Abstract
There is increasing evidence that interindividual epigenetic variation is an etiological factor in common human diseases. Such epigenetic variation could be genetic or non-genetic in origin, and epigenome-wide association studies (EWASs) are underway for a wide variety of diseases/phenotypes. However, performing an EWAS is associated with a range of issues not typically encountered in genome-wide association studies (GWASs), such as the tissue to be analyzed. In many EWASs, it is not possible to analyze the target tissue in large numbers of live humans, and consequently surrogate tissues are employed, most commonly blood. But there is as yet no evidence demonstrating that blood is more informative than buccal cells, the other easily accessible tissue. To assess the potential of buccal cells for use in EWASs, we performed a comprehensive analysis of a buccal cell methylome using whole-genome bisulfite sequencing. Strikingly, a buccal vs. blood comparison reveals > 6X as many hypomethylated regions in buccal. These tissue-specific differentially methylated regions (tDMRs) are strongly enriched for DNaseI hotspots. Almost 75% of these tDMRs are not captured by commonly used DNA methylome profiling platforms such as Reduced Representational Bisulfite Sequencing and the Illumina Infinium HumanMethylation450 BeadChip, and they also display distinct genomic properties. Buccal hypo-tDMRs show a statistically significant enrichment near SNPs associated to disease identified through GWASs. Finally, we find that, compared with blood, buccal hypo-tDMRs show significantly greater overlap with hypomethylated regions in other tissues. We propose that for non-blood based diseases/phenotypes, buccal will be a more informative tissue for EWASs.
Introduction
There is increasing evidence supporting a role for interindividual epigenetic variation in the etiology of a variety of common non-malignant diseases and phenotypes in humans.Citation1-Citation6 Such variation was originally studied with a view to explaining the non-genetic etiological component, but more recent large-scale analyses that integrate epigenetic and genetic variation show that a significant proportion of these genetic variants most likely exert their effects via modulation of epigenetic states.Citation7,Citation8 Consequently, a variety of epigenome-wide association studies (EWASs) for a variety of human diseases and phenotypes have been published in recent years,Citation9-Citation20 and many more are underway (e.g., www.roadmapepigenomics.org/participants).
Although EWAS and GWAS designs may appear ostensibly similar, the dynamic nature of epigenomic landscapes poses challenges for designing a successful EWAS that are not encountered in GWASs.Citation1-Citation5 Key issues include, (1) type of cohort since a standard ‘unrelated cases vs. controls’ design cannot establish causality, (2) which epigenomic mark to profile, although DNA methylation is by far the most studied mark in EWASs due to the practical difficulties of studying chromatin state and non-coding RNA in large numbers of live individuals, (3) the number of individuals required for adequate statistical power in an EWAS since the extent of interindividual epigenetic variation in human populations is poorly understood, (4) platform to use since a balance has to be struck between cost-effectiveness and genome coverage and (5) the tissue to assay since epigenetic landscapes are tissue-specific and genomic context-dependent. This problem is further compounded by the fact that in most cases the target tissue for non-blood disease/phenotypes are not readily available from significant numbers of live human individuals (a few large-scale adipose tissue and muscle biopsy collections notwithstanding, such as those profiled inCitation21,Citation22). Therefore, surrogate tissues are used in most EWASs, with blood being the strongly preferred option. But what is the evidence that blood is the most suitable tissue for non-blood based diseases/phenotypes compared with buccal cells, the other easily accessible tissue? To assess the potential of buccal cells for use in EWASs, here we report a comprehensive analysis of a buccal cell methylome using whole-genome bisulfite sequencing. Our data suggest that for EWASs of non-blood based diseases/phenotypes, buccal will be a significantly more informative surrogate tissue.
Results
Generation of a human buccal cell DNA methylome
To create a human buccal methylome, we BS-seq profiled buccal DNA samples from 14 different individuals of European ancestry (age range of 20–79 y.o.) (Table S1). Bisulfite conversion rates were > 95% for all BS-seq libraries (Table S2). The final data set for each individual corresponded to ~4X coverage so we pooled the 14 different data sets to increase depth (~60X) and reduce the effects of individual variation. Composite plots of a variety of genomic features revealed the expected profiles e.g., CpG islands were significantly hypomethylated, gene-bodies methylated, and known imprinted regions partially methylated (). It is important to note that we profiled buccal epithelial cells specifically, obtained by sterile brushes, and not saliva that can contain significant amounts of leukocyte contamination. To further ensure that the buccal cells used in our study were not contaminated with blood sub-types, we used the Illumina 450K array to generate additional DNA methylomic data for buccal cells and a variety of other blood and non-blood cell types from adults: CD4+ T-cells, CD14+ monocytes, CD34+ hematopoietic stem cells, mature spermatozoa, full term placenta, and pancreas (Methods and Table S3). The Illumina 450K array contains > 450,000 CpG sites associated with nearly all annotated human promoters, CpG islands, imprinted regions and a variety of other regions including gene bodies, enhancers and non-CG sites.Citation23 We then called tissue-specific differentially methylated regions (tDMRs) between all blood subtypes vs. all non-blood cell types, but excluding buccal cells at this stage (Methods). This identified 12 different tDMRs that were highly specific for blood subtypes (). We then looked at the buccal Illumina450K profiles and found that in all cases, methylation levels at these tDMRs were highly concordant with the non-blood cell types only, suggesting very little, if any, contamination of the buccal cells with blood cell types ().
