Abstract
Lysine methylation mediated by methyltransferase enzymes is present on multiple proteins throughout the cell; however, methods to uncover and characterize global protein lysine methylation patterns do not readily exist. Here we developed pan-specific methyl lysine antibodies that we utilized in immunoprecipitation experiments coupled with mass spectrometry to yield one of the first large-scale surveys of protein lysine methylation in vivo. In total, 552 different lysine methylation sites were determined, making this one of the most comprehensive global studies published to date. The large majority of these sites have not been yet reported. These sites showed significantly enriched sequence motifs and resided in proteins that are involved in diverse biological processes, particularly in chromatin organization. Our data provide a comprehensive view of lysine methylation in human cells and a powerful resource to facilitate investigations into the function of lysine methylation on non-histone proteins.
Introduction
Methylation on histone proteins plays a key role in genome stability, chromatin remodeling and gene expression.Citation1-Citation3 Protein lysine methyltransferases (PKMTs) and demethylases are responsible to maintain the dynamic balance of histone methylation in vivo. A large number of investigations have revealed that the aberrant methylation on histones induced by abnormalities in these enzymes are directly associated with cancers, inflammation and other diseases.Citation4 For example, the PKMT EZH2 controls H3K27 methylation and its overexpression has been linked to several types of cancers, including prostate, breast and lung cancers, as well as lymphomas.Citation5
It has been predicted that there are up to 52 genes that encode PKMTs in humans.Citation6 Numerous studies on these PKMTs have focused mainly on their regulation of histone methylation. Nevertheless, it is known that the protein targets of many of these enzymes extend beyond histones. An increasing number of non-histone proteins, such as the tumor suppressor p53, tyrosine kinase VEGFR1 and transcription factor TAF10, have been reported to serve as the substrates of some well-known PKMTs.Citation7-Citation10 These methylation sites are involved in diverse biological events, and different lysine methylation sites on the same protein correlate with distinct biological consequences. One of the best examples is found with p53. K370 monomethylation of p53 by the PKMT SMYD2 was demonstrated to inhibit transcriptional activity via decreasing recruitment of p53 to DNA, while neighboring K372me1 by the PKMT Set7/9 promoted p53 activation via increasing p53 stability.Citation7,Citation8 Additionally, methylation on these non-histone proteins can mediate other posttranslational modifications (PTMs). For instance, methylation at p53 K372 by Set7/9 was required for the binding and subsequent acetylation of p53 by acetyltransferase Tip60.Citation11
Systematic strategies have been applied to characterize new targets of lysine methyltransferases. For example, Rathert et al. utilized peptide array screening to determine the sequence specificity profile of the PKMT G9a and then screened an entire protein database to search for potential substrates of this methyltransferase.Citation12 Levy et al. adopted protein arrays in vitro to identify novel candidate substrates of the PKMT SETD6, in the end finding over one hundred proteins targeted by SETD6.Citation13 Together, these studies indicated that there were abundant non-histone candidate substrates of these methyltransferases in cells. However, due to the limitation of the technologies employed, very few actual methylation sites were demonstrated in any of the aforementioned studies. Therefore, the determination of methylation sites in vivo on a global scale has remained a great unmet challenge.
Immunoprecipitation of modified peptides by pan-specific antibodies coupled with mass spectrometry identification has been successfully applied to the large-scale interrogation of some PTMs, such as tyrosine phosphorylation, lysine acetylation and ubiquitylation.Citation14-Citation17 However, to date, similar analyses have not yet been performed for protein lysine methylation owing to lack of effective antibodies against the three degrees (mono-, di- and tri-) of methylation. Here we present our work toward the first global comprehensive large-scale identification of protein lysine methylation sites by combining peptide immunoprecipitation with pan-specific anti-methyl lysine antibodies with mass spectrometry detection. We identified 552 lysine mono- (me1), di- (me2) and tri- (me3) methylation sites on 413 human proteins. Our data provide a holistic view of protein lysine methylation in vivo and a resource for future functional investigation of lysine methylation in human cells.
Results and Discussion
Pan-specific anti-mono-, di- and tri-methyl lysine polyclonal antibodies were custom produced by Proteintech Group Inc. The specific antigen design (see Materials and Methods) resulted in the antibodies possessing high specificity for the particular degrees of mono-, di- and tri-methyl lysine, respectively (). These antibodies also revealed effective immunoprecipitation of methylated proteins extracted from HeLa cells, not surprisingly particularly histones (). Western blotting was performed to profile the three types of lysine methylation in 13 different types of cell lines (). In general, the methylation patterns in these cell lines were similar, indicating that the recognition of roughly the most abundant methylated proteins across the diverse cells was consistent. Histones were also the most abundant methylated proteins observed in vivo across these cells lines. Histones H3 and H4 have similar dimethylation abundance levels in most of these cells, as dimethylation is the most abundant degree of methylation on several residues, especially H3K9 and H4K20. H4K20 dimethylation in fact has been found to be present on as much as 80% of bulk chromatin extracted H4.Citation18 H3 monomethylation and trimethylation are much more abundant than the respective methylation states on H4, as there are several lysines that can be mono- and tri-methylated on H3, namely K4, K9, K18, K23, K27, K36 and K79, but only one main methylation site on histone H4, namely K20.
