methylation in line 72 after infection (). In line 63, about two thirds of the methylated CpGs became unmethylated, and the proportion of unmethylated CpGs was increased 1.5 times after MDV infection.
The average methylation of CGIs was reduced in line 63 from 0.16 to 0.09 after MDV infection, and was slightly increased in the infected line 72 from 0.16 to 0.20. The methylation levels of promoter CGIs were reduced in line 63 after MDV infection, from 0.13 to 0.06. Overall promoter CGI methylation was stable in line 72, with an average methylation level of 0.16 before and after infection. Methylation levels were similar between infected and uninfected line 72 at promoter regions, and were higher in infected line 72 than in uninfected chickens in the gene body (). In the resistant line 63, methylation across gene bodies was reduced 0.2 after infection (). For example, the methylation of 19 CpGs ~200 bp upstream of GATA2 (GATA binding protein 2) decreased from 53.1% to 34.6% in line 63 after MDV infection, and only reduced 2% in line 72 (Fig. S1A and B). In the MHCII loci, methylation was increased in region 1, and reduced in region 2 in line 72 after the infection. These two regions did not show methylation variations in line 63 after infection (). Examination of repetitive DNA methylation revealed hypomethylation in line 63 (0.24) and increased methylation in line 72 (0.5). For example, methylation in region 3 was dramatically upregulated in line 72, but was stable in line 63 after infection. In the CGI nearby, methylation was downregulated in both lines after MDV infection, and line 72 chickens showed higher methylation than line 63.
Differential methylation regions (DMRs) in non-infected chickens
To identify methylation variation, we defined differential methylation regions (DMRs) as those regions in which the methylation difference was greater than 0.3 in a 200–300 bp window size with more than 3 CpG sites. Prior to infection, 3,080 DMRs were discovered between two lines, and methylation levels were about 15% higher in line 72 than in line 63. These DMRs covered 530 RefSeq genes and 478 were found in repetitive DNA. To understand their potential function, pathway analysis was applied to DMR-associated genes. We found that these genes were enriched in pathways, including interferon signaling, iNOS signaling and cell cycle, and diseases such as inflammatory diseases and cancer (). To explore the regulatory function of DMRs, DMRs were mapped to promoters of RefSeq genes. Ninety-nine DMRs were located in the promoters of 97 genes, which suggest they may negatively control transcription. For example, a DMR was identified upstream of CDC42 (cell division cycle 42), a protein involved in cell cycle regulation, which was 10% less methylated in line 63 (Fig. S2A and B). Correspondingly, the mRNA level of CDC42 was 12% higher in line 63 than in line 72 (Fig. S2C).
Table 1. Before infection DMRs associated biofunction
Infection induced differential methylation regions (iDMRs)
To better understand methylation variation at individual loci, we determined infection induced differential methylation regions (iDMRs). In line 63, there were 3,180 iDMRs, which overlapped with 1,247 RefSeq genes and 524 were found in repetitive DNA. In line 72, 8,332 iDMRs were identified, which covered 3,079 genes and 1,568 repeats. Methylation levels were decreased about 5% in line 63 and increased 25% in line 72, respectively, after MDV infection. Among iDMRs, 94.6% of iDMRs in lines 63 overlapped with iDMRs in line 72, which contains 884 genes and 1,496 repeated sequences. Of the iDMRs that were different between the two lines, many were at repeats such as the iDMR in line 72 found at a CR1-B repeat on chromosome 3 (Fig. S3A and B). We also identified an iDMR in the promoter of GH (growth hormone), a previously identified candidate gene of MD resistance. The methylation level was higher in line 72 than in line 63 before MDV infection, and methylation was reduced ~0.52 in line 72 and decreases ~0.43 in line 63 after infection (Fig. S4). The iDMR upstream of CDC42 (cell division cycle 42) existed in line 72, with methylation decreasing from 64.5% to 31% (Fig. S2A and B). The same region did not show any methylation differences in line 63 after infection. CDC42 transcription was upregulated by ~24% in line 72 after MDV infection (p < 0.05), and was similar in line 63 (Fig. S2C). iDMR-related genes were enriched in different pathways in line 63 and 72, such as the IL-4 signaling, Myc mediated apoptosis signaling, and IGF-1 signaling in line 63, and PDGF pathway and erythropoietin signaling in line 72 (). Ingenuity systems pathway analysis (IPA) showed that genes associated with iDMRs were enriched in inflammatory response in line 63, while iDMRs identified line 72 were associated with genes functioning in cancer ().
Table 2. Top Canonical Pathways
Table 3. Top diseases and disorders
DNA demethylation
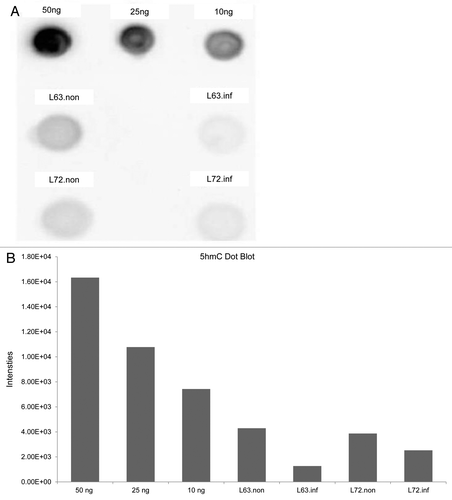
It is possible that, in addition to the decrease in DNMT expression, active demethylation could contribute to the observed reduction in global methylation levels observed in line 63 after MDV infection. To explore such a possibility, we measured global levels of 5-hmC, the intermediate of a potential active demethylation pathway (). 5-hmC content in 10 µg DNA sample from any of the chicken lines was found to be lower than the content of 5-hmC in 10 ng of positive control. 5-hmC content decreased slightly in both 63 and 72 infected chickens, indicating that this is not likely to contribute to the mechanism of demethylation observed in line 63 infected chickens.