Figure 1. (A) Canonical methylation profiles calculated for BS-Seq data of blood and buccal for CpG Islands (extracted from UCSC Genome Browser), Gene Body (from transcription start position to end position) and Promoter (2kbp upstream of the transcription start position). The methylation fraction (number of cytosines recorded/number of reads for that position) for each CpG contained within the genomic feature was calculated and the distribution of methylation was calculated. The discrete nature of the blood profile is due to the reduced coverage. (B) Several examples of regions on the 450K that are specifically hypomethylated or hypermethylated in blood compared with placenta, pancreas, sperm or buccal. Plotted is the methylation fraction (β value) for each probe contained on the array within this region. (C) A contour plot of the F measure for validation of the BS-Seq tDMRs using Illumina 450K data for different methylation and region size cut-offs. Pink represents a high F measure and hence good validation while blue represent a low F measure. (D) An example of a validated tDMR where each point represents the methylation fraction of the BS-Seq data for each CpG in this region for blood (red) and buccal (black). The called tDMR is highlighted as a transparent blue rectangle. Also plotted are the 450K probes for this region for blood (red square) and buccal (black square) which show good agreement with the BS-Seq data.
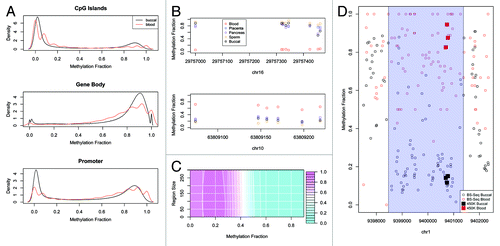
Comparative analysis of BS-seq-based blood and buccal DNA methylomes
To directly compare the buccal and blood methylomes, an ~20X fold coverage blood BS-Seq data set was obtained fromCitation24 and processed in the same manner as the buccal BS-seq data. Blood vs. buccal tDMRs were called using a ‘windowless’ approach that allows for region sizes to be automatically determined (Methods). The Cochran-Mantel-Haenszel test was used to initially define tDMRs at a genome-wide p < 0.05. To further filter the tDMRs, we only selected regions that were > 200bp in size and with > 50% methylation difference. To select these cut-offs we first called differences between buccal vs. blood using Illumina 450K data (described above). For a filtered list of tDMRs based on any methylation difference and region size cut-off we could then calculate a true positive i.e., how many of the 450K tDMRs did we capture with our filtered BS-Seq tDMRs, and false positive rate i.e., how many of the filtered BS-Seq tDMRs were not called as 450K tDMRs. is a surface plot of the harmonic mean (F measure) of this true positive and false positive rate for varying cut-offs. An example of a region that is validated by Illumina 450K data is shown in . The combination of > 200bp in size and > 50% methylation difference yielded a low false positive rate (10%), meaning that we have high confidence of the validity of the original BS-seq tDMRs. Using this filter does however lead to a low true positive rate meaning that we are very likely missing many true tDMRs. Therefore we also performed various analyses using 50bp minimum size and a 30% methylation difference i.e., the highest harmonic mean of true positive and false positive (Table S4). The main results of the paper are similar using either set of tDMRs (data not shown).
Buccal cells are significantly hypomethylated relative to blood
Using the above strategy, we called 33,998 autosomal buccal vs. blood tDMRs (sex chromosomes were not included). Strikingly, 29,418 were hypomethylated in buccal and only 4,580 hypomethylated in blood (). It may be possible that this skew is due to the mixed cell nature of whole blood. That is, the various blood subsets could all harbor different subset-specific low methylated regions, but in a mixed cell population such as whole blood, these appear as regions of intermediate to high methylation. However, an analysis of the Illumina450K we generated (described above) revealed buccals to be significantly hypomethylated relative to whole blood and a variety of sorted blood sub-types: CD14+, CD4+ and CD34+ (). The Illumina 450K analysis also proves that the significant hypomethylation observed in buccals is not due to a difference in fold coverage between the buccal and blood BS-seq methylomes. An example of a buccal-specific hypomethylated region is shown in .