Figure 1. Characterization of the antibodies against mono-, di- and tri-methyl lysine. (A) Dot blot analysis of the pan anti-methyl lysine antibodies. Peptides with un- (Kun), mono- (Kme1), di- (Kme2) and tri- (Kme3) methyl lysine were blotted onto nitrocellulose membranes and probed with the antibodies, respectively. (B) Immunoprecipitation (IP) analysis of lysates prepared from HeLa cells using the antibodies. Lanes A, B and C are input, IP eluates with normal rabbit IgG (negative control) and IP eluates with anti-methyl lysine antibodies, respectively. The concentration of SDS-PAGE was 15%. (C) Lysine methylation profiles in 13 human cell lines. The protein lysates of 13 cell lines were separated by 12.5% SDS-PAGE, transferred to PVDF membranes and probed with the antibodies, respectively.
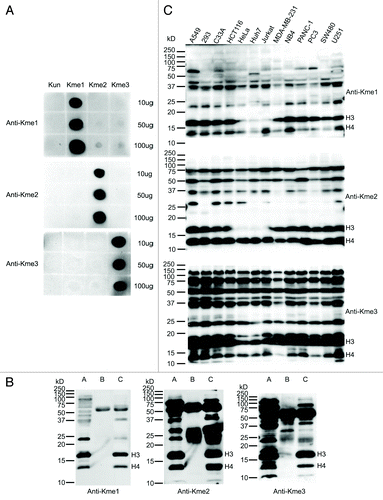
An in vitro methylation assay was performed using recombinant histone octamer and recombinant methyltransferase G9a (Fig. S1). As expected, monomethylation and dimethylation of histone H3 by G9a could be robustly detected by the anti-monomethyl lysine and anti-dimethyl lysine antibodies, respectively. Meanwhile, trimethylation of H3 and H4 were found unexpectedly. However, previously, some in vitro experiments has indicated that G9a is capable to catalyze H3 trimethylation in vitro, though G9a has been mainly considered as a histone mono- and di-methylation methyltransferase in vivo.Citation19-Citation21 In addition, we tested the antibodies on protein extracts from H3K27 methyltransferase EZH2-knockdown human embryonic kidney 293 cells and the corresponding shRNA control 293 cells (Fig. S2). Methylation levels of a couple proteins (especially dimethyl) were found to decrease in EZH2 knockdown cells compared with those in control cells, indicating that EZH2 may have some limited non-histone H3 substrates. In particular, we did not see the differences of H3 methylation levels between these cells, but saw the apparent differences of H4 monomethylation and trimethylation levels. This is interesting since EZH2 has been always considered only as histone H3K27 methyltransferase. With H3 containing many more methylation sites than just K27, we suspect that the observed non-decrease in global H3 methylation is masked by the non-EZH2 affected methyl sites, while H4 contains only one main methylation site (K20). Therefore, the decreased methylation level by EZH2 knockdown could be seen for H4 more easily, but not for H3, and brings up the interesting point of crosstalk between EHZ2, H3K27 methylation and H4 modifications, which has not been previously explored. As a consequence, these pan methyl lysine antibodies were proven to be effective to detect methylation states and their dynamic changes both in vitro and in vivo.
Next, we utilized these antibodies for global identification of the lysine methylation sites in HeLa cells. In brief, after trypsin digestion of total protein extracts, the methylated peptides were enriched by the antibodies through immunoprecipitation at the peptide level, respectively, and were subsequently subjected to mass spectrometry (). Meanwhile, suspecting that the antibodies could be quenched by a high abundance of methylated histone peptides, we also sub-cellularly fractionated the HeLa cells to isolate cytoplasmic and nuclear fractions. In addition, strong cation-exchange (SCX) separation was applied to improve the MS dynamic range and to increase peptide identifications. Lastly, we also employed in vivo heavy-methyl isotope labeling using 13CD3-methionine as a supplement to detect the methylation sites.Citation22
Figure 2. Illustration of the study strategy and characterization of the methylation sites. (A) Illustration of the study strategy. HeLa cells cultured in normal or 13CD3-methionine medium were lysed, or sub-cellularly fractionated to separate cytoplasm and nucleus, and their protein lysates were digested by trypsin. The methylated peptides were enriched through immunoprecipitation and underwent strong cation-exchange (SCX) separation. Each fraction was desalted and subjected to mass spectrometry. (B) MS/MS spectra of the methylated peptide, TKme1AAAAAAAAAPAAAATAPTTAATTAATAAQ. The upper and lower spectra come from normal and heavy methyl-labeled samples, respectively. (C) Heat map of relative distribution of amino acids around the methylated lysine sites. Amino acids from -6 to +6 positions adjacent to the methylated lysine sites and all lysine residues were extracted, respectively. At each position, the frequency of amino acid around methylated lysine sites was divided by that around all lysine residues, and the ratios were visually represented after logarithmic transformation (log2). Color scale reflects the degree of enrichment (red) or depletion (blue). (D) Evolutionary conservation of the methylated lysine sites and all lysines throughout the eukaryotic species. “Kme” and “K” denote methylated lysines and all lysines, respectively. (E) Distribution of methylated lysines and all lysines in protein secondary structure. “Kme” and “K” denote methylated lysines and all lysines, respectively.
