Abstract
Epigenetic mechanisms involved in primary hyperparathyroidism are poorly understood as studies are limited. In order to understand the role of aberrant DNA promoter methylation in the pathogenesis of parathyroid tumors, we have quantified the CpG island promoter methylation density of several candidate genes including APC (promoter 1A and 1B), β-catenin (CTNNB1), CASR, CDC73/HRPT2, MEN1, P16 (CDKN2A), PAX1, RASSF1A, SFRP1 and VDR in 72 parathyroid tumors and 3 normal parathyroid references using bisulfite pyrosequencing. Global methylation levels were assessed for LINE-1. We also compared methylation levels with gene expression levels measured by qRT-PCR for genes showing frequent hypermethylation. The adenomas displayed frequent hypermethylation of APC 1A (37/66; 56%), RASSF1A (34/66; 52%) and β-catenin (19/66; 29%). One of the three atypical adenomas was hypermethylated for APC 1A. The three carcinomas were hypermethylated for RASSF1A and SFRP1, and the latter was only observed in this subtype. The global methylation density was similar in tumors (mean 70%) and parathyroid reference samples (mean 70%). In general, hypermethylated genes had reduced expression in the parathyroid adenomas using qRT-PCR. Among the adenomas, methylation of APC 1A correlated with adenoma weight (r = 0.306, p < 0.05). Furthermore, the methylation status of RASSF1A correlated with each of APC 1A (r = 0.289, p < 0.05) and β-catenin (r = 0.315, p < 0.01). Our findings suggest a role for aberrant DNA promoter methylation of APC 1A, β-catenin and RASSF1A in a subset of parathyroid tumors.
Introduction
Three types of parathyroid tumors underlie the development of primary hyperparathyroidism (PHPT). Parathyroid adenoma is by far the most common. Parathyroid carcinoma is rare, but represents an important clinical challenge since diagnostic shortcomings are still associated with suboptimal treatment and fatal outcomes. Atypical adenoma is an intermediate form of presently unknown malignant potential.Citation1
Most of the molecular studies in parathyroid tumors have focused on the genetic mechanisms involved in PHPT aiming at the identification of aberrant molecules and pathways. These studies have identified two important tumor suppressor genes, MEN1 and CDC73/HRPT2, which are frequently mutated in parathyroid tumors, specifically in parathyroid adenomas and carcinomas, respectively.Citation2-Citation4 Other studies have reported reduced expression of VDR and CASR in parathyroid tumors; mutations in these two genes are rarely identified.Citation5-Citation7
An increasingly recognized mechanism of gene transcription regulation is epigenetic modification. In many pathological conditions including cancer, altered methylation patterns at gene-specific and/or global genome level are detected.Citation8-Citation10 In general, a state of global genome hypomethylation coupled with gene-specific hypermethylation is a common finding in cancer.Citation11
Epigenetic studies in parathyroid tumors have so far been limited. Previous studies have identified a number of genes to be frequently hypermethylated in parathyroid tumors: among them, APC, RASSF1A, P16 (CDKN2A) and SFRP1.Citation12,Citation13 While one study has reported hypermethylation of CDC73 in a subset of parathyroid carcinomas,Citation14 more recent reports excluded epigenetic silencing of CDC73 gene in parathyroid tumors.Citation15,Citation16 To further elucidate the role of DNA methylation in parathyroid tumors and as an extension to our previous study,Citation12 we have quantified the methylation density at the promoter of a number of plausible candidate genes, such as APC 1B, CASR, CDC73, MEN1, PAX1, SFRP1 and VDR, in addition to the previously analyzed genes, such as APC 1A, β-catenin (CTNNB1), P16 and RASSF1A, using a different and larger panel of parathyroid tumors. We also estimated the global methylation level by assessing LINE-1 methylation.
The selection of genes for methylation analysis was based either on their direct involvement in the pathophysiology of parathyroid tumors, such as, CDC73, MEN1, CASR and VDR or on their involvement in important cellular processes such as proliferation and growth, which are regulated by methylation in other tumor types (P16 (CDKN2A),Citation17,Citation18 PAX1,Citation19 RASSF1A).Citation20,Citation21 In addition, we have investigated genes from the Wnt signaling pathway, a pathway frequently disrupted in many tumor types including parathyroid tumors.Citation22,Citation23 The Wnt signaling pathway genes included, in addition to APC, the Wnt antagonist SFRP1Citation24 and the central player, β-catenin with oncogenic properties.Citation25,Citation26
Results
Gene-specific promoter methylation in parathyroid adenomas, atypical adenomas and carcinomas
The CpG island promoter methylation density was quantified for 10 selected genes in 72 parathyroid tumors using pyrosequencing, and compared with three normal parathyroid reference samples. For each individual tumor and gene, MetI was calculated. The overall results are summarized in and the MetI for each gene analyzed in each individual tumor along with the reference samples are summarized in Table S1.