Figure 2. (A) The methylation difference between blood and buccal for the BS-Seq tDMRs which were filtered for methylation differences > 50%. A large proportion are less methylated in buccal (Blood-Buccal > 0) (B) An example of a buccal specific hypomethylated region on the Illumina 450K when compared with Blood, CD14, CD4, CD34+ and CD34-. (C) A buccal hypomethylated tDMR which overlaps with epithelial DNaseI hotspots but does not overlap with any blood DNaseI hotspots. The top panel shows the methylation fraction for blood (red) and buccal (black) as measured by BS-Seq, with the called tDMR highlighted using a blue rectangle. The bottom panel shows regions of DNaseI hotspots for various different cell types as downloaded from ENCODE. Those cell types that are associated with blood have been highlighted in red while those associated with epithelial cells are highlighted in black.
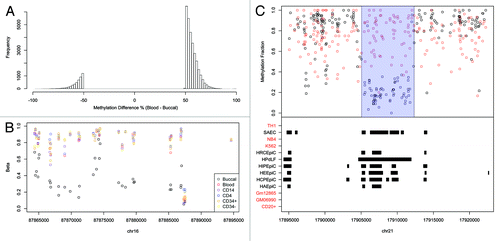
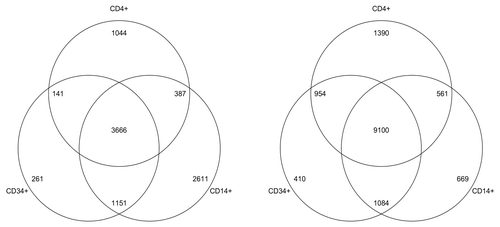
Figure 3. For each cell type (CD14+,CD4+ and CD34+) we called differences between each one and the buccal 450K and calculated the number of regions that were hypomethylated in buccal (right Venn diagram) and those that were hypomethylated in each of the cell type (left Venn diagram). The Buccal cells consistently contained more hypomethylated regions than that of the other cell types.
We then wanted to obtain additional evidence that the significant hypomethylation in buccal is both real and likely to be biologically relevant. We therefore considered the human whole-genome DNase-seq profiles generated as part of the recently released ENCODE data.Citation25 DNaseI sites are a strong predictor of regulatory activity and are often observed at low methylated regions.Citation25 Although buccal and whole blood per se weren’t profiled as part of ENCODE, a variety of other epithelial and blood subsets were subjected to DNase-seq. Indeed, the ENCODE project reported significantly more DNaseI hotspots in epithelial cells compared with all blood subsets (Mann-Whitney U test, p = 0.0025, ). Furthermore, we found that epithelial hotspots were enriched at buccal-hypomethylated tDMRs (). Next, since ENCODE did not profile buccal or whole blood, we identified DNaseI hotspots common to all epithelial cells, and separately DNaseI hotspots common to all blood subsets, the rationale being that these sites are very likely to also exist in buccal and whole blood respectively. In total, we found 17,635 epithelial-specific DNaseI hotspots and 1,717, blood-specific hotspots. Of the epithelial hotspots, 1,283 overlapped with buccal-hypomethylated tDMRs compared with only 14 that overlapped blood-hypomethylated tDMRs (Permutation test; Fold Change = 10.3, p < 2.2e-16). Similarly, of the blood-specific DNaseI hotspots, 55 overlapped with blood-hypomethylated tDMRs compared with only 12 that overlapped with buccal-hypomethylated tDMRs (Permutation test; Fold Change = 35.5, p < 2.2e-16). Overall, these analyses demonstrate that buccal cells contain significantly more hypomethylated regions relative to blood, and these sites are likely to be active regulatory elements as suggested by the comparative analysis with DNase-seq data.
Table 1. Extracted from Table S1Citation25 showing the number of DNaseI hotspots of blood and epithelial cell types. Epithelial cell types have a much greater number of hotspots compared with blood, which is inline with the increased number of HMRs found
Most buccal vs. blood tDMRs overlap non-canonical regulatory elements
We next sought to elucidate key genomic properties of these tDMRs. Given the strong correlation with DNaseI sites, it was not surprising to observe that over 50% of the BS-seq tDMRs overlap sites of putative regulatory activity as defined by the RegFeature track in the ENSEMBL database, and a variety of histone modification combinations typically associated with ‘active’ states (). Gene bodies were also strongly represented, consistent with the emerging idea that gene-body DNA methylation is far more dynamic than previously appreciated and a feature of eukaryotic DNA methylation systems that is even more evolutionarily conserved than methylation dynamics at mammalian genomic elements such as CpG islands.Citation26-Citation28 Analysis of spatial proximity of the buccal vs. blood tDMRs to annotated CpG Islands showed that 26,787 of the tDMRs are > 10 kb away, and only 7% are within 2 kb (). Consistent with this observation we found the vast majority of buccal vs. blood tDMRs to be CpG poor (CpGo/e < 0.3, ).