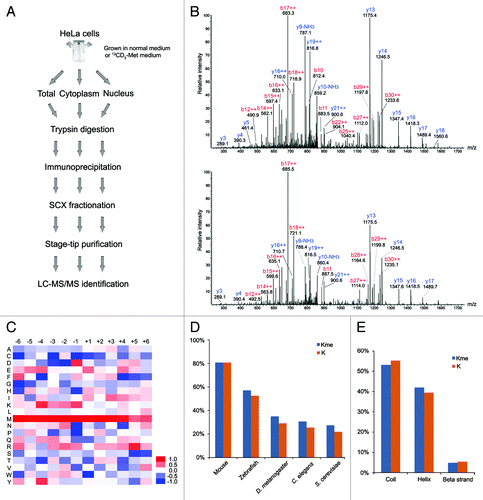
In total, we identified 493 different methylated peptides and determined 552 distinct lysine methylation sites, including 323 monomethylation sites, 127 dimethylation sites and 102 trimethylation sites (, S1 and S2). These peptides and sites were mapped to 413 distinct human proteins. Among them, 410 peptides with 484 sites were identified in the unlabeled samples, while 83 peptides with 84 sites were identified in the heavy 13CD3-methyl isotope labeled prepared samples. The identification of modified peptides in the heavy isotope-labeled sample was lower compared with that in the normal sample, as we did not run as many fractions on the MS instrument for these labeled samples. As an example, the MS/MS spectra derived from the mono-methylated C-terminal peptide of the Signal Recognition Particle 14 kDa protein (SRP14), TKme1AAAAAAAAAPAAAATAPTTAATTAATAAQ, is displayed in . This is the first time that SRP14 was discovered to be methylated. The methylated peptide was identified concurrently in both the normal (upper) and heavy isotope-labeled (lower) cell samples, having almost the same MS/MS fragment ion pattern with the appropriate 4 Da shift on b series of ions due to the heavy methyl group incorporation.
Table 1. Overview of the protein lysine methylation identification in HeLa cells. Normal (normal medium-cultured sample) and heavy (13CD3-methionine medium-cultured sample), respectively
The work presented here demonstrates one of the most in depth analyses of global protein lysine methylation to date. Most of the methylation sites identified in this work have never been reported previously. According to the UniportKB database, only 17 of the identified methylation sites were known to exist.Citation23 As a positive control, eight known histone methylation sites were determined in this work, including H3K27 mono-, di- and tri-methylation, K36 mono- and di-methylation and H4K20 mono-, di- and tri-methylation. Intriguingly, we also seemed to identify a novel monomethylation site at H2BK6. This lysine residue has been previously reported to undergo acetylation, but has not been known to be methylated.Citation17,Citation24 In support, there are several residues on histones that are known to be both acetylated and methylated (i.e., H3K9, H3K27 or H3K4) and these modifications serve antagonistic functions. Therefore, similar dual antagonistic marks may exist on H2B, although the function remains to be determined. In addition to these histone methylation sites, we also observed a few known methylation sites on non-histone proteins. These include K161me1 on Heterogeneous nuclear ribonucleoprotein D-like, K53me1 on 60S ribosomal protein L36a-like, K1162me3 on Wiz and K737me1 on Heat shock protein 90-α (HSP90α).Citation12,Citation25,Citation26 HSP90α K737 is known to be monomethylated by methyltransferase SMYD2.Citation25 The methylation at this site contributed to the formation of SMYD2 protein complex, so as to alter muscle function in muscle cells. Apart from this known site, a novel dimethylation site at HSP90α K615 was detected, suggesting there might be another biological mechanism to regulate HSP90α through lysine methylation.
