Abstract
The potential importance of DNA methylation in the etiology of complex diseases has led to interest in the development of methylome-wide association studies (MWAS) aimed at interrogating all methylation sites in the human genome. When using blood as biomaterial for a MWAS the DNA is typically extracted directly from fresh or frozen whole blood that was collected via venous puncture. However, DNA extracted from dry blood spots may also be an alternative starting material. In the present study, we apply a methyl-CpG binding domain (MBD) protein enrichment-based technique in combination with next generation sequencing (MBD-seq) to assess the methylation status of the ~27 million CpGs in the human autosomal reference genome. We investigate eight methylomes using DNA from blood spots. This data are compared with 1,500 methylomes previously assayed with the same MBD-seq approach using DNA from whole blood. When investigating the sequence quality and the enrichment profile across biological features, we find that DNA extracted from blood spots gives comparable results with DNA extracted from whole blood. Only if the amount of starting material is ≤ 0.5µg DNA we observe a slight decrease in the assay performance. In conclusion, we show that high quality methylome-wide investigations using MBD-seq can be conducted in DNA extracted from archived dry blood spots without sacrificing quality and without bias in enrichment profile as long as the amount of starting material is sufficient. In general, the amount of DNA extracted from a single blood spot is sufficient for methylome-wide investigations with the MBD-seq approach.
Introduction
Epigenetic modifications to chromatin provide stability and diversity to the cellular phenotype. One of the most intensively studied modifications is the methylation of DNA cytosine residues at the carbon 5 position. DNA methylation studies are a promising complement to genetic studies of variation in DNA sequence. First, because methylation can directly affect gene expression, it may capture additional individual variation in disease susceptibility.Citation1 Indeed, dysregulation of DNA methylation has been associated with a wide variety of human diseases.Citation2-Citation7 Second, methylation can account for a wide variety of phenomena that characterize complex diseasesCitation7,Citation8 such as sex differences,Citation9,Citation10 genotype-environment interactions,Citation11,Citation12 and age-related patterns associated with the disease course.Citation13 Third, as methylation is modifiable by environmental factors, including pharmaceutical interventions,Citation14,Citation15 methylation sites are potentially important new drug targets.Citation16
The potential importance of DNA methylation in the etiology of complex diseases has led to interest in methylome-wide association studies (MWAS)Citation17,Citation18 aimed at interrogating all methylation sites in the human genome.Citation19 Such a genome-wide approach proved fruitful in the context of genome-wide association studies (GWAS) with SNPs where even the first generation of GWAS identified susceptible variants for common diseases.Citation20-Citation23
When using blood as biomaterial for a MWAS, the DNA is typically extracted directly from fresh or frozen whole blood that was collected via venous puncture. However, dry blood spots may also be an alternative starting material.Citation18 Indeed, studies show that in the context of targeted methylation studies, DNA extracted from dry blood spots gives highly reliable results.Citation24,Citation25 Two recent studies successfully used DNA from blood spots to investigate the methylation level of 27K and 450K CpGs, respectively, using arrays.Citation26,Citation27 One of these studiesCitation26 also used the enrichment based sequencing approach, MeDIP-seq, to investigate 200ng DNA from blood spots for two individuals. According to the authors the MeDIP-seq approach was successful.Citation26 However, there was no overlap between the samples used on the array and for MeDIP-seq. This lack of overlap in combination with that only two assays were conducted, prevented rigorous quality evaluation from being performed. Taken together, these studies indicate that DNA from blood spots may be useful for MWAS.
There are a number of advantages with blood spots. Blood spots are collected from finger-pricks or, for infants and young children, from heel-pricks.Citation28 This fairly non-invasive blood collection procedure causes minimal risks to the participant and may therefore be of particular interest for studies involving young children and infants, where regular venous punctures may be challenging. Other benefits with blood spots are that they are easy to store, ship and handle. For example, blood collected in tubes should ideally be stored in +4°C and processed as soon as possible to obtain maximum DNA yield and quality. In contrast, the collected blood spots can be dried in room temperature and shipped without refrigeration. Furthermore, blood spots can be kept in a long-term storage facility (< -20°C) for years prior to DNA extraction without significant loss in quality.Citation29,Citation30 For example, DNA extracted from blood spots, stored for up to 25 y, has successfully been used in whole genome amplification for various genetic investigations including direct sequencing and GWAS.Citation29-Citation31 Finally, the simple collection procedure and the stability of the DNA in blood spots would, for example, allow the collection to be self-administrated by adults without requiring the participants to visit a medical facility. This may be of particular use in large epidemiological studies where participants are asked to give biosamples at multiple time points and/or when study participants are geographically widespread.
