Abstract
Histone deacetylases (HDACs) and acetyltransferases have important roles in the regulation of protein acetylation, chromatin dynamics and the DNA damage response. Here, we show in human colon cancer cells that dietary isothiocyanates (ITCs) inhibit HDAC activity and increase HDAC protein turnover with the potency proportional to alkyl chain length, i.e., AITC < sulforaphane (SFN) < 6-SFN < 9-SFN. Molecular docking studies provided insights into the interactions of ITC metabolites with HDAC3, implicating the allosteric site between HDAC3 and its co-repressor. ITCs induced DNA double-strand breaks and enhanced the phosphorylation of histone H2AX, ataxia telangiectasia and Rad3-related protein (ATR) and checkpoint kinase-2 (CHK2). Depending on the ITC and treatment conditions, phenotypic outcomes included cell growth arrest, autophagy and apoptosis. Coincident with the loss of HDAC3 and HDAC6, as well as SIRT6, ITCs enhanced the acetylation and subsequent degradation of critical repair proteins, such as CtIP, and this was recapitulated in HDAC knockdown experiments. Importantly, colon cancer cells were far more susceptible than non-cancer cells to ITC-induced DNA damage, which persisted in the former case but was scarcely detectable in non-cancer colonic epithelial cells under the same conditions. Future studies will address the mechanistic basis for dietary ITCs preferentially exploiting HDAC turnover mechanisms and faulty DNA repair pathways in colon cancer cells vs. normal cells.
Introduction
According to the American Cancer Society, about 142,820 people will be diagnosed with colorectal cancer and almost 51,000 people will die of the disease in 2013.Citation1 Cruciferous vegetables such as broccoli, Brussels sprouts, cabbage, cauliflower and watercress protect against colorectal cancer and other leading causes of cancer-related death.Citation2 The beneficial effects of cruciferous vegetables have been attributed, at least in part, to their content of isothiocyanates (ITCs).Citation3 Dietary ITCs and their metabolites act via multiple mechanisms,Citation4 including epigenetic changes at the level of DNA methylation and histone modifications.Citation5,Citation6
Histone deacetylase (HDAC) activity and chromatin remodeling affect DNA damage and repair pathways.Citation7-Citation9 HDACs are chromatin modifiers that alter gene expression, but also exert a broader range of functions by deacetylating non-histone proteins.Citation7,Citation10 HDACs overexpressed in cancer cells have been implicated in protecting such cells from genotoxic insults.Citation8 HDAC inhibitors such as trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA) and valproic acid (VPA) trigger cancer cell death by removing the protective effects of HDACs on DNA.Citation7,Citation11-Citation13 Open chromatin can provide greater access to genotoxins, while DNA repair mechanisms may be inhibited due to the altered acetylation status of key repair proteins.
Sulforaphane (SFN) and related ITCs inhibit HDAC activity and cause histone hyperacetylation in cancer cells.Citation14-Citation19 Recently, we showed that SFN decreased HDAC protein expression in human colon cancer cells, with HDAC3 identified as an early “sentinel” HDAC.Citation20 Here, we sought to examine the structure-activity relationship among ITCs with respect to HDAC changes and DNA damage/repair pathways in human colon cancer cells, including the role of CtBP-interacting protein (CtIP). The latter protein is a key player in homologous recombination,Citation21 it influences cellular tolerance to anti-cancer drugs,Citation22 and recent evidence points to acetylation as a critical regulator of CtIP activity.Citation7,Citation9 Our findings provide clear evidence for a differential effect of ITCs toward DNA repair events in colon cancer cells vs. non-cancer cells, with implications for improving upon current therapeutic strategies.
Results
ITCs inhibit HDAC activity and expression
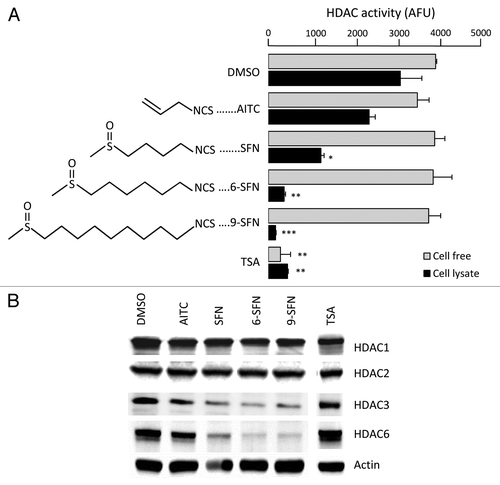
ITCs that occur naturally in mustard, broccoli, wasabi and watercress were examined for effects on HDAC activity and expression (). HDAC activity was reduced significantly in whole cell lysates of HCT116 colon cancer cells after treatment with SFN, 6-SFN and 9-SFN, the potency increasing with alkyl chain length (). When ITCs were added directly to HeLa nuclear extracts, HDAC activity was not affected (). Loss of HDAC activity was dose- and time-dependent (Fig. S1). Immunoblotting of whole cell lysates revealed a marked loss of HDAC3 and HDAC6 (), with little or no changes in other class I and II HDACs. The positive control, TSA, inhibited HDAC activity in cell free assays and whole cell lysates (), without loss of HDAC protein expression (). We focused on HDAC3 due to its key role in human colon cancerCitation23,Citation24 and our identification of HDAC3 as an early target for SFN-induced HDAC turnover mechanisms.Citation20
Figure 1. Alkyl chain length increases ITC-induced loss of HDAC activity and expression. (A) HCT116 cells were treated with vehicle (DMSO), ITC (15 μM) or TSA (1 μM) and 24 h later HDAC activity was measured in whole cell lysates (black bars). Compounds also were directly incubated with HeLa nuclear extracts in a cell-free assay (gray bars). The chemical structure of each ITC is shown. *p < 0.05, **p < 0.01, ***p < 0.001 vs. vehicle controls. (B) Whole cell lysates were immunoblotted for selected HDACs; β-actin, loading control. Data are representative of at least three independent experiments
ITCs induce DNA damage in colon cancer cells
HDAC3 is important for maintaining genomic stabilityCitation25 and DNA damage control,Citation26 and its inhibition has been shown to induce DNA damage.Citation27 Therefore, under the same conditions as described above, DNA damage was assessed in the ITC-treated colon cancer cells using the comet assay. Tail intensity was increased in cells treated with SFN, 6-SFN and 9-SFN (), whereas AITC was similar to controls (data not shown). Phosphorylated histone H2AX (pH2AX, also known as γH2AX) localizes to double-strand breaks within minutes of their formation and is considered a sensitive DNA damage marker.Citation28 Immunocytochemistry studies revealed increased nuclear pH2AX after treatment with SFN, 6-SFN and 9-SFN (), the order of potency corresponding with that observed in the HDAC activity ( and Fig. S2 for the corresponding DAPI counterstaining of nuclei).