In addition, we found novel non-histone methylation sites at the lysine residues that had been initially reported to possess other methylation degrees or types of PTMs. Elongation factor 1-α 2 (EEF1A2) K55 and K165 are known to contain trimethylation by similarity with its ortholog in rabbit,Citation27 while they were identified as dimethylation sites in our studies. We compared our data to the two other published large-scale lysine acetylation and ubiquitylation data sets.Citation15,Citation16 Six reported non-histone protein lysine acetylation sites were also found here to have different methylation states, including EEF1A2 K55me2, Isoform 1 of Myosin-9 (MYH9) K8me3, U2 small nuclear ribonucleoprotein A' (SNRPA1) K172me1, Isoform 1 of Serine/threonine-protein phosphatase 4 regulatory subunit 3A (SMEK1) K655me1, SUMO-activating enzyme subunit 2 (UBA2) K271me1 and EH domain-containing protein 4 (EHD4) K327me1. Meanwhile, 12 novel non-histone methylation sites had been previously identified to undergo ubiquitylation at those same sites, including Isoform 1 of LIM and SH3 domain protein 1 (LASP1) K75me1, Isoform 1 of Heterogeneous nuclear ribonucleoprotein D-like (HNRPDL) K161me1 and EEF1A2 K165me1.Citation16 This observation suggests that there could be extensive crosstalk between lysine methylation and other lysine PTMs that could have antagonistic effects to the other PTMs at the same lysine residues.
We sought to determine if there were any potential motif sequences for the methylation sites, so amino acids from -6 to +6 positions adjacent to the methylated lysine sites were extracted to calculate the enrichment degree of 20 canonical amino acids (). Some amino acids, especially the basic amino acids lysine and arginine, showed significant over-representation around the methylated lysine. Lysine was enriched at the upstream positions of the methylated lysine, from -4 to -1. Arginine was enriched at the upstream positions from -3 to -1 and at the downstream +4 and +5 positions. Among them, RK (p value = 0.02) motif was consistent with the previous work, which was considered as the main target motif of the methyltransferase G9a.Citation12 Additionally, methionine was over-represented at almost all the positions from -6 to +4, particularly at -2 (p value = 7.51E-06) and -1 (p value = 8.07E-06) positions. MK motif appears to be evolutionarily conserved through eukaryotic cells, and Pang et al. also noticed the same motif for methylated lysine from a much smaller protein data set in Saccharomyces cerevisiae.Citation28 Lysine and arginine methyltransferases seem to have distinct protein target specificities. In contrast to arginine methylation, we did not find over-represented glycine at any position around the lysine methylation sites, as the RG motif seems to be a dominant sequence for arginine methylation.Citation29 The methylated lysines were a little more conserved compared with the general lysine residues (). This characteristic was consistent with those found for protein lysine acetylation and even for protein phosphorylation.Citation15,Citation30 In addition, methylation preferred to occur in the ordered secondary structure, α-helix (42% vs. 39%) ().
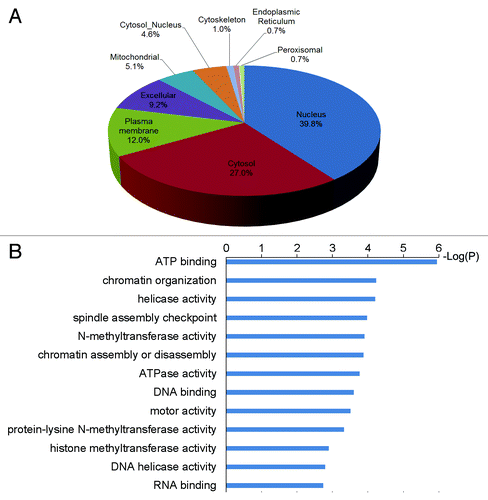
The identified methylated proteins were widely distributed in cell organelles (), and they were particularly enriched in the nucleus (p value = 5.85E-6). This was consistent with the fact that many established histone lysine methyltransferases are also known to be nuclear localized. Gene Ontology (GO) was used to analyze biological functions among the methylated proteins. Over-represented GO terms included chromatin organization, N-methyltransferase activity, DNA binding and motor activity (). A large number of the methylated proteins in our study have been previously found with at least one other type of PTM. Therefore, it is possible that there could be crosstalk between methylation and other PTMs even though they may not be found in close proximity in primary structure. We revealed novel lysine methylation sites in some important regulatory proteins, particularly proteins involved in epigenetic regulation. Taking the serine/threonine protein kinase ATM as an example, it is critical in DNA repair, cell cycle arrest and apoptosis, and it phosphorylates histone H2A after DNA damage.Citation31,Citation32 ATM has previously been confirmed to be phosphorylated and acetylated in response to DNA damage.Citation33,Citation34 Acetylation of ATM K3016 has been shown to activate its kinase activity, and we now found a monomethylation site at ATM K2643. Although our identification came from essentially normal asynchronous cells, it would be interesting to investigate if this methylation is regulated and has the influence on the ATM kinase activity during DNA damage repair and if it interplays with other PTMs. It was also noted that two other proteins involved in histone modifications, histone acetyltransferases MYST2 and MYST3, were detected to undergo methylation. These two acetyltransferases are responsible for the acetylation of H4 and H3 in vivo, respectively, and have been previously found to contain multiple phosphorylation and acetylation sites.Citation15,Citation33,Citation35-Citation37 The discovery of novel methylation sites on these proteins lends insight into complex regulation to these proteins as well as potentially modulation to gene expression. Remarkably, seven predicted protein methyltransferases including five histone lysine methyltransferases, SETDB1, PRMT3, DOT1L, WHSC1, MLL4, MLL2 and NNMT, were found to carry methylation sites. These findings suggest that methylation could possibly act as an activating or inhibiting mechanism to regulate activity or specificity of these enzymes. It might also be possible for the existence of methylation cascades to regulate these enzyme activities by other methyltransferases or in automethylation format, such as what has been described for G9a.Citation19
Figure 3.(A) Subcellular localization of methylated proteins. (B) Over-represented GO terms of methylated proteins.