A challenge when using blood spots for MWAS is the low yield of DNA that typically can be extracted. Whole genome amplification, which is applied for GWAS to generate sufficient DNA from blood spots, cannot be applied for MWAS because the methylation signal is lost if DNA is directly amplified. In this study we first modified the DNA extraction protocol to allow for maximum yield of high quality DNA from a complete blood spot in one single reaction. Next, we conducted methylome-wide profiling using next-generation sequencing to evaluate the use of DNA from archived dry blood spots for MWAS. In this evaluation we compared methylome-wide data from DNA extracted from blood spots with DNA extracted from fresh whole blood.Citation32 We also compared the effect of different amounts of starting material by performing multiple assays from the same individual starting with 0.25 µg–2 µg DNA.
Results
We have used a modified protocol to extract DNA from a complete blood spot in a single reaction (see supplementary material for details). Largely dependent on the diameter of the blood spots, which ranged from approximately 11–15 mm, we extracted 0.8–1.6 µg genomic DNA per blood spot. The 260/280 ratio ranged from 1.6–2.3 with a mean ratio of 1.9 and a standard deviation of 0.26, suggesting a high degree of purity of the extracted DNA. Furthermore, the Bioanalyzer DNA 1000 kit (Agilent) showed that RNA contamination was not present in our DNA samples. Finally, the agarose gel electrophoresis showed that the DNA degradation was low with the majority of DNA from each sample being greater than 10 kb in size.
To investigate the quality of the methylome-wide data we first studied possible differences in sequence reads between DNA extracted from blood spots vs. whole blood. Results are shown in . The measures for the percentages of non-duplicate reads, correctly matched reads, and uniquely aligned reads vs. multireads were similar for DNA extracted from whole blood and from DNA extracted from blood spots when using 2 µg of starting material. For the percentage aligned reads blood spots performed slightly better (i.e., more aligned reads) than whole blood.
Figure 1. Sequence quality of methylome-wide data. Data quality outcome variables (y-axis) are given for the samples from whole blood (averages from all samples are shown) and from all blood spots with 2 µg starting material (averages from all samples are shown) as well as for all conditions for the two samples (sample A and B) where different amounts of starting material were used (x-axis).
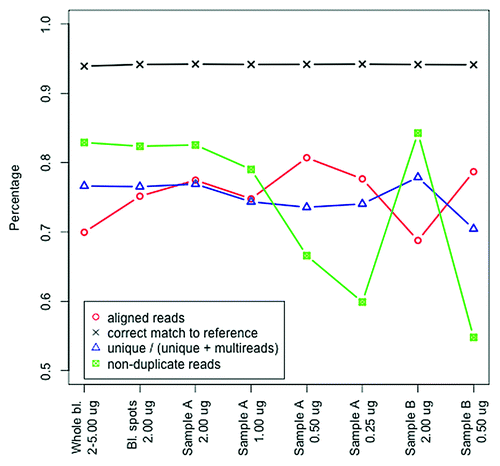
Next, we investigated whether the amount of starting material affected sequence data quality. In , the fairly straight lines for percentage aligned reads, uniquely mapped reads, and correct matches suggest that the amount of starting material does not affect the quality of the reads themselves. However, when using low amounts of starting material we observe more duplicate reads.
We also studied whether there were systematic differences in terms of enrichment profiles. shows that when comparing the percentage of covered CpGs overlapping with a number of biological features, including, regions masked by repeat masker, introns, exons, 5′ and 3′ untranslated regions and CpG dense CpG islands as well as isolated CpG sites we noticed a very similar distribution of overlap between the samples that used DNA extracted directly from whole blood and the blood spot samples with 2µg DNA starting material. This suggests that the type of starting material (blood spots vs. whole blood) does not dramatically affect the enrichment procedure of methylated fragments ().