Figure 6. Quantification of 5-hmC content by anti-5-hmC box blot. (A) Dot blot of 10 ug of DNA from thymus with 5-hmC positive controls of 50ng, 25ng and 10 ng DNA. (B) Quantitative measurement of dot blots in (A) using densitometry.
Methylation inhibition and MDV infection in vitro
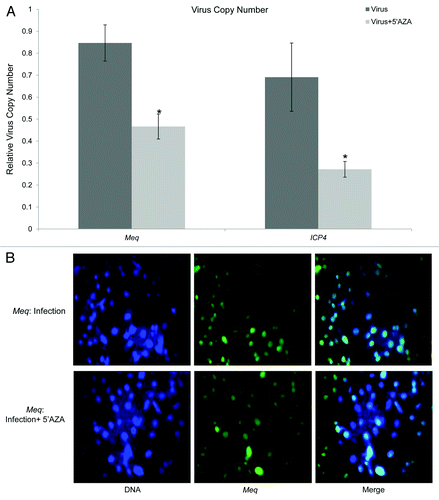
Due to the decreased DNA methylation on MD-resistant line 63, it is reasonable to speculate that methylation was involved in the MD resistance phenotype. To explore the role of DNA methylation on MDV infection, the methylation inhibitor 5-azacytidine (5-AZA) was used to treat MDV infected DF-1 cells. As shown in , we found that the virus genome contents declined 40–57% in infected cells in the presence of 5-AZA relative to the untreated control cells (p < 0.05) (). In addition, the MDV oncoprotein Meq, which is expressed in most cells in the control group, was absent in some drug treated cells ().
Figure 7. (A) Quantification of viral genome copy numbers. The virus copy numbers were evaluated based on viral gene Meq and ICP4 in MDV infected DF-1 cells with or without 5-AZA treatment, and normalized to a single copy gene, Vim. Quantitative results are represented as Mean ± STD (n = 3). (B) The expression of MDV oncogene Meq. The left panel showed the location of nuclei using DAPI to stain DNA (blue). The middle panel was the location and expression MDV oncoprotein Meq (green). The right panel indicated Meq expression in the MDV infected DF-1 cells with or without 5-AZA.
Global mapping of DNA methylation in chickens
Methyl-MAPS was used to uncover the methylome in MD resistant and susceptible chickens before and after MDV infection. The methylated compartment in the chicken was detected by digestion with five methyl-sensitive restriction enzymes, and the unmethylated part was investigated by the methyl-dependent enzyme McrBC. Using in silico analysis, Methyl-MAPS method theoretically covered 3,317,773 CpGs (~32%) in the chicken genome, which are recognized by both methyl-dependent and methyl-sensitive restriction endonucleases (MR sites). From each sequencing library, ~26–90 million reads were obtained, with ~50% of them mapping to chicken genome. For each sample, methylation information was obtained for 2.7 to 3.1 million CpG sites. Of those, 66–76% of them had coverage greater than 10 reads/site, which were used for further analysis (Table S2). Most of the CpGs were covered by 20–200 reads, and some had a coverage greater than 200 (Fig. S5). The MR and CpG site coverage were similar among all four samples. With a cutoff of a minimum 10 reads/site, 57.83–72.11% of MR sites were detected, which included 17.03–21.23% of chicken CpG sites (Table S3).
In total, 3,261,294 CpGs were detected by Methyl-MAPS in four samples. Among these CpGs, a total of 1,372,926 CpGs were annotated as either CGI_promoter, CGI_others, non_CGI promoter, gene body, or repeats. As shown in Figure S6, CpGs were unevenly distributed in different genomic compartments. Nearly one third of annotated CpGs were found in CpG islands. About 18.43% of CpGs were in the annotated promoter regions, including 16.73% of them in promoters within CGIs and 1.70% in the promoters lacking CGIs. We identified 26.95% of CpG sites located in gene bodies and 14.67% in repetitive DNA regions. Annotated CpG distributions were similar among the four samples (Table S4), which allowed us to compare methylation alterations induced by MDV infection in these two chicken lines.