Table 1. Results from gene specific and global methylation analyses in the 72 parathyroid tumors
Overall, hypermethylation was observed for APC 1A, β-catenin (CTNNB1), RASSF1A and SFRP1 with different frequencies in tumor subgroups. However, the differences in methylation level between tumor subgroups and normal parathyroid samples did not reach a statistical significance. Differential methylation was very rarely observed for APC 1B, CASR, CDC73, MEN1, P16, PAX1 and VDR ().
Figure 1. Individual value plot showing gene-specific MetI for individual samples. For each gene, MetI in three normal references (N) and 72 parathyroid tumors (T) are shown next to each other.
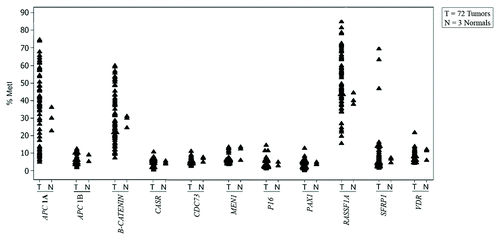
Promoter APC 1A was frequently hypermethylated in 37/66 (56%) of the adenomas with an overall MetI ranging from 5% to 75% compared with the reference samples that had average MetI of 30% (). Similarly, RASSF1A was frequently hypermethylated in 34/66 (52%) of the cases with MetI ranging from 16% to 79%. The promoter of the β-catenin gene was hypermethylated in 19/66 (28%) of cases with MetIs ranging from 7% to 60%. Representative pyrograms for hypermethylated APC 1A, β-catenin and RASSF1A are shown in Figure S1. The overall MetIs of the remaining genes (APC 1B, CASR, CDC73, MEN1, PAX1, P16, SFRP1 and VDR) were similar to the three normal references and they were all unmethylated (). Only one adenoma (T56) had high MetI (15%) for P16 compared with 4% MetI in the reference samples. This adenoma was also hypermethylated for both APC 1A (75%) and RASSF1A (56%). A similar finding was observed for adenoma (T3), which was the only adenoma with hypermethylation of the VDR gene promoter, with a MetI of 22% compared with MetI of 10% in reference samples. This adenoma also showed hypermethylation of APC 1A (MetI 61%) and RASS1A (MetI 59%).
Figure 2. Gene-specific MetI in the parathyroid tumor subgroups studied. (A) Parathyroid adenomas (n = 66). (B) Atypical parathyroid adenomas (n = 3). (C) Parathyroid carcinomas (n = 3). (D) MEN1 and CDC73 mutated adenomas. For each gene, MetI in normal references (N) and parathyroid tumors are shown next to each other. T refers to tumor samples, N to normal samples, M to MEN1 mutated and H to CDC73 mutated tumors.
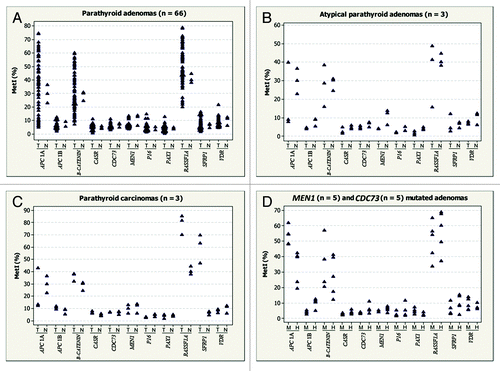
Among the 3 atypical adenoma samples analyzed, only one tumor (T69) was hypermethylated for APC 1A with MetI of 40% (, Table S1).
All three carcinoma cases (T70, T71 and T72) were hypermethylated for RASSF1A and SFRP1. The MetI ranged from 70–85% for RASSF1A and 47–70% for SFRP1. However, only one carcinoma (T72) was methylated at the APC 1A promoter with MetI of 43% (, Table S1).