Figure 2. Overlap with biological features. The percentage of covered CpG sites overlapping with biological features are given for the whole blood (averages from all samples are shown) and from all blood spots with 2 µg starting material (averages from all samples are shown) as well as for all conditions for the two samples (sample A and B) where different amounts of starting material were used (x-axis).
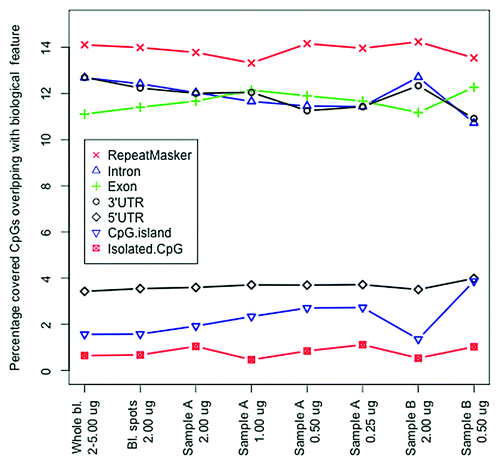
Neither did we observe any major differences in the distribution of enrichment profiles across biological features when altering the amount of starting material (, sample A and B). However, we noticed a lower ratio of percentage overlap (0.29 vs. 0.47, respectively) between CpG-rich regions (CpG islands) and CpG-poor regions (isolated CpGs) with lower amounts of starting material (0.5 µg) as opposed to higher amounts (e.g., 2.0 µg).
Discussion
In this study we found that the overall quality of methylome-wide investigations and the enrichment profile of the methylome using the MBD-seq approach is highly comparable for DNA extracted from blood spots and whole blood. Our data shows that as little as ~1 µg of starting material can be used without sacrificing the quality of the sequencing data or without biasing the methylation profile. However, if the starting material was ≤ 0.5µg we notice a decrease in quality, as more duplicate reads are then present in the NGS data. The explanation may be that when fewer fragments (lower amount of starting material) are competing for the reagents the likelihood that a single fragment generates multiple amplicons may increase. When the same amplicons are sequenced they would yield reads starting at exactly the same location (duplicate reads). As these duplicate reads are excluded in the quality control they will not affect the final statistical results. However, avoiding sequencing duplicate reads to begin with saves resourses and makes the sequencing procedure more cost effective.
We also noticed that the ratio of methylated fragments overlapping GC-rich and GC-poor regions shifted, suggesting that the enrichment profile was altered if the amount of starting material was ≤ 0.5µg. This difference potentially indicates that low amounts of starting material slightly bias the enrichment procedure as it may favor extraction of fragment from densely methylated regions. This suggests that the amount of starting material is important as less starting material might increase the variation between samples due to less complete enrichment for methylated fragments. Therefore too low amounts of starting material may cause false positive findings in MWAS.
Enrichment based methylome-wide profiling using an approach similar to MBD-seq called MeDIP-seq, where an antibody is used instead of the MBD protein to capture the methylated fraction of the genome, have been reported using as little as 200ng of DNA from blood spots.Citation26 The authors performed the MeDIP assay in two different individuals without any duplicates and, thus, the quality of their data could not be properly investigated. Therefore, before performing large-scale investigations with MeDIP-seq the quality of the data when using blood spots and the boundaries of the amount of starting material from blood spots should be evaluated in a similar way as it has been for MBD-seq.
It is important to note that while 1 µg can be considered as a fairly small amount of DNA it is typically the entire DNA yield extracted from a complete blood spot. Therefore, if the amount of biomaterial is very limited a complete blood spot may still be considered as too valuable for a single investigation. On the other hand, the knowledge that high quality methylome-wide assays can be conducted with DNA from blood spots opens for the opportunity to collect blood spots instead of whole blood in new sample collections.
In conclusion, our study suggests that for the vast majority of standard blood spots, one blood spot will suffice for methylome-wide investigation using MBD-seq. Furthermore, we show that high quality methylome-wide investigations using MBD-seq can be conducted in DNA from archived blood spots without sacrifice in sequence quality or methylome-wide enrichment profile. These results in combination with the simple collection procedure, the stability of the DNA and the straightforward handling and storage requirements for blood spots enable large-scale methylome-wide investigations in new and existing sample collections for a large set of phenotypes.