We also calculated methylation based on the genomic location of CpGs. A total of 1,622,778 CpGs were located among the 22,806 CGIs in the chicken genome, and one third of these CpGs (540,390) were directly probed, including 16,927 (about 74%) of CGIs. CGIs showed low average levels of methylation (< 0.20). The average CpG distribution and DNA methylation levels across all gene features are plotted in . As in mammalian genomes, we found that the CpG density gradually increased toward transcriptional start site (TSS). Methylation shows a negative association with CpG density in this region. With increased CpG density toward the TSS, methylation levels declined. CpG density reached the highest level at the 5′ and 3′ of splice sites of the first exons, and these promoter-associated CGIs were unmethylated. Within the coding region, compared with the low CpG density and methylation in introns, the internal exons and the last exon were enriched on methylated CpGs, and both CpG density and methylation reached a maximum level at 5′ and 3′ ends. CpGs were poor in regions coding for 3′UTRs and Poly (A) tails, and methylation level in these regions was similar to the level found in gene bodies. Repetitive DNA regions contain 950,055 CpGs, or about 27.05% of annotated chicken CpGs. In the four groups, about 12.35% to 14.78% of detected CpGs were found in repetitive DNA sequences, with average methylation at 0.3 (Table S4)
To validate the genome-wide methylation results, we used pyrosequencing and bisulfite sequencing to quantify methylation levels in several regions. Five CpG sites in a CGI on chromosome 2 (29, 264–29, 264, 968), upstream of HDAC9 (histone deacetylase 9), were analyzed by pyrosequencing (Fig. S7A and B). Average methylation levels were similar between infected and non-infected line 63; in line 72, methylation was significantly increased in the infected group. Methyl-MAPS also identified enhanced methylation in line 72, which showed methylation levels that were almost doubled. A CGI located upstream of FABP3 (Fatty acid binding protein 3) was detected as hypomethylated, with methylation levels lower than 0.1 among the four groups by both methods (Fig. S8A and B)—consistent with the hypomethylation of the region detected by Methyl-MAPS. Detected by both Methyl-MAPS and bisulfite sequencing, CR1-B methylation levels were similar in line 63 before and after infection, showing moderate methylation level (~50% methylation). In line 72, the highly methylated CR1-B (0.935 by Methyl-MAPS and 75% by bisulfite sequencing) became hypomethylated after infection (0.281 by Methyl-MAPS and 25% by bisulfite sequencing).
Discussion
DNMTs expression and DNA methylation variations
Virus infection and tumorigenesis change DNMTs transcriptional patterns. Compared with the adjacent normal tissues, tumor tissues often show abnormal DNMTs mRNA levels.Citation17-Citation19 Infection with EBV upregulated DNMT3a expression and decreased expression of DNMT1 and DNMT3b.Citation20 Additionally, our lab found that DNMT1 was activated in the spleen of susceptible chickens and DNMT3b was silenced in resistant chickens after MDV infection during MD development.Citation6 These results suggest that expression variation of DNMTs is dependent on disease progression, tissue and cell type, as well as on the genetics of the host. DNMT3a and DNMT3b, two methyltransferases responsible for de novo methylation, interact with DNMT1, the methyltransferase for maintaining methylation, to establish and spread methylation.Citation21 Changes in DNMTs expression can influence DNA methylation levelsCitation22,Citation23; therefore, it is reasonable to speculate that decreases in DNMT3a in infected line 63 may result in the reduction of DNA methylation. The overall hypomethylation in MDV-infected line 63 was caused by methylation decreases in CGIs, repetitive regions and genes. DNMT3a-dependent gene body methylation has been shown to promote expression of tissue-specific genes in mouse,Citation22 indicating that repressed DNMT3a may be responsible for the methylation reduction in gene body in line 63 and a factor contributing to infection resistance. DNMT3b expression was induced in line 72 after MDV infection, which was consistent with the increased methylation levels observed. The slight increase in genomic methylation levels in infected line 72 appeared mostly due to a slight upward shift in the scores rather than due to a large number of unmethylated CpGs becoming completely methylated. Intricate differences in DNA methylation existed between MD resistant and susceptible chickens. In this study, we found that global methylation levels were higher in line 72 than in line 63 prior to MDV infection. More CpGs fell into the unmethylated group and fewer were in the methylation and intermediate methylation classes in uninfected line 63 than in line 72, leading to lower methylation levels in line 63 before MDV infection. The lower methylation in line 63 was presumably attributed to lower expression of DNMT1 and DNMT3b. Additionally, the higher methylation levels in line 72 may be established during selection and inbreeding, and was line specific, suggesting that initial methylation levels may contribute to MD resistance and susceptibility.
Methylation variations and MD resistance/susceptibility
We also found methylation changes at individual loci that may contribute to resistance susceptibility. For example, we found differential methylation at the MHCII locus between line 63 and line 72 before viral infection. Since the MHC locus contains several immune-related genes, we believe that the pre-established methylation pattern in lines 63 and 72 may influence their immunity to MDV infection, and also contribute to the infection-induced methylation alterations in the two chicken lines. To further screen for potential loci of interest, we identified DMRs and iDMRs that potentially contribute to disease resistance or susceptibility and respond to MDV infection. For example, we discovered an iDMR at the promoter of MD resistance candidate gene GH.Citation24,Citation25 In addition, the enhanced transcription levels of CDC42 were coordinated with an observed decline in DNA methylation in a nearby DMR before and after infection, consistent with the fact that methylation silenced gene transcription.Citation26 Methylation may also regulate the transcription of genes involved in the cell cycle pathway, to further influence disease resistance and susceptibility. CDC42, a small GTPase protein, works with other proteins to modulate cell cycle and adhesion.Citation27 The decreased methylation and higher transcription level of CDC42 in the infected line 72 suggest it may contribute to MD susceptibility by deregulation of the cell cycle and proliferation. Pathway analysis reveals that DMRs were enriched in genes involved in inducible nitric oxide synthase (iNOS) signaling, which have been shown to produce nitric oxide (NO) to reduce viral replication.Citation28 Biofunctional analysis also demonstrated that different pathways may be provoked after infection in each line via changes in methylation status. Myc is known to regulate the immune response to infection, and is associated with human cancer and tumor development.Citation29 Myc-mediated apoptosis pathway were exclusively found in line 63, suggesting line-specific DNA methylation variation might affect disease resistance via regulating cell death. Given the fact that methylation inhibits gene expression and the reduction of DNA methylation in line 63, line 63 specific iDMRs may activate genes controlling inflammatory response, which would help eliminate the virus from the host. Line 72 iDMRs were associated with genes involved in cancer, suggesting DNA methylation may participate in this process, although the contribution of DNA methylation in MDV infection-induced tumorigenesis needs to be further elucidated.