Figure 2. ITCs trigger DNA damage and ATR signaling in colon cancer cells. HCT116 cells were treated as in , and DNA damage was assessed (A) in the comet assay or (B) via pH2AX immunocytochemistry. *p < 0.05, **p < 0.01, ITC vs. vehicle. DAPI stained nuclei are shown in Figure S2. (C) Phosphorylation of H2AX, ATR and CHK2, as determined by immunoblotting. Data are representative of at least two independent experiments.
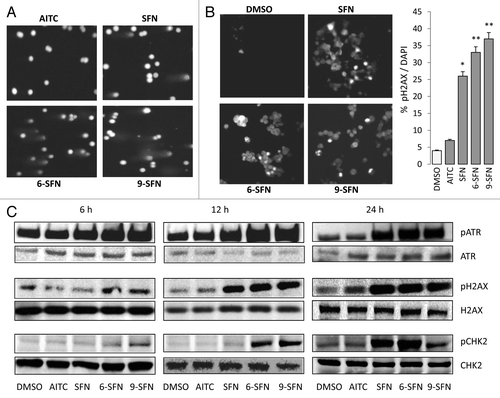
To better understand the time-course of ITC-induced DNA damage, effector kinases were examined by immunoblotting (). Increased phosphorylation of ATR was observed at around 6 h post-treatment with SFN, 6-SFN and 9-SFN, followed by H2AX phosphorylation at 6–12 h and then checkpoint kinase (Chk2) phosphorylation at 12–24 h. Notably, AITC, which had little effect on HDAC activity (), also had minimal impact on ATR, H2AX or Chk2 phosphorylation status under the same assay conditions (). Similar results were obtained in other colon cancer cell lines (data not shown); the SFN-induced DNA damage response was augmented in p21−/− cells but was decreased in p53−/− cells, compared with wild type (Fig. S3).
ITCs induce cell cycle arrest and apoptosis
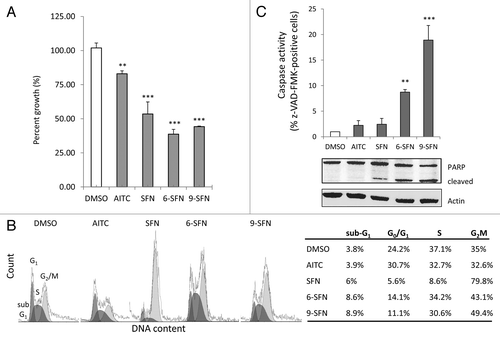
ITCs decreased the viability of HCT116 cells (), with SFN, 6-SFN and 9-SFN being highly significant (p < 0.001). Loss of cell viability was observed in other colon cancer cell lines treated with ITCs (Table S1). Fluorescence-activated cell sorting revealed no significant impact of AITC on cell cycle kinetics, compared with the vehicle controls (, lower left). However, HCT116 cells treated for 24 h with SFN were arrested in G2M, as reported.Citation20,Citation29 Interestingly, 6-SFN and 9-SFN also increased the proportion of cells in G2M, but to a lesser degree than SFN (43.1% and 49.4% vs. 79.8%, respectively). Notably, 6-SFN- and 9-SFN-treated cells had increased multi-caspase activity and PARP cleavage, indicative of greater apoptosis ().
Figure 3. Alkyl chain length increases ITC-induced loss of viability, cell cycle arrest and apoptosis. HCT116 cells treated for 24 h with 15 μM ITC, as in , were examined for (A) cell viability by CCK-8 assay, (B) DNA content via flow cytometry or (C) caspase activity and PARP cleavage. **p < 0.01, ***p < 0.001 vs. vehicle controls.
ITCs enhance CtIP acetylation and turnover
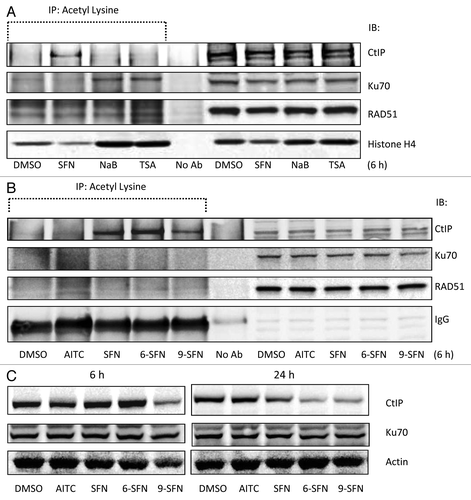
HDAC inhibitors alter the acetylation status of key DNA repair proteins,Citation8 including CtIP, Ku70 and RAD51. Under the same experimental conditions as in , SFN increased the acetylation status of CtIP at 6 h without affecting Ku70 or RAD51 acetylation (). Interestingly, the HDAC inhibitors TSA and sodium butyrate increased Ku70 acetylation, without affecting CtIP or RAD51 acetylation levels (). 6-SFN and 9-SFN also increased the acetylation of CtIP, whereas AITC lacked this activity (). CtIP immunoprecipitation followed by immunoblotting for acetyl-lysine confirmed these findings (data not shown). Loss of CtIP protein expression was not observed at 6 h, except in the case of 9-SFN treatment (, left panel), whereas SFN, 6-SFN and 9-SFN attenuated CtIP levels at 24 h (, right panel), without affecting Ku70 expression.