Materials and Methods
Study design
In this study we are investigating 2–3 blood spots/individual ascertained at a single time point from four individuals. The oldest blood spots were collected 19 y prior to DNA extraction. With this DNA we performed methylome-wide profiling using a next generation sequencing based approach.Citation32 To evaluate if DNA extracted from blood spots is equivalent with DNA extracted directly from fresh whole blood, we investigated measures of data quality and deviations in the enrichment profile across biological features. As starting material for the blood spots we used 2 µg of DNA/individual and compared this with methylome-wide data from whole blood where 2–5 µg DNA/individual was used.
To study the lower bound for the amount of required starting material, methylation assays were performed multiple times with different amounts of DNA from the same individuals. Using DNA from the same individual ensures that any detected differences are caused by technical differences (i.e., amount of starting material) and not by biological variation. A summary of the study design with the amount of starting material used for each methylome-wide profiling from blood spots is shown in .
Table 1. Study samples and amount of starting material used for each methylome-wide profiling
Blood spot samples
The blood spots used in this investigation were collected as part of the Great Smokey Mountain Study, which is a longitudinal study of the development of psychiatric disorders in youth collected in western North Carolina, USACitation33 that started in 1993. A trained staff member collected blood from two finger-pricks (yielding 10 spots total per visit) onto Schleicher and Schuell (S&S) filter paper number 903.Citation34 The samples were dried in room temperature and temporarily (less than two weeks) stored in refrigeration before shipped (without refrigeration) to the laboratory for long-term storage at -28°C. This protocol is consistent with the rigorous quality control program developed for newborn screening programs.Citation28 All procedures were approved by ethical committees in the US, and all subjects provided written informed consent (or legal guardian consent and subject assent).
DNA from the blood spots was extracted using a modified version of the QIAamp DNA Mini Kit (QIAGEN). By altering the buffer volumes and the incubations the modified protocol allows for DNA extraction of a complete blood spot within a single reaction column without sacrifice of DNA yield. Full details of the modifications are given in the supplementary materials. The quantity and quality of the extracted DNA was investigated with NanoDrop 1000 (Thermo Scientific), run on 1% agarose gel and examined using Bioanalyzer DNA 1000 kit (Agilent).
Whole blood samples
The methylome-wide data from DNA extracted directly from fresh whole blood used in this study originates from an existing methylome-wide investigation of 1500 schizophrenia case-control samples.Citation32 These samples were assayed, with high consistency in sequence data quality and distribution of methylation signal, using 2–5 µg of DNA as starting material (mean = 4.1 µg).
Methylome-wide profiling
We used MethylMiner (Invitrogen), which employs methyl-CpG binding domain (MBD) protein to enrich for the methylated genomic DNA fraction, followed by single-end next-generation sequencing (NGS) on the Applied Biosystems SOLiD platform (Life Technologies). Methods were standard and based upon manufacturers’ recommendations. With the exception that blood spots were sequenced using the SOLiD 5500xl instrument and whole blood samples were sequenced using SOLiD4 instruments, the same protocol was applied for both sample types. Briefly, genomic DNA was fragmented with ultra-sonication to a median fragment size of 150bp. The methylated fraction of the genome was extracted using MethylMiner and used as input material for NGS SOLiD libraries. The samples were barcoded and pooled in equal molarities prior to emulsion PCR and attached to beads. The beads were deposited to slides and the first 50bp of each fragment were sequenced. This enrichment in combination with NGS (MBD-seq) has already been demonstrated to be highly specific, sensitive and applicable to identify differently methylated regions.Citation35-Citation40
We have recently developed a data analysis pipeline that is specifically designed for MBD-seq.Citation32 In the present investigation we are following the analysis and quality control steps from that pipeline. In short, the sequenced reads were aligned to the human genome (build hg19/GRCh37) using BioScope 1.2 (Life Technologies). In the case of MBD-seq, only fragments with methylated CpGs can be extracted. Given that we know exactly where the CpGs are located, there is no need to search for read peaks to find methylated sites.Citation41,Citation42 We therefore simply calculated coverage for the ~27 million autosomal CpG sites in the reference genome (hg19/GRCh37). A standard procedure is to count the sequence reads covering the CpG. Because the methylation of any CpG in the entire fragment could lead to its capture, the read length is sometimes extended to the expected fragment length. However, because not all fragments have exactly the same size, there may be variation between samples and the fragment pool obtained after shearing may not be identical to the pool that gets successfully sequenced (e.g., smaller fragments may be more likely to get extracted by the enrichment protocol), this procedure can be imprecise. Thus, rather than assuming an identical pre-determined fragment size for all fragments and samples, we estimated the fragment size distribution for each sample from the empirical sequencing data.Citation43 The sample specific estimated fragment size distributions were used to calculate the probability for each read that the fragment it is tagging covers the CpG under consideration. Coverage for each CpG can then be calculated by taking the sum of the probabilities that all fragments in its neighborhood cover the CpG.