Methylation alterations in repetitive regions
MDV infection substantially altered the methylation status of repetitive DNA sequences. A large reduction in methylation was observed on repetitive DNA in line 63, indicating that methylation of these elements may be sensitive to viral infection. Repetitive DNA can exert regulatory functions on gene activity, and some cis-elements have originated from transposable elements (TEs) in humans, including LINEs and LTRs.Citation30,Citation31 DNA methylation is a determinant of TE mobility, which can affect nucleosome binding and regulate nearby gene expression.Citation32,Citation33 Moreover, repetitive regions are often hypomethylated in cancer cells, resulting in genomic instability.Citation26 In the chicken genome, less than 9% of the genome is comprised of interspersed elements. This is markedly lower than in the mammalian genome, which contains 40–50%.Citation34 The lower number of TEs is probably due to the lack of active elements in the chicken genome.Citation35 Therefore, it is reasonable to speculate that methylation variation at chicken TEs potentially influences gene transcription rather than TE retrotransposition.Citation35 It has been shown that CR1 elements upstream of STAT1 (signal transducer and activator of transcription 1) and IL12A (interleukin 12A) contain potential binding sites for GATA1 (GATA binding protein 1). The methylation levels on these two elements were downregulated after MDV infection, which was coordinated with the upregulation of mRNA levels. Since methylation inhibits transcription factor binding, decreased methylation at repetitive regions may up- or downregulate gene expression through the interaction between transcriptional activators or repressors and DNA after MDV exposure in both chicken lines. Our results also suggest that MDV infection changed the methylation levels at repetitive DNA, such as CR1 and LTR, which might mediate regulatory functions in response to infection.
DNA methylation in chickens
Previously, the chicken methylome was characterized using MeDIP-seq, an antibody binding affinity-based method.Citation36 However, this method is biased toward highly methylated CpG-rich regions and provides lower resolution and coverage.Citation12,Citation36,Citation37 In the current study we applied Methyl-MAPS, which allowed quantification of methylation at single copy and repetitive regions and avoided the bias due to the differences in methylation level or CpG density.Citation13 The MeDIP method only analyzed about 9–13% methylated CGIs in chicken genome, and was unable to detect CGIs with low methylation level.Citation36 The coverage of CGIs was improved to 74% by the current study, including CGIs at the promoters, intragenic and intergenic regions with high, low and intermediate methylation. Through intensive analysis, we found a negative correlation between CpG density and DNA methylation at the TSS region and a positive association at other regions in chicken, which is similar to the methylation patterns identified in human and other species.Citation13,Citation36 DNA methylation was depleted at CGI associated promoter regions, which has been shown in other vertebrates to allow transcription factors and RNA polymerase II to initiate transcription.Citation38 The analysis and global maps of methylation obtained in the current study provide a more comprehensive understanding and are a source of baseline methylation levels in the chicken genome.
Methylation inhibition and MDV replication
From our results, it seems that the disturbance of DNA methylation after MDV infection did not involve active DNA demethylation through the conversion to 5-hmC. Together with the downregulated DNMT3a expression, decreased methylation in line 63 is likely controlled by passive demethylation. Since methylation levels were lower in line 63 than line 72, DNA methylation inhibition presumably improves MD resistance. Methylation inhibitor treatment limited MDV replication in vitro, which was confirmed by the independent estimation of virus copy numbers using ICP4 and Meq. Lower expression of Meq in the drug-treated infected cell suggested that 5-AZA either limited viral replication and spread or inactivated Meq transcription. However, there is a debate about the function of 5-AZA on MDV infection. Meq expression was elevated in MDV-infected B lymphocytes transformed with avian leukosis virus (ALV).Citation39 This disagreement may be explained by the differences between ALV-transformed B cells and the chicken model used. Line 63 chickens are resistant to MD but susceptible to ALV, whereas line 72 chickens are resistant to ALV and susceptible to MD.Citation40 Resistance to ALV and MDV may be mediated by different mechanisms. Methylation inhibition may enhance ALV propagation, which may further assist MDV infection. In addition, the promoter of Meq was absent of DNA methylation in MDV-infected cells,Citation6 and the methylation inhibitor was unable to directly regulate Meq expression, increasing the possibility that ALV infection favors secondary infection with MDV.