Figure 4. (A) The breakdown of the overlap for tDMRs with various genomic annotations. Promoters were calculated as being 2kbp upstream of the transcription position and Gene Body was defined as being from the transcription start position to the transcription end position. Histone marks were downloaded as BED files and overlapped with tDMRs (See supplementary information for full details of the data) (B) CpG Island locations were downloaded from UCSC Genome Browser and the overlap of tDMRs with these CpG Islands was calculated for increasing window sizes of inclusion. Only 7% of tDMRs are within 2kbp of a CpG Island. (C) The CpGoe for BS-Seq tDMRs which either overlapped with 450K or RRBS-Seq(red) or not (black). In blue is the distribution for CpG Islands, which is significantly greater than either of the tDMRs. Highlighted in yellow is the region plotted in of.Citation38 (D) The methylation state in buccal cells vs those of blood for the BS-Seq tDMRs. Those points in the top left of the diagram are regions which are hypomethylated in buccal and the points in the bottom right of the diagram are regions which are hypomethylated in blood. The points are colored either in yellow if the methylation state in sperm (top panel) or ES cells (bottom panel) are less than 30% methylated or in blue if the methylation state is > 70%.
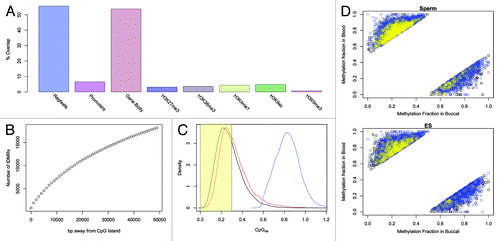
We next investigated the developmental dynamics of the buccal vs. blood tDMRs. We obtained raw BS-seq data for human sperm and ES cells from published studies,Citation29,Citation30 and processed the data sets in line with the buccal and blood BS-seq data (Methods). An integrated analysis of all four different BS-seq data sets revealed that the vast majority of either buccal- or blood-specific hypo-methylated regions are methylated in both sperm and ES cells (Mann-Whitney U test, p = 0.029) (). In other words, these tDMRs specifically lose methylation in the relevant developmental lineage, as opposed to starting from an unmethylated state and gaining methylation. The hyper-methylated state of the tDMRs in sperm is also consistent with their CpG-poor status, given the mutagenic effects of methylation and likely subsequent gradual loss of cytosines over evolutionary time-scales. The developmental DNA methylation dynamics of the tDMRs we describe here stands in contrast to canonical CpG-rich regulatory elements that are predominantly unmethylated in germ cells and early development.Citation30-Citation32
Disease-associated SNPs are enriched near buccal vs. blood tDMRs
The integration of EWAS and GWAS data has the potential to delineate the functional consequence of genetic variation. A few recent studies have used functional genomic data e.g., histone modifications and DNaseI hotspots, to identify active regulatory elements in a given tissue, and ask whether there is any correlation between such sites and previously identified GWAS hits.Citation7,Citation8 Indeed, there seems to be a significant enrichment of GWAS hits near regulatory elements, suggesting that in many cases germline genetic variants may act via modulating the activity of linked regulatory elements. Given the generally accepted view that unmethylated regions are often associated with ‘active’ regulatory sites, we wondered if similar analyses can be performed using DNA methylation data since, for practical reasons, it will not be possible to interrogate chromatin state in many tissues. We performed spatial correlations of hypo-tDMRs with statistically significant SNPs from published GWASs (Methods) by adapting the ‘Genomic Association Tester’ (GAT) method available from www.cgat.org/~andreas/documentation/gat. GAT works by calculating the expected overlap of genomic regions based on a sampling algorithm. The actual overlap can then be contrasted with the expected overlap and an empirical p-value is calculated. Blood hypo-tDMRs were found to be strongly associated with a variety of autoimmune diseases/phenotypes e.g., celiac disease and Graves disease ( lists the top 5 associations). Buccal hypo-tDMRs were associated with diseases/phenotypes strongly linked with, importantly, epithelial and not just buccal function, such as bladder cancer and Immunoglobulin A that is produced by epithelial cells.