In sum, lysine methylation, especially on non-histone proteins has received increased attention for its role in regulating protein function and could possibly be linked to human disorders. More and more evidence has linked aberrant protein lysine methylation with different types of cancers.Citation5,Citation38,Citation39 Therefore, building human lysine methylome atlases in different cell and tissue types, especially during disease states will be needed as a first pass to build testable hypotheses and design experiments to probe this phenomenon. Our current approaches and data have provided methods and a foundation for assessing the methylome and an initial glimpse into the global view of lysine methylation in human cells. We anticipate that our first study in this area will be a useful resource across many biological fields to investigate the functions of in vivo protein lysine methylation in future experiments.
Materials and Methods
Antibodies
Lys (meX)-Ahx (aminocaproic acid)-Cys (X = 1, 2 and 3) short peptides were synthesized by Proteintech Group Inc. and were conjugated to carrier protein keyhole limpet hemocyanin (KLH) using MBS (m-maleimidobenzoyl-N-hydoxysuccinimide ester), respectively. The conjugated proteins were injected to rabbits to produce antibodies in Proteintech Group Inc. Other antibodies used in this study are following: EZH2 (Cell Signaling); GAPDH (Sigma-Aldrich); H2B (Cell Signaling).
Cell culture, lysis and subcellular fractionation
All cell lines except HeLa S3 cell were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and antibiotics. HeLa S3 cells were maintained in minimum essential Joklik modified media (Sigma Aldrich) as previously described.Citation40 The homemade Joklik medium supplemented with 13CD3-methionine (Sigma) and 10% dialyzed FBS was used to maintain 13CD3-methionine-labeled HeLa cells. After washed in PBS twice, cells were lysed in the lysis buffer containing 8 M urea, 50 mM TRIS-HCl (pH 8.3), 50 mM NaCl and 1 × Halt protease and phosphatase inhibitor cocktail (Thermo Scientific). Cytoplasmic and nuclear fractions from HeLa cells were prepared using the method previously described.Citation41
Protein immunoprecipitation (IP)
HeLa cells were lysed in 1% NP40 lysis buffer (20 mM TRIS-HCl, pH 7.5, 150 mM NaCl, 1% NP40 and 1 × Halt protease and phosphatase inhibitor cocktail). One mg total lysate was incubated with 10 μg antibody or Rabbit IgG (Sigma Aldrich) coupled to protein A/G agarose beads (Thermo Scientific) overnight at 4°C. The beads were washed with the NP40 lysis buffer three times, and then proteins were eluted from the beads with SDS buffer (2% SDS, 10 mM DTT and 50 mM TRIS-HCl, pH 6.8).
In vitro methylation assay
Methylation reactions were conducted as described.Citation42 10 ug recombinant human histone octamers (EpiCypher) were incubated overnight with 10 ug GST or recombinant G9a in the reaction buffer containing 0.1 mM S-adenosyl-methionine (SAM, Sigma-Aldrich), 50 mM TRIS-HCl (pH 8.0), 10% glycerol, 20 mM KCl, 5 mM MgCl2, 1 mM DTT and 1 mM PMSF. One ug histone octamers were subjected to western blotting.
EZH2 knockdown assay
EZH2 shRNA lentiviral transduction particles and shRNA control transduction particles were ordered from Sigma-Aldrich and were transfected into human embryonic kidney 293 cells according to the manufacture’s protocol. The cells were lysed in the lysis buffer containing 2% SDS, 50 mM TRIS-HCl (pH 7.5), 50 mM NaCl and 1 × Halt protease and phosphatase inhibitor cocktail (Thermo Scientific).
Immunoblot analysis
Different amount of short peptides (Lys (meX)-Ahx-Cys, X = 0, 1, 2 and 3, Proteintech Group Inc.) were spotted on nitrocellulose membranes (Invitrogen). 50 μg total lysates of cell lines or IP-eluted samples were separated by 12.5% or 15% SDS-PAGE gels and subsequently transferred to PVDF membranes (Millipore). Membranes were blocked in 5% milk in TBST (0.05% Tween20) buffer 1 h at room temperature and then incubated with the primary antibodies in 5% milk in TBST buffer overnight at 4°C. After washed in TBST 3 × 5min, the membranes were incubated with the secondary antibody goat anti-rabbit IgG conjugated with HRP (Thermo Scientific) (Clean-Blot IP Reagent (Thermo Scientific) was used to IP samples) 1h at room temperature. After washed in TBST 4 × 10 min, the membranes were incubated with ECL reagent (GE Healthcare) and exposed to X-ray film in the dark room.