In summary, the genome-wide quantitative DNA methylation analysis in the current study provides more comprehensive DNA methylation maps for chicken as well as global maps of methylation changes related to MDV infection. Our data suggests that lower global methylation levels in the host may be related to MD resistance by activating genes with antiviral and antitumor functions. Higher methylation in MD-susceptible chickens might favor viral replication and spread by the disruption of normal growth and immune responses. These results suggest a mechanism of disease resistance determined by DNA methylation patterns, and support the notion that methylation variation could be both a cause and consequence of viral infection. Further work is required to functionally link observed gene expression and DNA methylation changes. Future combination of this work with histone modification analysis will provide a foundation to understand the role of epigenetics in the crosstalk between virus and host through the regulation of gene expression. Additionally, the comprehensive integration of both genetic and epigenetic information will help identify candidate genes implicated in disease resistance or susceptibility, improve our understanding of epigenetic predisposition to viral infection, and give clues for potential epigenetic therapies.
Materials and Methods
Experimental Animals and Sample Preparation
Line 63 and line 72 (USDA-ARS Avian Disease and Oncology Laboratory) are two highly inbred lines of specific-pathogen-free white leghorn chickens that are resistant and susceptible, respectively, to MD tumors. Chickens from each line were separated into two groups. One group was infected with a very virulent (vv+) strain of MDV (648A passage 40) at day 5 after hatching, while the other group did not receive MDV. Four chickens were selected from each group at 21 DPI, and none of them developed tumors during the experiment period. Fresh thymus samples were harvested individually and stored in RNAlater solution (QIAGEN) at -80°C for DNA or RNA extraction. The entire animal experiment was conducted following the procedures and guidelines described in the “Guidelines for Animal Care and Use” manual approved by the Animal Care and Use Committee, the USDA-ARS, and the Avian Disease and Oncology Laboratory (Approval ID 111–26).
DNA preparation and endonuclease digestion
Genomic DNA from four samples of each treatment group was extracted using the Wizard Genomic DNA purification kit (Promega). RNase treatment was performed to remove RNA. DNA concentration was measured by the Qubit dsDNA Broad-Range Assay (Invitrogen).
Four DNA samples from each group were pooled together with equal amounts. Methyl-MAPS libraries were made as in Edwards et al. with slight modification. In brief, the pooled samples were separated into two parts, and were digested by methyl-dependent (McrBC) or 5 methyl-sensitive restriction enzymes (HpaII, HpyCH4VI, AciI, HhaI and BstUI), respectively (New England Biolabs, referred to as RE). The amount of starting DNA was 15 ug for both RE and McrBC digestions. Three rounds of McrBC digestion were performed to assure complete digestion of methylated DNA. For the first two rounds, DNA was extracted using phenol-chloroform extraction and ethanol precipitation. After the last round of digestion, DNA was purified using QIAquick spin columns (QIAGEN) to remove residual GTP. Four rounds of RE digestion were conducted and followed by phenol-chloroform extraction and ethanol precipitation. The first round of RE was set up with HpaII and HpyCH4IV, the second round with AciI and HhaI, the third round with BstUI and the fourth with HpaII and HpyCH4IV. Samples were purified by phenol-chloroform extraction and ethanol precipitation between each round.
SOLiD mate-pair library construction
Mate-pair libraries were prepared with slight variation to the SOLiD library preparation guide according to the protocol in Edwards et al. In brief, fractionated DNA was repaired using End-It DNA End-repair kit (Epicenter), subsequently purified using QIAquick spin columns (QIAGEN) to remove dNTPs, and quantified by Qubit dsDNA Broad-Range Assay (Invitrogen). Endogenous EcoP15I sites were then methylated using EcoP15I enzyme with 1 × NEB buffer 3, 1 × BSA, 360 µM SAM. The methylation reaction was performed at 37°C for 2–3 h, and then boosted with additional EcoP15I, NEB buffer 3, BSA, and SAM for another 2–3 h at 37°C. The DNA was purified using QIAquick spin columns (QIAGEN). The EcoP15I cap adaptor (50 pmol/µl) was ligated to the end-repaired and EcoP15I methylated DNA using Quick Ligation Kit (NEB), and purified using MinElute (QIAGEN) to remove excess adapters.
DNA samples were size fractionated using a 1% agarose gel. DNA was selected based on 7 fragment sizes: 0.8–1.1 kb, 1.1–1.5kb, 1.5–2 kb, 2–3kb, 3–5 kb, 5–8kb and > 8kb. Each fraction was purified using QIAquick Gel Extraction kit (QIAGEN). DNA from each fraction was then circularized with a biotin labeled T30 sticky linker using the Quick Ligation Kit (NEB) and purified using QIAquick spin columns (QIAGEN). Fractions were combined into two tubes, one for < 2 kb and the other for > 2 kb. Circularized DNA was isolated using ATP-dependent Plasmid-safe DNase (Epicenter) to degrade the linear DNA. The purified DNA samples were digested with EcoP15I to release mate-pair tagged fragments. The digested DNA was repaired by Klenow (NEB), and then heat inactivated at 65°C for 20 min. P1 and P2 adapters were added to the end repaired DNA using Quick Ligation Kit (NEB). P2 adapters contained distinct barcodes for each library so libraries can be later pooled for sequencing. The < 2 kb and > 2kb fractions were combined together for library purification.