Global methylation analysis of LINE-1
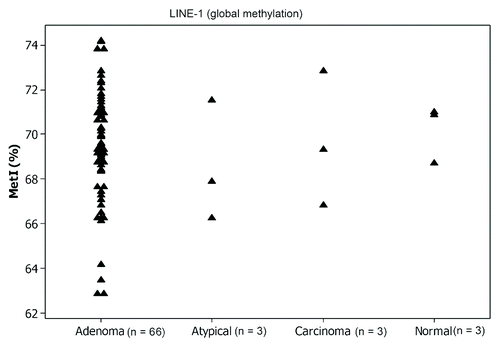
Estimation of the methylation density at the repeated sequences such as LINE-1 can indicate the overall global methylation status for the analyzed tumors. The global methylation analysis in our tumor panel revealed no altered global methylation in any of the tumors (T1-T72) and all the tumors were methylated with a MetI of about 70% (ranged from 63–74%), similar to those of the reference samples that had a MetI of 70% ().
Figure 3. Global methylation of LINE-1 in parathyroid tumors and normal references. The MetI is shown for each sample in the different tumor subgroups. (n) refers to total number of cases.
qRT-PCR gene expression analysis
In order to investigate the effect of hypermethylated genes on their transcription, we studied the expression of APC 1A, APC 1B, β-catenin and RASSF1A in 52/66 of the parathyroid adenomas along with the three normal reference samples using qRT-PCR. About 25% of the APC 1A hypermethylated parathyroid adenomas had reduced expression of APC 1A transcript, while no altered expression of APC 1B promoter could be detected, as all 52 parathyroid adenomas have expressed APC 1B transcript similar to the normal parathyroid samples (). The expression of β-catenin was reduced in about 25% of the hypermethylated tumors, while 48% of the RASSF1A hypermethylated adenomas displayed reduced RASS1A gene expression ().
Table 2. MetI and mRNA expression in 52 adenomas for genes showing promoter hypermethylation
Correlation between methylation status, mRNA expression and clinical/biochemical parameters
We studied the correlation between methylation status of parathyroid tumors for hypermethylated genes including APC 1A, β-catenin and RASSF1A by measuring mRNA expression level in the parathyroid adenomas as well as recoding the corresponding clinical and biochemical parameters, such as age at diagnosis, sex, parathyroid gland weight, serum levels of parathyroid hormone (PTH), calcium (S-Ca2+) and phosphate. Methylation of APC 1A was significantly correlated with adenoma weight (r = 0.306, p < 0.05). However, no statistically significant correlation could be detected between the methylation level and the mRNA expression level.
Studying the correlation between the differentially methylated genes revealed a statistically significant positive concordant methylation between RASSF1A and APC 1A (r = 0.289, p < 0.05) and between RASSF1A and β-catenin (r = 0.315, p < 0.01) genes. Furthermore, comparing the methylation status of the analyzed genes between MEN1 and CDC73 mutated cases did not reveal any statistically significant differences ().
Discussion
Gene silencing by DNA promoter CpG island methylation is the main and most well-studied epigenetic mechanism in humans.Citation27 This mechanism is an important component of tumorigenesis and has been described in many tumor types.Citation28-Citation30 In the present study, using pyrosequencing technique, we have quantified the methylation density at the promoter of several candidate genes, including APC (promoter 1A and 1B), β-catenin, CASR, CDC73, MEN1, P16, PAX1, RASSF1A, SFRP1 and VDR, as well as the global methylation level using LINE-1, in a parathyroid tumor panel consisted of 66 parathyroid adenomas, 3 atypical adenomas and 3 carcinomas. We found frequent hypermethylation of APC 1A, β-catenin and RASSF1A promoters in all the subgroups with the exception of the carcinomas that were not methylated for β-catenin, but exclusively hypermethylated for the SFRP1 promoter. In general, hypermethylated genes in the adenoma cases showed reduced mRNA expression. No alterations in global methylation level could be detected.