Figure 4. ITC-induced CtIP acetylation and loss of CtIP protein expression. (A, B) HCT116 cells were incubated with 15 μM ITC, 10 mM sodium butyrate (NaB) or 1 μM TSA for 6 h and whole cell lysates were immunoprecipitated with anti-acetyl lysine antibody, followed by immunoblotting for CtIP, Ku70, RAD51 or histone H4, as indicated. IgG was used occasionally as a loading control. (C) Nuclear lysates (no acetyl-lysine IP step) were immunoblotted directly for CtIP and Ku70 at 6 h and 24 h, with β-actin as loading control.
HDAC3 levels influence CtIP acetylation and turnover
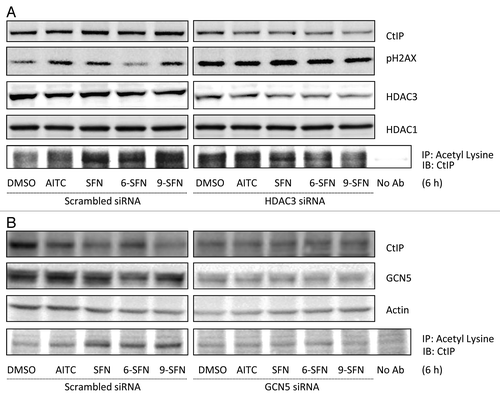
To study the role of HDAC3 in SFN-induced DNA damage and repair processes, HDAC3 knockdown experiments were performed (). Reduced HDAC3 expression following siRNA treatment recapitulated ITC effects with respect to pH2AX induction, CtIP acetylation and attenuated CtIP protein levels. On the other hand, HDAC3 overexpression rescued cells from ITC-induced CtIP acetylation and turnover (Fig. S4). Knockdown of GCN5, a histone acetyltransferase (HAT) involved in CtIP acetylation,Citation7 also rescued the ITC-induced acetylation of CtIP (). Interestingly, GCN5 knockdown did not restore CtIP protein expression to the levels seen in vehicle-treated scrambled siRNA controls (); suggesting additional CtIP turnover pathways were activated independent of acetylation.
Figure 5. HDAC3 knockdown recapitulates ITC-induced CtIP acetylation and turnover, while GCN5 knockdown rescues cells. (A) HCT116 cells transfected with non-specific scrambled siRNA or HDAC3 siRNA for 24 h or (B) GCN5 siRNA for 48 h, followed by ITC treatment for 6 h. Whole cell lysates were immunoprecipitated with anti-acetyl lysine antibody and immunoblotted for CtIP.
Autophagy in ITC-treated cells
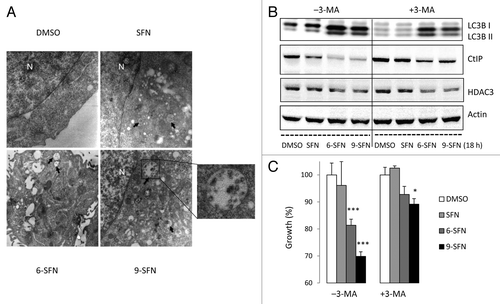
SFN has been reported to cause autophagy,Citation30 which plays a role in CtIP turnover following acetylation.Citation7 Electron microscopy studies revealed that 6-SFN and 9-SFN strongly induced the appearance of autophagosomes (). In addition to numerous double-membrane vacuoles, some of which contained cellular debris, swollen mitochondria and ER were abundant in cells treated with 6-SFN and 9-SFN, and to a lesser extent SFN. Treatment with 3-methyladenine (3-MA), an inhibitor of autophagy, partially or completely blocked cleavage of the autophagy marker LC3B and attenuated ITC-induced CtIP turnover () and cell growth inhibition (). HDAC3 loss was evident in the presence and absence of 3-MA ().
Figure 6. Induction of autophagy by SFN and related ITCs. (A) Representative transmission electron micrographs ( × 10 000) showing autophagosomes (arrows) in HCT116 cells, 24 h after treatment with SFN, 6-SFN and 9-SFN; N, nucleus. A high magnification image (x 35 000) reveals autophagosomes with cellular debris and degraded organelles. (B) Immunoblotting of whole cell lysates for LC3B, CtIP and HDAC3 confirmed that the autophagy inhibitor 3-methyladenine (3-MA) attenuated ITC-induced LC3B cleavage and CtIP loss, which coincided with enhanced cell growth and viability (C). *p < 0.05, ***p < 0.001 vs. the respective vehicle controls.
Differential responses of cancer and non-cancer cells
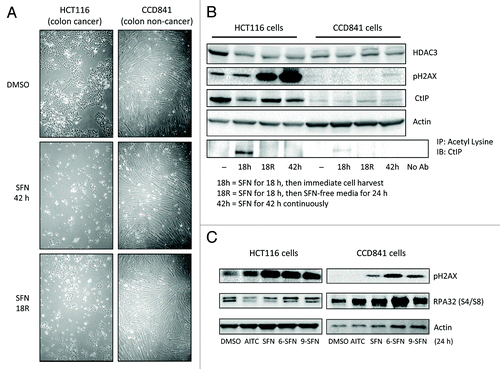
HCT116 colon cancer cells were more sensitive to SFN-induced phenotypic changes, such as cell rounding and colony formation, than CCD841 non-cancer colon epithelial cells (). Constitutive HDAC3 levels were higher in cancer cells than in non-cancer cells, and in the former case HDAC3 protein expression was reduced by continuous or discontinuous SFN treatment (). Continuous SFN treatment for 18 h led to CtIP acetylation and turnover in HCT116 cells and correlated with HDAC3 loss (). No CtIP acetylation was detected at 42 h, possibly due to loss of SFN metabolites or HAT turnover during autophagy/apoptosis. HCT116 cells treated for 18 h with SFN and then replaced with SFN-free media for an additional 24 h continued to express reduced levels of HDAC3 and CtIP (, lane 18R). Prior studies showed that full recovery of HDACs occurred after 72 h.Citation20 pH2AX induction increased even after SFN removal in HCT116 cells, whereas little or no pH2AX was detected in CCD841 cells under the same conditions (). The reduced levels of pH2AX in ITC-treated CCD841 cells compared with HCT116 cells correlated with RPA phosphorylation on Ser4/8, indicating active DNA repair in normal cells (). RPA32 phosphorylation was reduced when CtIP was knocked down by siRNA and was further attenuated by SFN treatment (Fig. S5). The latter experiments also showed that loss of CtIP does not directly induce DNA damage, since CtIP knockdown did not increase pH2AX levels compared with negative siRNA controls. Collectively, these findings provide evidence both at the morphological and molecular level that DNA repair was compromised in cancer cells but not in non-cancer cells following ITC treatment.