Libraries were bound to M280 streptavidin beads (Invitrogen) through the biotin on the T30 internal adaptor. Washed library-bound beads were nick translated. Trial PCR was performed by serial dilution of beads to ensure the existence of 156-bp products and to determine the sufficient cycles to obtain enough products. Final libraries were amplified using all the beads and Phusion High Fidelity DNA Polymerase (NEB) with 19–23 cycles. The PCR products were combined together for ethanol precipitation.
A 6% DNA PAGE gel (Lonza) was used to purify the 156-bp library DNA. The final library DNA concentration was measured using the Qubit HS assay kit (Invitrogen), and the quality and quantity was assessed by Bioanalyzer DNA 1000 LabChip (Agilent).
DNA sequencing
The DNA libraries were sequenced by EdgeBio on an ABI SOLiD 4 system according to the manufacturers instructions for 25 base-pair mate-pair sequencing. Eight libraries from 4 samples were pooled and sequenced on a single chip using the standard SOLiD protocol.
Tag mapping and data analysis
Barcoded sequences were separated using a custom perl script that allows for error checking in the barcode sequence subsequent analysis steps were performed as in Edwards et al. In brief, sequence tags were mapped using SOLiD system software analysis package Corona-lite (Applied Biosystem). Two mismatches in each 25 bp read to the chicken genome (galGal3, http://genome.ucsc.edu) were allowed. Reads were filtered to remove fragments without at least one enzyme cut site on the fragment end. Read counts were normalized as in Edwards et al.Citation13 The methylation level for each CpG site was calculated as the number of reads from methylation (RE) library divided by the number of reads from both RE and McrBC libraries.
Each CpG site was annotated according to its genomic location (promoter, gene body, intergenic, repetitive regions and CpG islands). Fisher’s Exact test was used to identify the differential methylation at individual CpG sites while controlling for an FDR less than 0.01. The annotation information, including Refseq, repeatMask, CpG islands were obtained from the UCSC genome browser (galGal3). Sites of differential methylation were defined as the absolute methylation level difference for each CpG between infected and non-infected sample greater than 0.3. Infection induced differential methylation regions (iDMRs) were discovered by merging adjacent sliding windows with at least 3 differentially methylated CpGs in a 2 kb window. DMRs were reported only if their length was greater than 3 kb.
Bisulfite conversion, pyrosequencing and bisulfite sequencing
Sodium bisulfite conversion reagents were used to treat 500 ng of DNA (EZ DNA Methylation Golden Kit) using the standard protocol provided by the manufacturer. PCR primers for methylation validation were listed in Table S4. For pyrosequencing, we used biotin labeled universal primer in the PCR reaction. The bisulfite PCR included 1 µl of 1:5 diluted bisulfite converted DNA, primers and PCR reagents from Hotstar Taq polymerase kit (QIAGEN) with four biological replicates. The methylation level was detected individually by Pyro Q-CpG system (PyroMark ID) using 20 µl of PCR products. For bisulfite sequencing, an equal amount of DNA from four samples of treated or control groups from each chicken line were pooled together, serving as a template for the bisulfite conversion and the bisulfite PCR, and then PCR products were purified (QIAquick Gel Extraction Kit, QIAGEN). The purified PCR products were ligated to pCR® 2.1 Vector (The Original TA cloning® Kit, Invitrogen), transformed to DH5α competent cells (ZYMO Research), and screened for successful insertions (blue-white selection) after incubation at 37°C overnight. Ten white colonies from each group were cultured in a 37°C shaker overnight. Plasmid DNA was isolated using the QIAprep® Miniprep Kit (QIAGEN), and M13 reverse primer was used for sequencing.
Purification and quantification of mRNA levels
RNA was extracted from four samples per group using the RNeasy Mini Kit (QIAGEN) and the standard method described by the manufacturer. An on-column DNase digestion was performed to remove any contaminant DNA. RNA concentration was measured by Nanodrop. Reverse transcription was performed on 1 μg of purified total RNA using SuperScript™ III Reverse Transcriptase (Invitrogen) with oligo (dT) 12–18 primers (Invitrogen); mRNA levels were quantified using SYRB Green PCR Kit (QIAGEN) with four biological replicates from each group. PCR primers for mRNA quantification are listed in Table S4.
DNA dot blot
DNA samples were mixed with 0.1 volume of 1M NaOH, denatured at 99°C for 5 min, and snap cooled on ice. The denatured DNA was neutralized with 0.1 volume of 6.6 M Ammonium acetate. DNA samples were then loaded on a nitrocellulose membrane (Bio-rad), and UV cross-linked. Blocking was performed using 10% non-fat milk. The membrane was incubated with primary antibodies against 5-mC (Active Motif) and 5-hmC (Diagenode), and diluted in blocking solution (1:5,000) at 4°C overnight. After 3 washes, the membrane was incubated with anti-rabbit IgG and anti-mouse IgG secondary antibodies (Santa Cruz) and diluted in TBST (1:5000) for 1 h at room temperature. Membranes were developed with ECL (Amersham) and measured using ChemiDoc XRS (Bio-Rad). Each dot was circled, and the average volume in the circle was exported to Excel to estimate 5-mC and 5-hmC contents using Quantity One software (Bio-rad). 5-mC and 5-hmC positive controls were synthesized by PCR using 5-mCTPs and 5-hmCTPs (ZMYO research).