Table 2. The top 5 disease associated SNPs which overlap with the BS-Seq tDMRs which are either hypomethylated in blood or buccal. Genomic Association Tester (GAT) was used to calculate a p-value and fold enrichment for each of the different disease associated SNPs
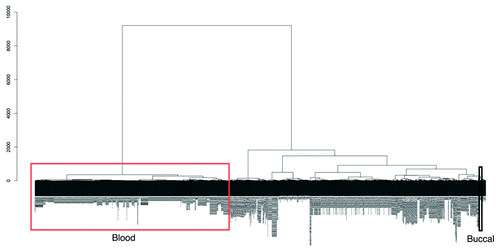
The main rationale of our study is to characterize the human buccal methylome with view to assessing its potential for EWASs since buccal, and blood, will in many cases be used as surrogate tissues as the target tissue will be inaccessible from large numbers of individuals. Assuming that hypomethylated regions are significantly stronger markers of regulatory activity compared with hypermethylated regions, we compared buccal and blood data to Reduced Representation Bisulfite Sequencing (RRB-seq) data of a variety of ‘non-accessible’ tissues such as brain and islets profiled by ENCODE.Citation25 In all cases, hypomethylated sites in buccal cells better captured hypomethylated regions in other tissues (). To investigate this further we extracted all publicly available Illumina450K data from the GEO database (on November 14, 2012), producing a data set of over 1,052 samples (after filtering, Methods) of various different tissue types and disease states. Using unsupervised clustering we found two distinct clusters that separate all blood samples (including various different blood subtypes) and all other samples including buccals, stem cells, transformed cells, brain, kidney, liver and even sperm (). This clustering was performed using all probes on the Illumina450K array and not just the buccal vs. blood tDMRs we define here. This further emphasizes the fact that, relative to blood, buccal methylation profiles are closer to all other non-blood cells considered in our analysis.
Table 3. RRBS-Seq data of different inaccessible tissues was downloaded from ENCODE project and the average methylation state for each of the tissues was calculated for data that overlapped with the BS-Seq tDMRs. We found that there was an increased amount of overlap between buccal hypomethylated tDMRs with regions that were < 30% methylated in the tissues than compared with blood
Figure 5. An unsupervised hierarchical cluster dendogram of 1,052 different Illumina 450K arrays. Due to the large size of the image the sample labels are not legible in the figure but a large version is available to download in the supplementary (Fig. S2). We have highlighted in the figure the location of blood cell types (red rectangle) and those of our buccal data (black rectangle). There are two main clusters, one of blood cell types and the other of all other tissue types including cell lines, somatic tissue and our buccal data. This suggests that blood cell types have a vastly different methylation state than those of all others. The dendrogram was calculated using agglomerative hierarchical clustering with the euclidian distance as the metric and the Ward linkage criteria. The root of the dendrogram is at the top and the y-axis represents the euclidian distance between the clusters at each splitting point.
Most buccal vs. blood tDMRs are not captured by commonly used platforms for DNA methylomic analyses
Our data also allowed us to address another key issue of EWAS design, namely which platform is most suitable. In recent years, a variety of methylome profiling strategies have been published based on immunoprecipitation, restriction enzyme digestion, and/or bisulfite conversion (systematic comparisons of these various methods were reported in two recent papersCitation33,Citation34). Of these, two platforms in particular have proven to be popular for EWASs as they provide significant genome-scale coverage, single cytosine resolution methylation levels, and low cost, (1) the Illumina 450K array (and the earlier version—the Illumina27K array) and (2) Reduced Representation Bisulfite Sequencing (RRB-seq), in which restriction enzymes whose recognition sites contain a CG site are used to first restrict the DNA, and then the resulting fragments are sequenced.Citation35 However, both platforms are generally biased toward either canonical genomic elements and/or CpG dense regions. An important question that arises from our analysis of buccal vs. blood tDMRs is what proportion is captured by RRBseq and/or Illumina450K arrays. A key point when doing this analysis is to ensure that a lack of overlap between BS-seq tDMRs and RRBseq and/or Illumina450K probes also means that the methylation dynamics are not being captured i.e., the RRBseq fragment/Illumina450K probes are not simply behaving as ‘surrogate’ tDMRs. To classify an RRBseq fragment or Illumina 450K probe as overlapping with the BS-Seq tDMRs, we allowed the probe to be up to 100bp from either end of the tDMR (). Thus we ensured that fragments/probes very close to the BS-seq tDMRs that capture similar methylation dynamics as the BS-seq tDMR were included despite not overlapping exactly. We found, however, that when increasing this value, the validation rate fell (Fig. S1) suggesting that these probes were no longer sampling the true difference, and hence 100bp was chosen to maintain a high validation rate. is an example of such a tDMR for which the closest Illumina probes are > 1kbp away from the edge of the tDMR and hence do not validate the tDMR.
Figure 6. (A) Diagram showing the definition of boundaries for calculating overlap between Illumina 450k probes and our tDMRs (B) BS-Seq data of blood (red) and buccal (black) for a tDMR region which is highlighted in blue. All Illumina 450K probes present in this region are plotted in black rectangles for buccal data and red rectangles for blood data. There is good agreement between the Illumina 450k data and BS-Seq and as the probes are 1kbp from the tDMR found in the BS-Seq data they find no difference between blood and buccal. (C) Over 75% of the tDMRs are not covered by either 450K or RRBS-Seq suggesting that a large proportion of possible variation may be missed by 450K or RRBS-Seq (D) The distribution of sizes in basepairs (bps) of BS-Seq tDMRs covered by 450K (red) and those not covered by 450K (black) (E) A histogram of the different transcription factors that were enriched greater than 2-fold in either BS-Seq tDMRs which overlapped Illumina 450k (black) probes or those that did not (gray).