Figure 7. Differential responses of non-cancer cells and cancer cells to ITC-induced DNA damage. (A) Phase contrast images of HCT116 cells and CCD841 cells treated with DMSO (vehicle) or 15 μM SFN for 42 h or incubated with SFN for 18 h followed by SFN-free media for 24 h (”R,” removal). (B) Under similar experimental conditions as in (A), HDAC3, pH2AX and CtIP expression were assessed by immunoblotting. Lysates also were immunoprecipitated with anti-acetyl lysine antibody, followed by immunoblotting for CtIP. (C) HCT116 and CCD841 cells were treated with vehicle or 15 μM ITC and whole cell lysates were immunoblotted at 24 h for pH2AX and phosphorylated RPA32 at S4/S8. Data are representative of at least two independent experiments.
Discussion
HDAC inhibitor drugs are thought to activate epigenetically-silenced genes by altering the acetylation of histone proteins.Citation10 However, these agents also affect the acetylation of non-histone proteins, including those with critical roles in DNA damage recognition and repair. The terms lysine deacetylase and “DAC inhibition” have been applied in situations where there is loss of activity and/or expression of deacetylase enzymes, regardless of the final outcomes for histones and chromatin remodeling.Citation5
Our original working hypothesis was that SFN metabolites bind to the HDAC pocket and inhibit HDAC activity.Citation14 A subsequent study identified turnover of HDACs at the protein level, in particular HDAC3 and HDAC6.Citation20 However, this model does not account for the results with TSA and butyrate, which despite their known binding to the HDAC pocket failed to similarly induce CtIP acetylation (). A possible clue came from molecular modeling studies of HDAC3 in association with its co-repressor partner SMRT. Thus, whereas TSA and ITC metabolites docked favorably within the HDAC3 pocket, a second site between HDAC3 and SMRT also demonstrated good affinity for ITC metabolites, but not TSA (Table S2). The –NAC metabolites of AITC, SFN, 6-SFN and 9-SFN interacted most favorably with the allosteric site, longer-chain ITCs having greater affinity (). To our knowledge, this is the first report to model such interactions with the allosteric site, providing new insights into the dissociation of HDAC3/SMRT complexes in colon cancer cells.Citation20 We speculate that binding of ITC metabolites to the allosteric site weaken interactions between HDAC3 and SMRT, which facilitates complex dissociation and GCN5 (HAT) recruitment on CtIP.
Figure 8. Molecular docking of ITCs in the site between HDAC3 and its co-repressor. (A) AITC-NAC, (B) SFN-NAC, (C) 6-SFN-NAC and (D) 9-SFN-NAC were docked into human HDAC3/SMRT inositol tetraphosphate binding pocket (ICM v3.5–1p). Docked ligands are displayed as sticks and colored by atom type, with carbon atoms in orange; residues K474 and K475 are colored in black; protein displayed as Connolly Surface, solid mode and colored by electro potential (ICM v3.5–1p).
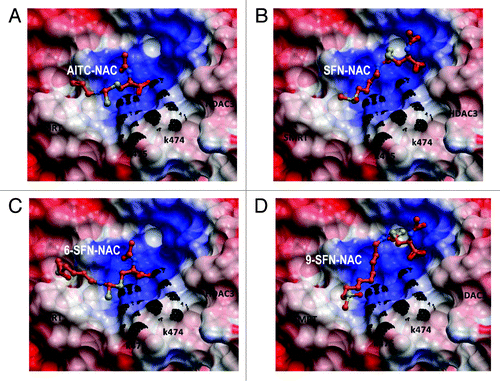
ITC-NAC metabolites oriented into the binding cleft with the negative-charged carboxylate group pointing toward the positively-charged surface between HDAC3 and SMRT (). The basic residues Lys 474 and Lys 475 (part of SMRT) were involved in hydrogen bonding. The binding site at the interface between the two proteins is mainly positively charged, and this surface attracted -S = O groups in the tail of SFN, 6-SFN and 9-SFN (). Increasing chain length generally enhanced interactions and produced additional favorable enthalpy. Future experiments will define the relative levels of AITC, SFN, 6-SFN and 9-SFN metabolites in cancer cells and normal cells and their possible contributions to allosteric site interactions.
HDACs have been implicated in DNA damage and/or repair,Citation9,Citation25,Citation26,Citation31 and HDAC3 knockdown recapitulated some of the changes associated with DNA damage. Notably, pH2AX induction occurred within 6 h, the same timeframe as HDAC3 turnover in SFN-treated colon cancer cells.Citation20 Sirtuin activity assays (data not shown) prompted immunoblotting studies of class III HDACs and the novel finding of nuclear SIRT6 turnover by SFN and other ITCs (Fig. S6). CtIP acetylation was evident following SIRT6 knockdown, as reported,Citation9 and this was enhanced by SIRT6+HDAC3 double knockdown (Fig. S7). Under the same conditions, Ku70 acetylation was not increased (Fig. S7). We are now studying the relative contributions of SIRT6 and HDAC3 toward CtIP stability and turnover, including protein-protein interactions and the key residues for post-translational modifications. A genetic screen provided initial insights into the genes required for ITC-induced DNA damage signaling (manuscript in preparation).