Cell culture and MDV infection
The chicken embryo fibroblast cell line DF-1 was grown in Dulbecco’s modified Eagle’s medium supplemented with 10%, fetal bovine serum, streptomycin (100 mg/ml) and penicillin (100 U/ml) (Invitrogen). Cells were maintained at 37°C in humidified 5% CO2 conditions. The very virulent + (vv +) strain of MDV (648A passage 40) was obtained from USDA-ARS Avian Disease and Oncology Laboratory. The DF-1 cells were inoculated with the virus at multiplicity of infection (M.O.I) of 0.1 for 3 d. The DNA methylation inhibitor 5-azacytidine (Fisher) was used to treat the infected cells at concentration of 5 µM.
Immunofluorescence
The DF-1 cells were fixed with 4% of paraformaldehyde for 15 min, and permeabilized using 0.5% Triton-X 100 in PBS for 15 min. Primary antibodies were prepared at 1:500 and 1:1000 dilutions in PBS, and incubated with cells for 1 h. After washing the cells 3 times, FITC-labeled donkey anti-mouse or donkey anti-rabbit IgG secondary antibodies were diluted 1:1,000 in PBS, and incubated with cells for 30 min. The nuclei were labeled with 4', 6-diamidino-2-phenylindole (DAPI) (Invitrogen). The immunoreactive complexes were visualized with a Zeiss LSM 510 confocal fluorescence microscope, and the images were processed using LSM Image Examiner software (Zeiss).
Additional material
Download Zip (2.6 MB)Acknowledgments
Thanks Dr. Yanghua He for figures and Dr. Hans H. Cheng for the precious discussion of this manuscript.
Author’s contributions
FT and JH performed Methyl-MAPS experiment, did the validation of the results by Q-PCR, analyzed some of the result and wrote the paper. FZ, NV and JE analyzed the data. ZH did the microarray experiment and collected the samples and helped revised the manuscript. ZKJ and JZS designed and revised this paper.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Fred Davidson VN, ed. Marek's disease: An evolving problem. London: Elsevier Academic Press, 2004.
- Burgess SC, Young JR, Baaten BJ, Hunt L, Ross LN, Parcells MS, et al. Marek’s disease is a natural model for lymphomas overexpressing Hodgkin’s disease antigen (CD30). Proc Natl Acad Sci U S A 2004; 101:13879 - 84; http://dx.doi.org/10.1073/pnas.0305789101; PMID: 15356338
- Davison TF, Nair V. Marek's disease: an evolving problem. Amsterdam; Boston: Elsevier, 2004.
- Brown AC, Nair V, Allday MJ. Epigenetic regulation of the latency-associated region of Marek’s disease virus in tumor-derived T-cell lines and primary lymphoma. J Virol 2012; 86:1683 - 95; http://dx.doi.org/10.1128/JVI.06113-11; PMID: 22090140
- Ferrari R, Berk AJ, Kurdistani SK. Viral manipulation of the host epigenome for oncogenic transformation. Nat Rev Genet 2009; 10:290 - 4; http://dx.doi.org/10.1038/nrg2539; PMID: 19290008
- Luo J, Yu Y, Chang S, Tian F, Zhang H, Song J. DNA Methylation Fluctuation Induced by Virus Infection Differs between MD-resistant and -susceptible Chickens. Front Genet 2012; 3:20; http://dx.doi.org/10.3389/fgene.2012.00020; PMID: 22363343
- Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 2010; 11:607 - 20; http://dx.doi.org/10.1038/nrm2950; PMID: 20683471
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008; 9:465 - 76; http://dx.doi.org/10.1038/nrg2341; PMID: 18463664
- Kim JH, Dhanasekaran SM, Prensner JR, Cao X, Robinson D, Kalyana-Sundaram S, et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res 2011; 21:1028 - 41; http://dx.doi.org/10.1101/gr.119347.110; PMID: 21724842
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4:143 - 53; http://dx.doi.org/10.1038/nrc1279; PMID: 14732866
- Grafodatskaya D, Choufani S, Ferreira JC, Butcher DT, Lou Y, Zhao C, et al. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines. Genomics 2010; 95:73 - 83; http://dx.doi.org/10.1016/j.ygeno.2009.12.001; PMID: 20005943
- Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 2010; 28:1106 - 14; http://dx.doi.org/10.1038/nbt.1681; PMID: 20852634
- Edwards JR, O’Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res 2010; 20:972 - 80; http://dx.doi.org/10.1101/gr.101535.109; PMID: 20488932
- Yu Y, Zhang H, Tian F, Zhang W, Fang H, Song J. An integrated epigenetic and genetic analysis of DNA methyltransferase genes (DNMTs) in tumor resistant and susceptible chicken lines. PLoS One 2008; 3:e2672; http://dx.doi.org/10.1371/journal.pone.0002672; PMID: 18648519
- Luo J, Yu Y, Zhang H, Tian F, Chang S, Cheng HH, et al. Down-regulation of promoter methylation level of CD4 gene after MDV infection in MD-susceptible chicken line. BMC Proc 2011; 5 Suppl 4:S7.