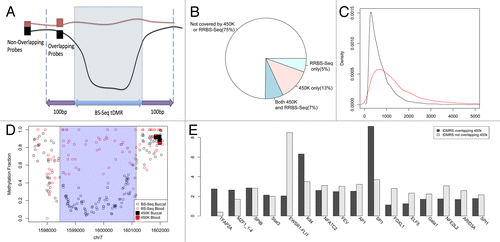
Strikingly we found that collectively only ~25% of all BS-seq-based buccal vs. blood tDMRs are captured by these two platforms (). More specifically, the Illumina 450K array contains ~20% of the regions compared with RRBS-Seq that profiles ~13%. We also calculated the overlap with enhanced versions of RRB-Seq, in which different combinations of enzymes and fragment distributions yields increased genomic coverage,Citation28,Citation36 but even this approach does not dramatically increase the percentage of buccal vs. blood tDMRs captured (). Further analysis comparing tDMRs captured by Illumina450K/RRB-seq with those tDMRs not captured by these platforms showed that the latter tend to be significantly smaller in size (Mann-Whitney U test p < 2.2e-16, ). Interestingly, an analysis of transcription factor (TF) binding site sequences revealed that in some cases, there is differential enrichment of TF binding sites between tDMRs captured by Illumina450K/RRB-seq with those tDMRs not captured by these platforms ().
Table 4. A number of different RRBS-Seq methods have been proposed in which different combinations of enzymes and fragment distributions yields increased genomic coverage. We have simulated the genomic coverage of each of these RRBS-Seq methods and calculated the possible overlap between the BS-Seq tDMRs
Discussion
The challenges associated with conducting a successful EWAS has been highlighted by several authors.Citation1-Citation5 One such challenge is the choice of tissue since in most cases the target tissue will not be accessible from significant numbers of live individuals. Consequently, surrogate tissues have to be used and blood has been the default option in the vast majority of cases, without any evidence to suggest that it is more informative than buccal, the other easily accessible tissue type, for non-blood based diseases/phenotypes. Based on the data presented here, we propose that buccals may be more informative for EWASs of non-blood cells. It is not in doubt that hypomethylated regions are strong markers of potential regulatory activity and variation at these sites, whether due to genetic or non-genetic influences, will have a bigger phenotypic impact. Furthermore, Feinberg and colleagues have provided evidence in several separate studies that tDMRs also show increased interindividual variability both in the context of disease and normal epigenetic variation.Citation37 Finally, the correlation we find between GWAS hits and hypo-tDMRs, including the observation that buccal hypo-tDMRs are associated with diseases/phenotypes strongly linked with epithelial and not just buccal function, further emphasizes the relevance of hypomethylated regions in human diseases. Although one could argue that the true value of buccals can only be known by actually using them in EWASs, decisions about which tissue to use need to be made at the outset. It is important to note that we are not suggesting that interindividual variation at normally methylated regions does not occur or is unimportant. But rather that the closer clustering of buccals with all other somatic tissues (using unsupervised clustering that utilizes all sites on the 450K array and not just hypomethylated sites) would suggest that buccals are more likely to display dynamics that are more representative of other tissues than blood.
Buccal cells cannot replace blood in a variety instances e.g., for blood-based conditions of course, but also in cases when chromatin profiling needs to be done (although the vast majority of EWASs in the coming years will focus on DNA methylation). Furthermore, in some instances only blood will be available as is the case for stored samples such as Guthrie cards.Citation38 Therefore, if possible, profiling both blood and buccals will not be redundant but rather provide complementary information. However, in the case of an EWAS of a non-blood disease/phenotype, if a choice has to be made between the two surrogate tissues, then buccal may be more informative.
Finally, the fact that commonly used methylomic platforms capture a relatively small fraction of buccal vs. blood tDMRs has potential implications for drawing conclusions from ongoing EWASs, especially in the case of negative results. A question in any profiling experiment where the entire genome is not being assayed is how much variation is potentially being missed? We believe that both the Illumina450K and RRB-seq methods are, at this stage, powerful and cost-effective methods for performing EWASs, but it should be kept in mind that most variable regions may lie outside of the genomic regions covered by these more focused methods. Similarly, blood-based EWASs for a non-blood based disease/phenotype are likely to benefit from the inclusion of buccals as the relevant variability may simply not be present in blood.
Materials and Methods
Samples
Buccal samples were from 14 different individuals of European ancestry (age range of 20–79 y.o.) (Table S1). All subjects gave informed consent and the study was approved by the Northern and Yorkshire Research Ethics committee (REC Reference Number: 06-MREO-3–22).