CpG sites in loci that are problematic in terms of alignment need to be eliminated prior to analyses, as coverage estimates will be confounded with alignment errors. Based on the results from a previous in silico experimentCitation32 using exactly the same alignment parameters as applied in this investigation we eliminated CpG sites with known alignment problems.
Outcome variables
Multi-reads (reads aligning to multiple locations), mismatches (non-perfect alignments) and duplicate reads (reads that start at the same nucleotide position) can be explained by biological reasons or be created by technical artifacts. If the same methylome (i.e., the same DNA sample) is investigated multiple times any variation caused by biological reasons can be excluded. Therefore, the increased occurrence of multi-reads, mismatches and duplicate reads are indications of decreased sequence quality. To compare the quality of the sequence reads, we used four outcome measures: (1) percentage of aligned reads, where fewer reads are expected to map if data quality is lower, (2) percentage of uniquely aligned reads vs. multi-reads, where we expect fewer uniquely aligning reads if data quality is lower, (3) percentage of non-duplicate reads that represent reads that start uniquely at a specific location, where we would expect a smaller percentage of unique start locations if data quality is lower and (4) the percentage of bases of a read that correctly match the sequence of the reference genome, where we expect that base call errors result in fewer matches with the reference.
Furthermore, we investigated the methylome-wide enrichment profile across biological features. Discrepancies in profiles, as compared with the DNA from whole blood, would indicate that data quality is decreased. For this purpose we annotated all ~27 million CpGs in the autosomal human reference genome. Annotation features included: regions overlapping with known repetitive elements (RepeatMasker), introns, exons, 3′ and 5′ untranslated regions, CpG islands (CGIs) and isolated CpGs (CpGs located at least 400 bp from any other CpG). Except for the isolated CpGs, which were extracted directly from the reference sequence, the features were downloaded from the UCSC genome browser (http://genome.ucsc.edu/).
| Abbreviations: | ||
| MWAS | = | methylome-wide association study |
| MBD | = | methyl-CpG binding domain |
| MBD-seq | = | methyl-CpG domain enrichment combined with next-generation sequencing |
| GWAS | = | genome-wide association study |
| SNP | = | single nucleotide polymorphism |
| MeDIP | = | methylated DNA immunoprecipitation |
| GC | = | guanine-cytosine |
| NGS | = | next-generation sequencing |
Additional material
Download Zip (95.8 KB)Acknowledgments
This work was supported by the National Institute of Mental Health (Grant RC2 MH089996). This paper is subject to the NIH Public Access Policy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature 1978; 274:775 - 80; http://dx.doi.org/10.1038/274775a0; PMID: 355893
- Kwok JB. Role of epigenetics in Alzheimer’s and Parkinson’s disease. Epigenomics 2010; 2:671 - 82; http://dx.doi.org/10.2217/epi.10.43; PMID: 22122050
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 2008; 82:696 - 711; http://dx.doi.org/10.1016/j.ajhg.2008.01.008; PMID: 18319075
- Hedrich CM, Tsokos GC. Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol Med 2011; 17:714 - 24; http://dx.doi.org/10.1016/j.molmed.2011.07.005; PMID: 21885342
- Ordovás JM, Smith CE. Epigenetics and cardiovascular disease. Nat Rev Cardiol 2010; 7:510 - 9; http://dx.doi.org/10.1038/nrcardio.2010.104; PMID: 20603647