Prolonged HDAC inhibition and an open chromatin configuration exposes DNA to the potential for increased damage from both exogenous and endogenous sources.Citation32-Citation36 The effects of SFN on CtIP acetylation and TSA/butyrate on Ku70 acetylation () point to differential roles in homologous vs. non-homologous repair, respectively.Citation37-Citation39 These findings may be significant, because SFN-induced DNA damage is repaired predominantly through homologous recombination,Citation40 and destabilizing a critical repair protein in this pathway, CtIP, provides an avenue for synthetic lethality.Citation41
HDACs maintain CtIP in the deacetylated state, whereas GCN5-mediated acetylation shunts CtIP into autophagy-mediated degradation.Citation7 We observed that ITC-induced CtIP acetylation and turnover coincided with the activation of an autophagic response, the degree of which increased with length of the alkyl side chain (). Although evidence for HDAC3 directly interacting with CtIP is still lacking, HDAC3 knockdown did not affect SIRT6 levels (Fig. S7), indicating a direct role for HDAC3 on CtIP deacetylation independent of SIRT6.
One hallmark of cancer is genomic instability.Citation42 Therapeutic approaches have sought to exploit the differences in DNA damaging signaling between cancer cells and non-cancer cells, often with mixed results. Because colon cancer cells overexpress HDAC3,Citation23,Citation43 we hypothesized that ITCs might preferentially target DNA damage/repair pathways in cancer cells, leaving non-cancer colonic epithelial cells less affected. In agreement with this hypothesis, ITCs reduced HDAC3 and CtIP levels and induced significant DNA damage which accumulated over time, whereas CCD841 non-cancer cells had little or no such damage (). Defects in double-strand break resection related to ITC-induced HDAC inhibition/turnover and CtIP loss might explain the low levels of pRPA32 in cancer cells, which were strongly increased in non-cancer cells, indicative of active DNA repair ().
Based on the collective results from this investigation, we propose a model for the differential effects in cancer cells vs. non-cancer cells of DAC inhibition and DNA damage/repair signaling following ITC treatment (Fig. S8). Further studies are needed to clarify the precise role of acetylation and other post-translational changes induced by dietary ITCs in non-histone proteins, including CtIP. A clear understanding of such effects should help to clarify the role of dietary ITCs as potential chemosensitizers. Preliminary findings (Fig. S9) showed synergy between low dose SFN and the DNA damaging agent Mitomycin C, with inhibition of HDAC3, decreased CtIP and enhanced apoptosis in colon cancer cells.
Materials and Methods
Cells and test compounds
HCT116, HT29, SW48 and SW480 (colon cancer cells) and CCD841 (non-cancer colonic epithelial cells) from ATCC were cultured in GIBCO-BRL growth medium containing 10% FBS/1% penicillin-streptomycin. Cell lines were authenticated and tested for mycoplasma by IDEXX RADILL. HCT116 p21−/− and p53−/− cells were courtesy of Bert Vogelstein and Kenneth W. Kinzler (Johns Hopkins University). Sulforaphane (SFN), 6-methylsulfinylhexyl isothiocyanate (6-SFN) and 9-methylsulfinylnonyl isothiocyanate (9-SFN) were from LKT laboratories. Allyl isothiocyanate (AITC), TSA, Mitomycin C and 3-methyladenine (3-MA) were from Sigma. Aliquots of the stock solutions were stored at -20°C and thawed for single use before each experiment. AITC was prepared directly in growth medium, whereas other ITCs (50 mM in DMSO) were diluted in growth medium and added to cells at a final concentration of 15 μM, unless indicated otherwise.
HDAC activity
HDAC activity of whole cell lysate was measured using the Fluor-de-Lys assay, as reported earlier.Citation20 Incubations were performed with whole-cell extract (10 μg protein) of HCT116 cells following treatment with DMSO/ITCs or with HeLa nuclear extract (cell free assays), using Fluor-de-Lys substrate in HDAC assay buffer for 30 min at 37°C followed by addition of developer for 30 min. Fluorescence was detected using a Spectra MaxGemini XS fluorescence reader (Molecular Devices), and results were expressed as AFU.
Overexpression and knockdown experiments
HDAC3, as transfection-ready DNA in pCMV6-XL4 vector, and siRNA (Trilencer-27) for HDAC3, GCN5, CtIP, SIRT6 and control siRNA were procured from Origene. Cells were transfected with Lipofectamine 2000 (Invitrogen) at a ratio of 1:3 or 1:4 in reduced-serum medium (OPTI-MEM, Invitrogen), for 24–48 h, using the manufacturer’s protocol. Immunoblotting was performed with whole cell extracts, prepared as reported earlier.Citation20
Flow cytometry
Cell cycle analysis was performed as reported before.Citation20 Briefly, cells in the exponential growth phase were seeded at 0.1 × 106 cells/60-mm culture dish and treated with vehicle or ITC test compounds. Adherent and non-adherent cells were collected at 24 h in cold PBS, fixed in 70% ethanol and stored at 4°C for at least 48 h. Fixed cells were washed with PBS and resuspended in propidium iodide (PI)/Triton X-100 staining solution containing RNaseA. Samples were incubated in the dark for 30 min before cell cycle analysis. DNA content was detected using a Guava-PCA instrument (Guava Technologies).
Cell growth
Cells in the exponential growth phase were plated at a cell density of 5,000 cells per well in 96-well tissue culture plates. After attachment overnight, cells were treated with ITCs for the indicated times. Cell viability was determined using the CCK-8 assay (Dojindo). The colorimetric CCK-8 assay assesses cell viability based on the ability of living cells to reduce soluble WST-8 to formazan.
Caspase activity
HCT116 cells (0.1 × 106) were treated with either DMSO (vehicle) or ITC and harvested after 24 h. Cell number was counted using a Neubauer chamber and adjusted to 5 × 105 cells/ml in 1X Apoptosis Wash Buffer, prior to assays using the MultiCaspase Detection Kit (Guava Technologies). Percent SR-VAD-FMK(+) cells, representing the total apoptotic population, was plotted for each treatment.