Generation and analysis of BS-seq data
DNA from Buccal cells was extracted using the Gentra Puregene Buccal cell kit (Oiagen). For each library, 50 ng of DNA was sonicated to median size of ~300 bp and libraries prepared according to the method described in the Supplementary section. Libraries were size selected and sequenced on an Illumina HiSeq2000 machine. BS-Seq data was mapped using BIFast with parameters –n 1 and –l 50 for blood, sperm, ES and buccal data (Lowe et al., submitted). BIFast is an efficient implementation of the BISMARK algorithimCitation39 written on top of BOWTIECitation40 and is freely available from https://bitbucket.org/xboxrob/bifast. Clonal reads were filtered where paired end mapping produced fragments at exactly the same location and one of the clonal fragments was chosen randomly. tDMRs were called using the windowless approach of BIFast. By looping through each of the chromosomes, we group CpGs that have the same directionality of methylation difference between the two tissue types. We then applied the Cochran-Mantel-Haenszel test to calculate a p-value for each region and tDMRs were filtered for p-value < 0.01, an average methylation difference > 50% and a minimum size of 200bp and stored as BED files. BEDTools was used to calculate the various different overlaps.
Generation and analysis of Illumina450K data
All array experimental procedures were performed according to the manufacturers instructions. Quantile normalization was performed on the intensity values of the red and green channels of type I and type II probes separately. Probes with a detection p-value < 0.01 or those mapping to more than one location or to chromosome X or Y were removed from further analysis. Intensities were then combined into the standard β number as a measure of methylation. All subsequent analyses were performed using custom scripts in R (available on request from the authors).
Unsupervised clustering of Illumina450K data obtained from GEO
Custom scripts were used to download all data available on 14th November 2012. Samples which contained β values were used and each sample was checked to make sure that β values were > = 0 and < = 1. Those that passed this initial qc were used and probes were then filtered so that each sample had a recorded measurement leaving 196,817 probes. These probes were then used as input to the unsupervised clustering contained in R of the 1057 samples as well as our blood and buccal samples. The resulting clusters were driven by tissue type rather than batch, allowing allowed us to investigate how similar different tissues were to each other in their methylation profiles.
Processing of publicly available RRB-seq data
A custom python script was used to download data from ENCODE and convert the format of the files. The sites which overlapped with each tDMR were identified and the average methylation across this region was calculated by summing up the methylation of each overlapping CpG and dividing by the total number of overlapping CpGs. We then defined regions as being hypomethylated in the RRB-seq data if the average methylation across the region was < 30%.
Transcription Factor motif analysis
Position Weight Matrices for vertebrate transcription factors were downloaded from JASPER. We used the BioStrings library (www.bioconductor.org/packages/2.10/bioc/html/Biostrings.html) to match PWMs to sequences of our tDMRs. tDMRs were split into two groups; those that overlapped 450K probes and those that did not. A background model of a zero order Markov Chain Model with equal probabilities for each nucleotide was used to calculate enrichments. Twenty was added to both the background and foreground counts to prevent over enrichment of a small number of hits and those transcription factors with greater than 2 fold enrichment in one of the two groups were reported.
Additional material
Download Zip (227.5 KB)Acknowledgments
VKR, CG, RDL are supported by the BBSRC, UK (BB/H012494/1). RL, VKR, MIH, RDL are also supported by the EU-FP7 ‘BLUEPRINT’ program (282510). RDL is also supported by Juvenile Diabetes Research Foundation International (JDRFI Award 5–2011–145). SVR is funded by the Multiple Sclerosis Society of the United Kingdom and the MRC, UK [G0801976]. HB was supported by EFSD/Novo Nordisk Programme Grant and Diabetes UK (10/0004107).