- Sarkies P, Sale JE. Cellular epigenetic stability and cancer. Trends Genet 2012; 28:118 - 27; http://dx.doi.org/10.1016/j.tig.2011.11.005; PMID: 22226176
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature 2007; 447:433 - 40; http://dx.doi.org/10.1038/nature05919; PMID: 17522677
- Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 2010; 465:721 - 7; http://dx.doi.org/10.1038/nature09230; PMID: 20535201
- Jost JP, Saluz HP, Pawlak A. Estradiol down regulates the binding activity of an avian vitellogenin gene repressor (MDBP-2) and triggers a gradual demethylation of the mCpG pair of its DNA binding site. Nucleic Acids Res 1991; 19:5771 - 5; http://dx.doi.org/10.1093/nar/19.20.5771; PMID: 1945854
- Yokomori N, Moore R, Negishi M. Sexually dimorphic DNA demethylation in the promoter of the Slp (sex-limited protein) gene in mouse liver. Proc Natl Acad Sci U S A 1995; 92:1302 - 6; http://dx.doi.org/10.1073/pnas.92.5.1302; PMID: 7877972
- Sutherland JE, Costa M. Epigenetics and the environment. Ann N Y Acad Sci 2003; 983:151 - 60; http://dx.doi.org/10.1111/j.1749-6632.2003.tb05970.x; PMID: 12724220
- Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition 2004; 20:63 - 8; http://dx.doi.org/10.1016/j.nut.2003.09.011; PMID: 14698016
- Cooney CA. Are somatic cells inherently deficient in methylation metabolism? A proposed mechanism for DNA methylation loss, senescence and aging. Growth Dev Aging 1993; 57:261 - 73; PMID: 8300279
- Boks MP, de Jong NM, Kas MJ, Vinkers CH, Fernandes C, Kahn RS, et al. Current status and future prospects for epigenetic psychopharmacology. Epigenetics 2012; 7:20 - 8; http://dx.doi.org/10.4161/epi.7.1.18688; PMID: 22207355
- Melas PA, Rogdaki M, Ösby U, Schalling M, Lavebratt C, Ekström TJ. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J 2012; 26:2712 - 8; http://dx.doi.org/10.1096/fj.11-202069; PMID: 22426120
- Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 2000; 24:88 - 91; http://dx.doi.org/10.1038/71750; PMID: 10615135
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 2010; 11:191 - 203; http://dx.doi.org/10.1038/nrg2732; PMID: 20125086
- Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011; 12:529 - 41; http://dx.doi.org/10.1038/nrg3000; PMID: 21747404
- Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics 2009; 1:177 - 200; http://dx.doi.org/10.2217/epi.09.14; PMID: 22122642
- Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science 2008; 322:881 - 8; http://dx.doi.org/10.1126/science.1156409; PMID: 18988837
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet 2009; 10:241 - 51; http://dx.doi.org/10.1038/nrg2554; PMID: 19293820
- Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, Illig T, et al. A common genetic variant is associated with adult and childhood obesity. Science 2006; 312:279 - 83; http://dx.doi.org/10.1126/science.1124779; PMID: 16614226
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005; 308:385 - 9; http://dx.doi.org/10.1126/science.1109557; PMID: 15761122
- Fowler KE, Reitter CP, Walling GA, Griffin DK. Novel approach for deriving genome wide SNP analysis data from archived blood spots. BMC Res Notes 2012; 5:503; http://dx.doi.org/10.1186/1756-0500-5-503; PMID: 22974252
- Xu H, Zhao Y, Liu Z, Zhu W, Zhou Y, Zhao Z. Bisulfite genomic sequencing of DNA from dried blood spot microvolume samples. Forensic Sci Int Genet 2012; 6:306 - 9; http://dx.doi.org/10.1016/j.fsigen.2011.06.007; PMID: 21737370
- Beyan H, Down TA, Ramagopalan SV, Uvebrant K, Nilsson A, Holland ML, et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res 2012; 22:2138 - 45; http://dx.doi.org/10.1101/gr.134304.111; PMID: 22919074