Immunoblotting
Whole cell extracts were prepared and immunoblotted as described previously.Citation20 Equal amounts of protein (20 μg/lane) were separated by SDS-PAGE on 4–12% Bis-Tris gel or 3–8% TRIS-acetate gel for larger proteins (NuPAGE, Invitrogen) and transferred to nitrocellulose membranes (Invitrogen). Membranes were saturated with 2% BSA for 1 h, followed by overnight incubation at 4°C with primary antibodies for HDAC1 (#7872), HDAC2 (#7899), HDAC3 (#11417), HDAC6 (#11420), pH2AX Ser139 (#101696), H2AX (#54607), CtIP (#22838), RAD-51 (#8349) and p53 (#126) from Santa Cruz; pATR Ser428 (#2853), pCHK2 Thr68 (#2661), ATR (#2790), CHK2 (#2662), p21WAF1 (#2947), PARP (#9542), GCN5 (#3305) and cleaved caspase-3 (#9661) from Cell Signaling; Ku70 (#K4763), LC3B (#L7543) and β-actin (#A5441) from Sigma; SIRT1 (#39353) and SIRT6 (#39911) from Active Motif; SIRT3 (#2860–1), SIRT4 (#T1295) and SIRT5 (#T1296) from Epitomics; and pRPA32 S4/S8 (#A300–245A) from Bethyl Labs. After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad) for 1 h. Bands were visualized using Western Lightning Plus-ECL Enhanced Chemiluminescence Substrate (Perkin Elmer, Inc.) and detected using FluorChem-8800 chemiluminescent imager (Alpha Innotech).
Immunoprecipitation (IP)
The IP methodology was performed as reported earlier.Citation20 Whole cell extracts from adherent and non-adherent cells were prepared as previously described. Cell extract (500 μg) was pre-cleared with 100 μl Protein A Sepharose CL-4B beads (GE Healthcare Life sciences) on a rotator at 4°C for 2 h. Pre-cleared supernatant was subjected to overnight IP with anti-acetyl lysine antibody (10 μg/mg protein, #AB3879, Millipore). Samples were incubated with 100 μl of beads on a rotator at 4°C for 2 h and acetylated proteins bound to the beads were washed 3 times with PBST, denatured in standard loading buffer and examined by immunoblotting with primary antibodies for CtIP (Santa Cruz, #22838), RAD-51 (Santa Cruz, #8349), Ku70 (Sigma, #K4763) and histone H4 (Cell Signaling, #2592) as described above.
Single cell gel electrophoresis
“Comet” assays were performed as reported earlier.Citation44 In brief, ~106 cells were mixed with low melting agarose to form a cell suspension. Slides were immersed in cold lysis solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris, pH 10.0, 1% sodium sarcosinate, 1% Triton X-100, 10% DMSO) overnight at 4°C followed by electrophoresis at 0.8 V/cm for 30 min. After rinsing at 4°C to neutralize excess alkali, slides were stained with ethidium bromide. Fifty randomly chosen nuclei per slide were analyzed using a Nikon E400 fluorescence microscope linked to Comet Assay III software (Perspective Instruments).
Immunofluorescence
Cells grown on glass coverslips (#1.5, VWR), pre-coated with poly-L-Lysine (Sigma, #P1399), were treated with vehicle or ITCs in 6-well plates. Following treatment, cells were fixed with 2% buffered formalin (10 min) and permeabilized with 0.5% Tween 20, 2.1% citric acid (10 min) at room temperature. Samples were blocked in 1% BSA and incubated overnight with pH2AX Ser139 antibody (Cell Signaling, #9718), followed by incubation with secondary antibody coupled to AlexaFluor 488 (1:250, Molecular Probes) for 1 h. DAPI (Prolong Gold antifade reagent, Molecular Probes) was used to counterstain the nuclei. Fluorescent images were captured on a Zeiss Axiovert 100S Widefield Microscope and MetaMorph Imaging Software (Zeiss) was used for image acquisition and analysis.
Electron microscopy
Cells treated with either DMSO (control) or ITCs were collected at 24 h and processed for transmission electron microscopy (TEM). Briefly, cells were fixed in 2.5% glutaraldehyde, 1% formaldehyde in 0.1 M cacodylate acid (pH 7.4) for 1.5 h at room temperature. Samples were rinsed with PBS and post-fixed in 2% osmium tetroxide in 0.1 M cacodylate acid (pH 7.4), dehydrated in acetone and embedded in Spurr’s resin polymerized at 60°C for 24 h. Ultrathin sections (65 nm) were stained with uranyl acetate and lead citrate and examined at 60 kV with a Philips CM12 TEM at x10,000 and x35,000 magnification.
Molecular modeling
The 3D coordinates of human HDAC3 bound to co-repressor SMRT in the presence of inositol tetraphosphate was based on a recent publicationCitation45 and was available from the Protein Data Bank (PDB 4A69). The model was energetically refined in the internal coordinate space using the program Molsoft ICM.Citation46 Docking protocols were initially validated by docking inositol tetraphosphate into the binding site of interest and reproducing the crystallographic orientation. For molecular docking, five types of interaction potentials were represented: (1) van der Waals potential for a hydrogen atom probe; (2) van der Waals potential for a heavy-atom probe (generic carbon of 1.7 Å radius); (3) optimized electrostatic term; (4) hydrophobic terms; and (5) loan-pair-based potential, which reflects directional preferences in hydrogen bonding. The energy terms were based on the Merck Molecular Force Field (MMRF) to account for solvation free energy and entropic contributions.Citation47 Modified intermolecular terms such as soft van der Waals and hydrogen-bonding, as well as a hydrophobic term were added. Conformational sampling was based on the biased probability Monte Carlo (BPMC) procedure, which randomly selects a conformation in the internal coordinate space and then makes a step to a new random position independent of the previous one, but according to a predefined continuous probability distribution. It has also been shown that after each random step, full local minimization greatly improves the efficiency of the procedure. In the ICM-VLS (Molsoft ICM) screening procedure, the ligand scoring was optimized to obtain maximal separation between the binders and non-binders. Each compound was assigned a score according to fit within the receptor; this ICM score accounted for continuum and discreet electrostatic, hydrophobic and entropy parameters.Citation47-Citation49 The binding energies were determined as reported previously.Citation50
Statistics
Results are representative of at least three independent assays unless otherwise indicated and expressed as mean ± SD. Differences between groups were determined by ANOVA followed by Bonferroni's Multiple Comparison Test using GraphPad Prism™ software version 5.04. Statistical significance was indicated in the figures as follows: p < 0.05 (*), p < 0.01 (**) or p < 0.001 (***).