Trypsin digestion and peptide immunoprecipitation
About 20 mg protein lysate per antibody were reduced and alkylated by DTT and iodoacetamide and then were diluted to a final urea concentration of 1.5 M using 50 mM TRIS-HCl (pH 8.3). Trypsin (Roche) was added to protein solutions at a ratio of 1:100 (w/w) and proteins were digested overnight at 37°C. After digestion, peptides were desalted using Sep-Pak C18 cartridges (Waters) as described previously and were lyophilized.Citation43 The lyophilized peptides were dissolved in 1 ml of IP loading buffer (50 mM MOPS, pH 7.2, 10 mM Na2HPO4, 50 mM NaCl) and centrifuged at the maximal speed for 5 min to remove any insoluble material. The supernatant was incubated with 50 μg antibody coupled to protein A/G agarose beads overnight at 4°C and washed with IP loading buffer three followed by MilliQ water three times. Peptides were eluted from beads using 50 μl of 0.1% TFA twice and the fractions were combined to lyophilize.
Strong cation-exchange (SCX) fractionation and Stage-tip purification
Lyophilized peptides were dissolved in SCX Buffer A (10 mM KH2PO4, 30% acetonitrile (ACN), pH 2.7) and centrifuged at the maximal speed for 5 min to remove any insoluble material. Peptides were loaded to a PolySULFOETHYL A™ column (4.6 mm I.D. × 250 mm, PolyLC) using System Gold HPLC (Beckman Coulter) and separated with a gradient of 2 min 100% Buffer A, 0–25% Buffer B (10 mM KH2PO4, 500 mM KCl, 30% ACN, pH 2.7) over 33 min and followed by 5 min 100% Buffer B. Fractions were collected and finally pooled to 5 (heavy-labeled sample) or 11 fractions (normal sample). Each fraction was frozen in liquid nitrogen and then lyophilized. The lyophilized peptides were redissolved in 0.1% acetic acid and subjected to desalting before LC-MS/MS analysis using homemade C18 STAGE tips as previously described.Citation44
NanoLC-MS/MS analysis
NanoLC-MS/MS was performed on a LTQ-Orbitrap XL (Thermo Scientific) mass spectrometer equipped with an AS2 autosampler (Eksigent) and an Agilent 1200 binary HPLC pump (Agilent). Lyophilized samples were dissolved in Buffer A (0.1 M acetic acid in water) and loaded to a homemade C18 analytical column (75 μm I.D. × 150 mm) packed with ReproSil-Pur C18-AQ 3 μm resin (Dr Maisch GmbH). A 90 min LC gradient from 5 to 35% Buffer B (98% ACN, 0.1 M acetic acid) was used to separate peptides at a flow rate of 200 nL/min (post-split). The full MS scan range was m/z 350–1600. The top 10 precursor ions were selected in the full MS scan in the Orbitrap with R = 60,000 (m/z 400) to perform MS/MS scans in the ion trap by collision-induced dissociation (CID). The automated gain control (AGC) values were 1E6 and 1E4 for full MS and MS/MS scans, respectively. Normalized CID energy was set to 35.0. Dynamic exclusion was enabled with +/− 1.5 mass window and exclusion time of 30 sec. Lock mass calibration in full MS scan was implemented using polysiloxane ions, 371.1012 and 445.1200.
Data analysis
pFind studio package was used for database searching and analyzing the results.Citation45 The acquired MS/MS spectra were searched against the target-decoy database consisting of forward and reversed protein sequences in the human IPI database (version 3.87).Citation46 The precursor ion tolerance was set to 10 ppm, and the fragment ion tolerance was set to 0.5 Da. Carbamidomethylation (57.0215) of cysteine was considered as a static modification, and mono- (14.0156), di- (28.0313) and tri-methylation (42.0469) of lysine and oxidation (15.9949) of methionine were considered as dynamic modifications. For heavy-isotope labeled sample, heavy mono- (18.0378), di- (36.0757) and tri-methylation (54.1135) of lysine, and unoxidized (4.0222) and oxidized (20.0171) types of heavy-isotope methionine were also set as dynamic modifications. A 1.0% False Discover Ratio (FDR) was used to filter the identified peptides.Citation47 All spectra of methylated peptides were manually checked. PSORT was used to predict protein subcellular localization.Citation48 SABLE was used for prediction of the protein secondary structure.Citation49 Cytoscape plugin BiNGO was used to assess overrepresentation of Gene Ontology (GO) categories.Citation50 The hypergeometric model and the Benjamini Hochberg false discovery rate correction were applied. A probability value of 0.05 was considered to be significant.