- Hollegaard MV, Grauholm J, Nørgaard-Pedersen B, Hougaard DM. DNA methylome profiling using neonatal dried blood spot samples: A proof-of-principle study. Mol Genet Metab 2013; 108:225 - 31; http://dx.doi.org/10.1016/j.ymgme.2013.01.016; PMID: 23422032
- Mei JV, Alexander JR, Adam BW, Hannon WH. Use of filter paper for the collection and analysis of human whole blood specimens. J Nutr 2001; 131:1631S - 6S; PMID: 11340130
- Hollegaard MV, Grove J, Grauholm J, Kreiner-Møller E, Bønnelykke K, Nørgaard M, et al. Robustness of genome-wide scanning using archived dried blood spot samples as a DNA source. BMC Genet 2011; 12:58; http://dx.doi.org/10.1186/1471-2156-12-58; PMID: 21726430
- Winkel BG, Hollegaard MV, Olesen MS, Svendsen JH, Haunsø S, Hougaard DM, et al. Whole-genome amplified DNA from stored dried blood spots is reliable in high resolution melting curve and sequencing analysis. BMC Med Genet 2011; 12:22; http://dx.doi.org/10.1186/1471-2350-12-22; PMID: 21306642
- Hollegaard MV, Thorsen P, Norgaard-Pedersen B, Hougaard DM. Genotyping whole-genome-amplified DNA from 3- to 25-year-old neonatal dried blood spot samples with reference to fresh genomic DNA. Electrophoresis 2009; 30:2532 - 5; http://dx.doi.org/10.1002/elps.200800655; PMID: 19639574
- Aberg KA, McClay JL, Nerella S, Xie LY, Clark SL, Hudson AD, et al. MBD-seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case--control samples. Epigenomics 2012; 4:605 - 21; http://dx.doi.org/10.2217/epi.12.59; PMID: 23244307
- Costello EJ, Angold A, Burns BJ, Stangl DK, Tweed DL, Erkanli A, et al. The Great Smoky Mountains Study of Youth. Goals, design, methods, and the prevalence of DSM-III-R disorders. Arch Gen Psychiatry 1996; 53:1129 - 36; http://dx.doi.org/10.1001/archpsyc.1996.01830120067012; PMID: 8956679
- Worthman CM, Stallings JF. Hormone measures in finger-prick blood spot samples: new field methods for reproductive endocrinology. Am J Phys Anthropol 1997; 104:1 - 21; http://dx.doi.org/10.1002/(SICI)1096-8644(199709)104:1<1::AID-AJPA1>3.0.CO;2-V; PMID: 9331450
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 2010; 28:1097 - 105; http://dx.doi.org/10.1038/nbt.1682; PMID: 20852635
- Hogart A, Lichtenberg J, Ajay SS, Anderson SM, Margulies EH, Bodine DM, NIH Intramural Sequencing Center. Genome-wide DNA methylation profiles in hematopoietic stem and progenitor cells reveal overrepresentation of ETS transcription factor binding sites. Genome Res 2012; 22:1407 - 18; http://dx.doi.org/10.1101/gr.132878.111; PMID: 22684279
- Lan X, Adams C, Landers M, Dudas M, Krissinger D, Marnellos G, et al. High resolution detection and analysis of CpG dinucleotides methylation using MBD-Seq technology. PLoS One 2011; 6:e22226; http://dx.doi.org/10.1371/journal.pone.0022226; PMID: 21779396
- Li N, Ye M, Li Y, Yan Z, Butcher LM, Sun J, et al. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods 2010; 52:203 - 12; http://dx.doi.org/10.1016/j.ymeth.2010.04.009; PMID: 20430099
- Nair SS, Coolen MW, Stirzaker C, Song JZ, Statham AL, Strbenac D, et al. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 2011; 6:34 - 44; http://dx.doi.org/10.4161/epi.6.1.13313; PMID: 20818161
- Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res 2010; 38:391 - 9; http://dx.doi.org/10.1093/nar/gkp992; PMID: 19906696
- Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods 2009; 6:Suppl S22 - 32; http://dx.doi.org/10.1038/nmeth.1371; PMID: 19844228
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008; 9:R137; http://dx.doi.org/10.1186/gb-2008-9-9-r137; PMID: 18798982
- van den Oord EJ, Bukszar J, Rudolf G, Nerella S, McClay JL, Xie LY, et al. Estimation of CpG coverage in whole methylome next-generation sequencing studies. BMC Bioinformatics 2013; 14:50; http://dx.doi.org/10.1186/1471-2105-14-50; PMID: 23398781