- Yu Y, Luo J, Mitra A, Chang S, Tian F, Zhang H, et al. Temporal transcriptome changes induced by MDV in Marek’s disease-resistant and -susceptible inbred chickens. BMC Genomics 2011; 12:501; http://dx.doi.org/10.1186/1471-2164-12-501; PMID: 21992110
- Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 1999; 27:2291 - 8; http://dx.doi.org/10.1093/nar/27.11.2291; PMID: 10325416
- Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res 1988; 48:1159 - 61; PMID: 3342396
- Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 2001; 97:1172 - 9; http://dx.doi.org/10.1182/blood.V97.5.1172; PMID: 11222358
- Leonard S, Wei W, Anderton J, Vockerodt M, Rowe M, Murray PG, et al. Epigenetic and transcriptional changes which follow Epstein-Barr virus infection of germinal center B cells and their relevance to the pathogenesis of Hodgkin’s lymphoma. J Virol 2011; 85:9568 - 77; http://dx.doi.org/10.1128/JVI.00468-11; PMID: 21752916
- Kim GD, Ni J, Kelesoglu N, Roberts RJ, Pradhan S. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J 2002; 21:4183 - 95; http://dx.doi.org/10.1093/emboj/cdf401; PMID: 12145218
- Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, et al. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 2010; 329:444 - 8; http://dx.doi.org/10.1126/science.1190485; PMID: 20651149
- Ostler KR, Davis EM, Payne SL, Gosalia BB, Expósito-Céspedes J, Le Beau MM, et al. Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene 2007; 26:5553 - 63; http://dx.doi.org/10.1038/sj.onc.1210351; PMID: 17353906
- Liu HC, Cheng HH, Tirunagaru V, Sofer L, Burnside J. A strategy to identify positional candidate genes conferring Marek’s disease resistance by integrating DNA microarrays and genetic mapping. Anim Genet 2001; 32:351 - 9; http://dx.doi.org/10.1046/j.1365-2052.2001.00798.x; PMID: 11736805
- Liu HC, Kung HJ, Fulton JE, Morgan RW, Cheng HH. Growth hormone interacts with the Marek’s disease virus SORF2 protein and is associated with disease resistance in chicken. Proc Natl Acad Sci U S A 2001; 98:9203 - 8; http://dx.doi.org/10.1073/pnas.161466898; PMID: 11470922
- Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358:1148 - 59; http://dx.doi.org/10.1056/NEJMra072067; PMID: 18337604
- Duncan MC, Peifer M. Regulating polarity by directing traffic: Cdc42 prevents adherens junctions from crumblin’ aPart. J Cell Biol 2008; 183:971 - 4; http://dx.doi.org/10.1083/jcb.200811057; PMID: 19064672
- Yu M, Zhang C, Yang Y, Yang Z, Zhao L, Xu L, et al. The interaction between the PARP10 protein and the NS1 protein of H5N1 AIV and its effect on virus replication. Virol J 2011; 8:546; http://dx.doi.org/10.1186/1743-422X-8-546; PMID: 22176891
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2002; 2:301 - 10; http://dx.doi.org/10.1038/nrc780; PMID: 12001991
- Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet 2008; 9:397 - 405; http://dx.doi.org/10.1038/nrg2337; PMID: 18368054
- Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet 2003; 19:68 - 72; http://dx.doi.org/10.1016/S0168-9525(02)00006-9; PMID: 12547512
- Huda A, Jordan IK. Epigenetic regulation of Mammalian genomes by transposable elements. Ann N Y Acad Sci 2009; 1178:276 - 84; http://dx.doi.org/10.1111/j.1749-6632.2009.05007.x; PMID: 19845643
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 2007; 8:272 - 85; http://dx.doi.org/10.1038/nrg2072; PMID: 17363976
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004; 432:695 - 716; http://dx.doi.org/10.1038/nature03154; PMID: 15592404
- Lee SH, Eldi P, Cho SY, Rangasamy D. Control of chicken CR1 retrotransposons is independent of Dicer-mediated RNA interference pathway. BMC Biol 2009; 7:53; http://dx.doi.org/10.1186/1741-7007-7-53; PMID: 19691826
- Li Q, Li N, Hu X, Li J, Du Z, Chen L, et al. Genome-wide mapping of DNA methylation in chicken. PLoS One 2011; 6:e19428; http://dx.doi.org/10.1371/journal.pone.0019428; PMID: 21573164
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 2010; 11:191 - 203; http://dx.doi.org/10.1038/nrg2732; PMID: 20125086
- Singh H. Teeing up transcription on CpG islands. Cell 2009; 138:14 - 6; http://dx.doi.org/10.1016/j.cell.2009.06.028; PMID: 19596228
- Fynan EF, Ewert DL, Block TM. Latency and reactivation of Marek’s disease virus in B lymphocytes transformed by avian leukosis virus. J Gen Virol 1993; 74:2163 - 70; http://dx.doi.org/10.1099/0022-1317-74-10-2163; PMID: 7691987
- Bacon LD, Hunt HD, Cheng HH. A review of the development of chicken lines to resolve genes determining resistance to diseases. Poult Sci 2000; 79:1082 - 93; PMID: 10947175