Additional material
Download Zip (763.6 KB)Acknowledgments
We are very grateful to Alex Wilkinson and Scott Carlson for performing the in vitro G9a methylation assay. We also thank members of the Garcia lab, Shu Lin, Rosalynn C. Molden, Barry M. Zee and also Prof. Or Gozani for reading the manuscript and giving constructive suggestions. This work was supported by funding from an NIH Innovator grant (DP2OD007447) from the Office of the Director, National Institutes of Health, a National Science Foundation (NSF) Early Faculty CAREER award and NSF grant CBET-0941143.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev 2002; 12:198 - 209; http://dx.doi.org/10.1016/S0959-437X(02)00287-3; PMID: 11893494
- Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol 2002; 14:286 - 98; http://dx.doi.org/10.1016/S0955-0674(02)00335-6; PMID: 12067650
- Sims RJ 3rd, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet 2003; 19:629 - 39; http://dx.doi.org/10.1016/j.tig.2003.09.007; PMID: 14585615
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012; 13:343 - 57; http://dx.doi.org/10.1038/nrg3173; PMID: 22473383
- Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res 2008; 647:21 - 9; http://dx.doi.org/10.1016/j.mrfmmm.2008.07.010; PMID: 18723033
- Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, et al. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des 2011; 78:199 - 210; http://dx.doi.org/10.1111/j.1747-0285.2011.01135.x; PMID: 21564555
- Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, et al. Regulation of p53 activity through lysine methylation. Nature 2004; 432:353 - 60; http://dx.doi.org/10.1038/nature03117; PMID: 15525938
- Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006; 444:629 - 32; http://dx.doi.org/10.1038/nature05287; PMID: 17108971
- Luttun A, Tjwa M, Carmeliet P. Placental growth factor (PlGF) and its receptor Flt-1 (VEGFR-1): novel therapeutic targets for angiogenic disorders. Ann N Y Acad Sci 2002; 979:80 - 93; http://dx.doi.org/10.1111/j.1749-6632.2002.tb04870.x; PMID: 12543719
- Kouskouti A, Scheer E, Staub A, Tora L, Talianidis I. Gene-specific modulation of TAF10 function by SET9-mediated methylation. Mol Cell 2004; 14:175 - 82; http://dx.doi.org/10.1016/S1097-2765(04)00182-0; PMID: 15099517
- Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, et al. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol Cell 2008; 29:392 - 400; http://dx.doi.org/10.1016/j.molcel.2007.12.025; PMID: 18280244
- Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, et al. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol 2008; 4:344 - 6; http://dx.doi.org/10.1038/nchembio.88; PMID: 18438403
- Levy D, Liu CL, Yang Z, Newman AM, Alizadeh AA, Utz PJ, et al. A proteomic approach for the identification of novel lysine methyltransferase substrates. Epigenetics Chromatin 2011; 4:19; http://dx.doi.org/10.1186/1756-8935-4-19; PMID: 22024134
- Ballif BA, Carey GR, Sunyaev SR, Gygi SP. Large-scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. J Proteome Res 2008; 7:311 - 8; http://dx.doi.org/10.1021/pr0701254; PMID: 18034455
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325:834 - 40; http://dx.doi.org/10.1126/science.1175371; PMID: 19608861
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 2011; 44:325 - 40; http://dx.doi.org/10.1016/j.molcel.2011.08.025; PMID: 21906983
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 2006; 23:607 - 18; http://dx.doi.org/10.1016/j.molcel.2006.06.026; PMID: 16916647
- Pesavento JJ, Bullock CR, LeDuc RD, Mizzen CA, Kelleher NL. Combinatorial modification of human histone H4 quantitated by two-dimensional liquid chromatography coupled with top down mass spectrometry. J Biol Chem 2008; 283:14927 - 37; http://dx.doi.org/10.1074/jbc.M709796200; PMID: 18381279
- Chin HG, Estève PO, Pradhan M, Benner J, Patnaik D, Carey MF, et al. Automethylation of G9a and its implication in wider substrate specificity and HP1 binding. Nucleic Acids Res 2007; 35:7313 - 23; http://dx.doi.org/10.1093/nar/gkm726; PMID: 17962312
- Patnaik D, Chin HG, Estève PO, Benner J, Jacobsen SE, Pradhan S. Substrate specificity and kinetic mechanism of mammalian G9a histone H3 methyltransferase. J Biol Chem 2004; 279:53248 - 58; http://dx.doi.org/10.1074/jbc.M409604200; PMID: 15485804
- Collins RE, Tachibana M, Tamaru H, Smith KM, Jia D, Zhang X, et al. In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J Biol Chem 2005; 280:5563 - 70; http://dx.doi.org/10.1074/jbc.M410483200; PMID: 15590646
- Ong SE, Mann M. Identifying and quantifying sites of protein methylation by heavy methyl SILAC. Curr Protoc Protein Sci 2006; Chapter 14:Unit 14 9.
- Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A. UniProtKB/Swiss-Prot. Methods Mol Biol 2007; 406:89 - 112; PMID: 18287689
- Beck HC, Nielsen EC, Matthiesen R, Jensen LH, Sehested M, Finn P, et al. Quantitative proteomic analysis of post-translational modifications of human histones. Mol Cell Proteomics 2006; 5:1314 - 25; http://dx.doi.org/10.1074/mcp.M600007-MCP200; PMID: 16627869
- Donlin LT, Andresen C, Just S, Rudensky E, Pappas CT, Kruger M, et al. Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization. Genes Dev 2012; 26:114 - 9; http://dx.doi.org/10.1101/gad.177758.111; PMID: 22241783
- UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res 2012; 40:Database issue D71 - 5; http://dx.doi.org/10.1093/nar/gkr981; PMID: 22102590
- Kahns S, Lund A, Kristensen P, Knudsen CR, Clark BF, Cavallius J, et al. The elongation factor 1 A-2 isoform from rabbit: cloning of the cDNA and characterization of the protein. Nucleic Acids Res 1998; 26:1884 - 90; http://dx.doi.org/10.1093/nar/26.8.1884; PMID: 9518480
- Pang CN, Gasteiger E, Wilkins MR. Identification of arginine- and lysine-methylation in the proteome of Saccharomyces cerevisiae and its functional implications. BMC Genomics 2010; 11:92; http://dx.doi.org/10.1186/1471-2164-11-92; PMID: 20137074
- Boisvert FM, Côté J, Boulanger MC, Richard S. A proteomic analysis of arginine-methylated protein complexes. Mol Cell Proteomics 2003; 2:1319 - 30; http://dx.doi.org/10.1074/mcp.M300088-MCP200; PMID: 14534352
- Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, et al. Systematic functional prioritization of protein posttranslational modifications. Cell 2012; 150:413 - 25; http://dx.doi.org/10.1016/j.cell.2012.05.036; PMID: 22817900
- Canman CE, Lim DS. The role of ATM in DNA damage responses and cancer. Oncogene 1998; 17:3301 - 8; http://dx.doi.org/10.1038/sj.onc.1202577; PMID: 9916992
- Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 2002; 4:993 - 7; http://dx.doi.org/10.1038/ncb884; PMID: 12447390
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160 - 6; http://dx.doi.org/10.1126/science.1140321; PMID: 17525332
- Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol 2007; 27:8502 - 9; http://dx.doi.org/10.1128/MCB.01382-07; PMID: 17923702
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006; 127:635 - 48; http://dx.doi.org/10.1016/j.cell.2006.09.026; PMID: 17081983
- Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A 2008; 105:10762 - 7; http://dx.doi.org/10.1073/pnas.0805139105; PMID: 18669648
- Mayya V, Lundgren DH, Hwang SI, Rezaul K, Wu L, Eng JK, et al. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal 2009; 2:ra46; http://dx.doi.org/10.1126/scisignal.2000007; PMID: 19690332
- Takawa M, Cho HS, Hayami S, Toyokawa G, Kogure M, Yamane Y, et al. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res 2012; 72:3217 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-11-3701; PMID: 22556262
- Komatsu S, Imoto I, Tsuda H, Kozaki KI, Muramatsu T, Shimada Y, et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 2009; 30:1139 - 46; http://dx.doi.org/10.1093/carcin/bgp116; PMID: 19423649
- Zee BM, Levin RS, Xu B, LeRoy G, Wingreen NS, Garcia BA. In vivo residue-specific histone methylation dynamics. J Biol Chem 2010; 285:3341 - 50; http://dx.doi.org/10.1074/jbc.M109.063784; PMID: 19940157
- Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, et al. Directed proteomic analysis of the human nucleolus. Curr Biol 2002; 12:1 - 11; http://dx.doi.org/10.1016/S0960-9822(01)00650-9; PMID: 11790298
- Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell 2007; 27:636 - 46; http://dx.doi.org/10.1016/j.molcel.2007.07.012; PMID: 17707234
- Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol 2005; 23:94 - 101; http://dx.doi.org/10.1038/nbt1046; PMID: 15592455
- Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 2003; 75:663 - 70; http://dx.doi.org/10.1021/ac026117i; PMID: 12585499
- Li D, Fu Y, Sun R, Ling CX, Wei Y, Zhou H, et al. pFind: a novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics 2005; 21:3049 - 50; http://dx.doi.org/10.1093/bioinformatics/bti439; PMID: 15817687
- Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. The International Protein Index: an integrated database for proteomics experiments. Proteomics 2004; 4:1985 - 8; http://dx.doi.org/10.1002/pmic.200300721; PMID: 15221759
- Elias JE, Haas W, Faherty BK, Gygi SP. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods 2005; 2:667 - 75; http://dx.doi.org/10.1038/nmeth785; PMID: 16118637
- Horton P, Nakai K. Better prediction of protein cellular localization sites with the k nearest neighbors classifier. Proc Int Conf Intell Syst Mol Biol 1997; 5:147 - 52; PMID: 9322029
- Adamczak R, Porollo A, Meller J. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins 2005; 59:467 - 75; http://dx.doi.org/10.1002/prot.20441; PMID: 15768403
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005; 21:3448 - 9; http://dx.doi.org/10.1093/bioinformatics/bti551; PMID: 15972284