| Abbreviations: | ||
| HDAC | = | histone deacetylase |
| HAT | = | histone acetyltransferase |
| ITC | = | isothiocyanate |
| SFN | = | sulforaphane |
| AITC | = | allyl isothiocyanate |
| 6-SFN | = | 6-methylsulfinylhexyl isothiocyanate |
| 9-SFN | = | 9-methylsulfinylnonyl isothiocyanate |
| DSB | = | double strand break |
| ATR | = | ataxia telangiectasia and Rad3-related protein |
| CHK2 | = | checkpoint kinase-2 |
| CtIP | = | c-terminal binding protein (CtBP) interacting protein |
| AFU | = | arbitrary fluorescence unit, PBS, phosphate buffered saline |
| PI | = | propidium iodide |
| CCK8 | = | cell Counting Kit-8 |
| WST8 | = | water soluble tetrazolium-8 |
| DMSO | = | dimethylsulfoxide |
| IP | = | immunoprecipitation |
| IB | = | immunoblotting |
| No Ab | = | no antibody |
| RAD-51 | = | RAD51 homolog (S. cerevisiae) |
| Ku70 | = | non-homologous end joining (NHEJ) factor |
| DAPI | = | 4'-6-diamidino-2-phenylindole |
| ANOVA | = | analysis of variance |
| comet | = | also known as single cell gel electrophoresis assay |
| γH2AX | = | phosphorylated histone H2AX |
| PARP | = | poly (ADP-ribose) polymerase |
| TSA | = | trichostatin A |
| SIRT6 | = | sirtuin 6 |
| 3-MA | = | 3-methyladenine |
| LC3B | = | light chain 3B |
| DAC | = | deacetylase |
| GCN5 | = | a ubiquitous histone acetyltransferase |
Additional material
Download Zip (748.3 KB)Acknowledgments
Supported by US National Institutes of Health grants CA090890, CA65525, CA122906, CA122959, CA80176 and ES00210. We thank Teresa Sawyer for help with EM, Andrew Hau for autophagy antibodies and the Cell Imaging and Analysis Core for assistance with immunocytochemistry.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- American Cancer Society. Cancer Facts & Figures 2012. Atlanta: American Cancer Society, 2013.
- Herr I, Büchler MW. Dietary constituents of broccoli and other cruciferous vegetables: implications for prevention and therapy of cancer. Cancer Treat Rev 2010; 36:377 - 83; http://dx.doi.org/10.1016/j.ctrv.2010.01.002; PMID: 20172656
- Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res 2007; 55:224 - 36; http://dx.doi.org/10.1016/j.phrs.2007.01.009; PMID: 17317210
- Keum YS, Jeong WS, Kong ANT. Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat Res 2004; 555:191 - 202; http://dx.doi.org/10.1016/j.mrfmmm.2004.05.024; PMID: 15476860
- Rajendran P, Williams DE, Ho E, Dashwood RH. Metabolism as a key to histone deacetylase inhibition. Crit Rev Biochem Mol Biol 2011; 46:181 - 99; http://dx.doi.org/10.3109/10409238.2011.557713; PMID: 21599534
- Hsu A, Wong CP, Yu Z, Williams DE, Dashwood RH, Ho E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin Epigenetics 2011; 3:3; http://dx.doi.org/10.1186/1868-7083-3-3; PMID: 22303414
- Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011; 471:74 - 9; http://dx.doi.org/10.1038/nature09803; PMID: 21368826
- Rajendran P, Ho E, Williams DE, Dashwood RH. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin Epigenetics 2011; 3:4; http://dx.doi.org/10.1186/1868-7083-3-4; PMID: 22247744
- Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 2010; 329:1348 - 53; http://dx.doi.org/10.1126/science.1192049; PMID: 20829486
- Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem 2009; 107:600 - 8; http://dx.doi.org/10.1002/jcb.22185; PMID: 19459166
- Kachhap SK, Rosmus N, Collis SJ, Kortenhorst MS, Wissing MD, Hedayati M, et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS One 2010; 5:e11208; http://dx.doi.org/10.1371/journal.pone.0011208; PMID: 20585447
- Rosato RR, Almenara JA, Maggio SC, Coe S, Atadja P, Dent P, et al. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol Cancer Ther 2008; 7:3285 - 97; http://dx.doi.org/10.1158/1535-7163.MCT-08-0385; PMID: 18852132
- Chen CS, Wang YC, Yang HC, Huang PH, Kulp SK, Yang CC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res 2007; 67:5318 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-06-3996; PMID: 17545612
- Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res 2004; 64:5767 - 74; http://dx.doi.org/10.1158/0008-5472.CAN-04-1326; PMID: 15313918
- Batra S, Sahu RP, Kandala PK, Srivastava SK. Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-kappaB turnoff in human pancreatic carcinoma cells. Mol Cancer Ther 2010; 9:1596 - 608; http://dx.doi.org/10.1158/1535-7163.MCT-09-1146; PMID: 20484017
- Lea MA, Randolph VM, Lee JE, desBordes C. Induction of histone acetylation in mouse erythroleukemia cells by some organosulfur compounds including allyl isothiocyanate. Int J Cancer 2001; 92:784 - 9; http://dx.doi.org/10.1002/ijc.1277; PMID: 11351296
- Ma X, Fang Y, Beklemisheva A, Dai W, Feng J, Ahmed T, et al. Phenylhexyl isothiocyanate inhibits histone deacetylases and remodels chromatins to induce growth arrest in human leukemia cells. Int J Oncol 2006; 28:1287 - 93; PMID: 16596246
- Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, et al. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol 2008; 33:375 - 80; PMID: 18636159
- Dashwood RH, Myzak MC, Ho E. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention?. Carcinogenesis 2006; 27:344 - 9; http://dx.doi.org/10.1093/carcin/bgi253; PMID: 16267097
- Rajendran P, Delage B, Dashwood WM, Yu TW, Wuth B, Williams DE, et al. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol Cancer 2011; 10:68; http://dx.doi.org/10.1186/1476-4598-10-68; PMID: 21624135
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 8; http://dx.doi.org/10.1038/nature08467; PMID: 19847258