Accession numbers
Sequencing and Illumina 450K data have been deposited into the NCBI Gene Expression Omnibus under accession GSE45529.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011; 12:529 - 41; http://dx.doi.org/10.1038/nrg3000; PMID: 21747404
- Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 2010; 465:721 - 7; http://dx.doi.org/10.1038/nature09230; PMID: 20535201
- Heijmans BT, Mill J. Commentary: The seven plagues of epigenetic epidemiology. Int J Epidemiol 2012; 41:74 - 8; http://dx.doi.org/10.1093/ije/dyr225; PMID: 22269254
- Bell JT, Spector TD. A twin approach to unraveling epigenetics. Trends Genet 2011; 27:116 - 25; http://dx.doi.org/10.1016/j.tig.2010.12.005; PMID: 21257220
- Relton CL, Davey Smith G. Epigenetic epidemiology of common complex disease: prospects for prediction, prevention, and treatment. PLoS Med 2010; 7:e1000356; http://dx.doi.org/10.1371/journal.pmed.1000356; PMID: 21048988
- Feinberg AP. Epigenomics reveals a functional genome anatomy and a new approach to common disease. Nat Biotechnol 2010; 28:1049 - 52; http://dx.doi.org/10.1038/nbt1010-1049; PMID: 20944596
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011; 473:43 - 9; http://dx.doi.org/10.1038/nature09906; PMID: 21441907
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012; 337:1190 - 5; http://dx.doi.org/10.1126/science.1222794; PMID: 22955828
- Joubert BR, Håberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 2012; 120:1425 - 31; PMID: 22851337
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 2011; 88:450 - 7; http://dx.doi.org/10.1016/j.ajhg.2011.03.003; PMID: 21457905
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013; 49:359 - 67; http://dx.doi.org/10.1016/j.molcel.2012.10.016; PMID: 23177740
- Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet 2011; 7:e1002300; http://dx.doi.org/10.1371/journal.pgen.1002300; PMID: 21980303
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 2010; 20:434 - 9; http://dx.doi.org/10.1101/gr.103101.109; PMID: 20219945
- Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, et al, MuTHER Consortium. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet 2012; 8:e1002629; http://dx.doi.org/10.1371/journal.pgen.1002629; PMID: 22532803
- Toperoff G, Aran D, Kark JD, Rosenberg M, Dubnikov T, Nissan B, et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum Mol Genet 2012; 21:371 - 83; http://dx.doi.org/10.1093/hmg/ddr472; PMID: 21994764
- Häsler R, Feng Z, Bäckdahl L, Spehlmann ME, Franke A, Teschendorff A, et al. A functional methylome map of ulcerative colitis. Genome Res 2012; 22:2130 - 7; http://dx.doi.org/10.1101/gr.138347.112; PMID: 22826509
- Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet 2011; 20:4786 - 96; http://dx.doi.org/10.1093/hmg/ddr416; PMID: 21908516
- Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 2010; 20:170 - 9; http://dx.doi.org/10.1101/gr.100289.109; PMID: 20028698
- Ellis JA, Munro JE, Chavez RA, Gordon L, Joo JE, Akikusa JD, et al. Genome-scale case-control analysis of CD4+ T-cell DNA methylation in juvenile idiopathic arthritis reveals potential targets involved in disease. Clin Epigenetics 2012; 4:20; http://dx.doi.org/10.1186/1868-7083-4-20; PMID: 23148518
- Waterland RA, Kellermayer R, Laritsky E, Rayco-Solon P, Harris RA, Travisano M, et al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet 2010; 6:e1001252; http://dx.doi.org/10.1371/journal.pgen.1001252; PMID: 21203497
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, et al. Genetics of gene expression and its effect on disease. Nature 2008; 452:423 - 8; http://dx.doi.org/10.1038/nature06758; PMID: 18344981
- Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, et al, MuTHER Consortium. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 2011; 7:e1002003; http://dx.doi.org/10.1371/journal.pgen.1002003; PMID: 21304890
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692 - 702; http://dx.doi.org/10.4161/epi.6.6.16196; PMID: 21593595
- Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol 2010; 8:e1000533; http://dx.doi.org/10.1371/journal.pbio.1000533; PMID: 21085693
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al, ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57 - 74; http://dx.doi.org/10.1038/nature11247; PMID: 22955616
- Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A 2010; 107:8689 - 94; http://dx.doi.org/10.1073/pnas.1002720107; PMID: 20395551
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010; 328:916 - 9; http://dx.doi.org/10.1126/science.1186366; PMID: 20395474
- Nanty L, Carbajosa G, Heap GA, Ratnieks F, van Heel DA, Down TA, et al. Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res 2011; 21:1841 - 50; http://dx.doi.org/10.1101/gr.121640.111; PMID: 21940836
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315 - 22; http://dx.doi.org/10.1038/nature08514; PMID: 19829295
- Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, et al. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 2011; 146:1029 - 41; http://dx.doi.org/10.1016/j.cell.2011.08.016; PMID: 21925323
- Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, Gräf S, et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res 2008; 18:1518 - 29; http://dx.doi.org/10.1101/gr.077479.108; PMID: 18577705
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457 - 66; http://dx.doi.org/10.1038/ng1990; PMID: 17334365
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 2010; 28:1097 - 105; http://dx.doi.org/10.1038/nbt.1682; PMID: 20852635
- Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 2010; 28:1106 - 14; http://dx.doi.org/10.1038/nbt.1681; PMID: 20852634
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008; 454:766 - 70; PMID: 18600261
- Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet 2012; 8:e1002781; http://dx.doi.org/10.1371/journal.pgen.1002781; PMID: 22737091
- Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci U S A 2010; 107:Suppl 1 1757 - 64; http://dx.doi.org/10.1073/pnas.0906183107; PMID: 20080672
- Beyan H, Down TA, Ramagopalan SV, Uvebrant K, Nilsson A, Holland ML, et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res 2012; 22:2138 - 45; http://dx.doi.org/10.1101/gr.134304.111; PMID: 22919074
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011; 27:1571 - 2; http://dx.doi.org/10.1093/bioinformatics/btr167; PMID: 21493656
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; http://dx.doi.org/10.1186/gb-2009-10-3-r25; PMID: 19261174