- Nakamura K, Kogame T, Oshiumi H, Shinohara A, Sumitomo Y, Agama K, et al. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet 2010; 6:e1000828; http://dx.doi.org/10.1371/journal.pgen.1000828; PMID: 20107609
- Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem 2006; 281:13548 - 58; http://dx.doi.org/10.1074/jbc.M510023200; PMID: 16533812
- Godman CA, Joshi R, Tierney BR, Greenspan E, Rasmussen TP, Wang HW, et al. HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biol Ther 2008; 7:1570 - 80; http://dx.doi.org/10.4161/cbt.7.10.6561; PMID: 18769117
- Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010; 18:436 - 47; http://dx.doi.org/10.1016/j.ccr.2010.10.022; PMID: 21075309
- Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell 2008; 30:61 - 72; http://dx.doi.org/10.1016/j.molcel.2008.02.030; PMID: 18406327
- Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, et al. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res 2010; 70:4470 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-09-3028; PMID: 20460513
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858 - 68; http://dx.doi.org/10.1074/jbc.273.10.5858; PMID: 9488723
- Pappa G, Bartsch H, Gerhäuser C. Biphasic modulation of cell proliferation by sulforaphane at physiologically relevant exposure times in a human colon cancer cell line. Mol Nutr Food Res 2007; 51:977 - 84; http://dx.doi.org/10.1002/mnfr.200700115; PMID: 17628879
- Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res 2006; 66:5828 - 35; http://dx.doi.org/10.1158/0008-5472.CAN-06-0139; PMID: 16740722
- Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci U S A 2010; 107:20003 - 8; http://dx.doi.org/10.1073/pnas.1013754107; PMID: 21037108
- Fimognari C, Turrini E, Ferruzzi L, Lenzi M, Hrelia P. Natural isothiocyanates: genotoxic potential versus chemoprevention. Mutat Res 2012; 750:107 - 31; http://dx.doi.org/10.1016/j.mrrev.2011.12.001; PMID: 22178957
- Sharma R, Sharma A, Chaudhary P, Pearce V, Vatsyayan R, Singh SV, et al. Role of lipid peroxidation in cellular responses to D,L-sulforaphane, a promising cancer chemopreventive agent. Biochemistry 2010; 49:3191 - 202; http://dx.doi.org/10.1021/bi100104e; PMID: 20205397
- Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009; 30:2 - 10; http://dx.doi.org/10.1093/carcin/bgn250; PMID: 18978338
- Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase inhibitors and genomic instability. Cancer Lett 2009; 274:169 - 76; http://dx.doi.org/10.1016/j.canlet.2008.06.005; PMID: 18635312
- Doyle K, Fitzpatrick FA. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem 2010; 285:17417 - 24; http://dx.doi.org/10.1074/jbc.M109.089250; PMID: 20385560
- Rossetto D, Truman AW, Kron SJ, Côté J. Epigenetic modifications in double-strand break DNA damage signaling and repair. Clin Cancer Res 2010; 16:4543 - 52; http://dx.doi.org/10.1158/1078-0432.CCR-10-0513; PMID: 20823147
- Subramanian C, Opipari AW Jr., Bian X, Castle VP, Kwok RPS. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A 2005; 102:4842 - 7; http://dx.doi.org/10.1073/pnas.0408351102; PMID: 15778293
- Zhang JX, Li DQ, He AR, Motwani M, Vasiliou V, Eswaran J, et al. Synergistic inhibition of hepatocellular carcinoma growth by cotargeting chromatin modifying enzymes and poly (ADP-ribose) polymerases. Hepatology 2012; 55:1840 - 51; http://dx.doi.org/10.1002/hep.25566; PMID: 22223166
- Sekine-Suzuki E, Yu D, Kubota N, Okayasu R, Anzai K. Sulforaphane induces DNA double strand breaks predominantly repaired by homologous recombination pathway in human cancer cells. Biochem Biophys Res Commun 2008; 377:341 - 5; http://dx.doi.org/10.1016/j.bbrc.2008.09.150; PMID: 18854174
- Chernikova SB, Game JC, Brown JM. Inhibiting homologous recombination for cancer therapy. Cancer Biol Ther 2012; 13:61 - 8; http://dx.doi.org/10.4161/cbt.13.2.18872; PMID: 22336907
- Cazaux C. [Genetic instability as a driver for oncogenesis]. Bull Cancer 2010; 97:1241 - 51; PMID: 21084240
- Spurling CC, Godman CA, Noonan EJ, Rasmussen TP, Rosenberg DW, Giardina C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol Carcinog 2008; 47:137 - 47; http://dx.doi.org/10.1002/mc.20373; PMID: 17849419
- Wang W, Jariyasopit N, Schrlau J, Jia Y, Tao S, Yu TW, et al. Concentration and photochemistry of PAHs, NPAHs, and OPAHs and toxicity of PM2.5 during the Beijing Olympic Games. Environ Sci Technol 2011; 45:6887 - 95; http://dx.doi.org/10.1021/es201443z; PMID: 21766847
- Watson PJ, Fairall L, Santos GM, Schwabe JWR. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012; 481:335 - 40; PMID: 22230954
- Cardozo T, Totrov M, Abagyan R. Homology modeling by the ICM method. Proteins 1995; 23:403 - 14; http://dx.doi.org/10.1002/prot.340230314; PMID: 8710833
- Totrov M, Abagyan R. Protein-Ligand Docking as an Energy Optimization Problem. In: Raffa RB, ed(s). Drug-Receptor Thermodynamics: Introduction and Experimental Applications. Vol 1, New York: John Wiley & Sons, 2001:603–24.
- Abagyan R, Totrov M, Kuznetsov D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comput Chem 1994; 15:488 - 506; http://dx.doi.org/10.1002/jcc.540150503
- Totrov M, Abagyan R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins 1997; Suppl 1 215 - 20; http://dx.doi.org/10.1002/(SICI)1097-0134(1997)1+<215::AID-PROT29>3.0.CO;2-Q; PMID: 9485515
- Bisson WH, Koch DC, O’Donnell EF, Khalil SM, Kerkvliet NI, Tanguay RL, et al. Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J Med Chem 2009; 52:5635 - 41; http://dx.doi.org/10.1021/jm900199u; PMID: 19719119