The frequent hypermethylation of APC 1A promoter detected in our study is in line with our previous studyCitation12 as well as with a recently published genome-wide methylation analysis in parathyroid tumors.Citation13 APC is an important component of the Wnt/β-catenin signaling pathway, which is frequently disrupted in many tumor types including parathyroid tumors.Citation22,Citation23 Along with two companion proteins namely Axin2 and GS3K, APC functions as a direct suppressor of β-catenin. Therefore, upon inactivation/silencing of APC, β-catenin is stabilized and exerts its growth and proliferation promoting effect, which in turn may lead to neoplastic transformation.Citation31 APC is frequently mutated in colorectal cancer as well as in other tumor types such as tumors of breast and thyroid.Citation32,Citation33 Although no APC mutations have been reported in parathyroid tumors, reduced APC immunoreactivity has been described and specifically linked to parathyroid carcinomas.Citation23 Furthermore, aberrant APC 1A promoter methylation has been described in other tumor types such as colorectal, breast and lung cancers.Citation25,Citation34,Citation35
The oncogene β-catenin was also frequently hypermethylated in our tumor panel, with the exception of the carcinoma cases. In parathyroid tumors, a rare, homozygous activating β-catenin (CTNNB1) mutation has been demonstrated for a few cases, suggesting a role for β-catenin in parathyroid tumorigenesis.Citation36 Subsequent studies regarding β-catenin expression in parathyroid tumors revealed discordant findings regarding sub-cellular localization and overall intensity, where some find nuclear accumulation of β-catenin in tumor cells while others observe no nuclear staining and little or no difference, as compared with the corresponding normal parathyroid rim.Citation23,Citation36,Citation37 Our study indicates that β-catenin is not hypomethylated in parathyroid tumors as compared with the normal samples, thereby excluding this potential epigenetic mechanism as a modus operandi for the suggested nuclear accumulation of β-catenin. Our finding of increased methylation in adenomas as compared with normal parathyroid samples was unexpected, as β-catenin is a well-characterized proto-oncogene. Interestingly, β-catenin has been shown to induce apoptosis in some cell systems,Citation38 suggesting a “just right hypothesis” for β-catenin expression, where high and low levels of β-catenin induces cell death while “just right” levels confer proliferation. It is therefore tempting to speculate that the increased methylation of β-catenin in our tumor series could perchance provide the tumors with a proliferative advantage even though it argues against the traditional role of a bona fide oncogene. This hypothesis remains to be investigated. Hypermethylation of β-catenin has been reported in other neuroendocrine tumors such as small intestinal carcinoids.Citation39
About half of the tumors in our study, including all three carcinomas, displayed hypermethylation of RASSF1A promoter. RAS association family domain 1 (RASSF1A) is an important tumor suppressor gene involved in the regulation of cell growth and proliferation as well as DNA repair.Citation40 Frequent RASSF1A hypermethylation was reported in head and neck cancersCitation20 and many other human tumorsCitation21,Citation41-Citation43 and in one study it was associated with high grade salivary gland carcinomas.Citation44 Frequent hypermethylation of APC and RASSF1A reinforces our previous resultsCitation12 and further supports the role of aberrant APC and RASSF1A genes in parathyroid tumors development.
Interestingly, among all tumors analyzed, hypermethylation of SFRP1 promoter was only detected in the carcinomas. SFRP1 is a potent antagonist of the Wnt signaling pathway and silencing of this gene, for example by methylation, can constitutively activate the Wnt signaling pathway.Citation24,Citation45 Hypermethylation of this gene as well as other related family members including SFRP2 and SFRP4 has been reported in parathyroid tumors in a genome-wide methylation analysis.Citation13 However, this study reported hypermethylation of the SRFP1 gene in both benign and malignant parathyroid tumors, in contrast to our study where hypermethylation of SFRP1 was only detected in the carcinomas. This discrepancy could be explained by variable position of CpG dinucleotides targeted for analysis, tumor heterogeneity as well as the cut offs and algorithms used for the detection of differential methylation implicated in both methods. Epigenetic inactivation of SFRP1 has been reported in colorectalCitation24 and renal cell carcinomasCitation45 as well as an independent poor prognostic feature in breast cancer.Citation46 Of more interest, while hypermethylation of SFRP1 promoter was detected in all the carcinomas, none were hypermethylated for β-catenin. This finding might suggest the constitutive activation of the canonical Wnt/β-catenin signaling in this subgroup. However, due to lack of tissue, we could not assess the effects of differentially methylated genes on gene expression. Moreover, SFRP1 could potentially be of interest as an immunohistochemical marker for the detection of malignancy in parathyroid tumors, as this gene was only epigenetically silenced in the carcinomas.
As for tumor suppressor genes, inactivation of the MEN1 or CDC73 genes by promoter methylation is a possible second hit mechanism. However, no MEN1 or CDC73 promoter hypermethylation could be detected in our tumor panel, including in those with established MEN1 or CDC73 mutations. While no studies investigated MEN1 methylation specifically in parathyroid tumors, lack of CDC73 promoter hypermethylation is in agreement with our own and others’ previous reports,Citation15,Citation16 further excluding promoter hypermethylation as an inactivation mechanism. In addition, the methylation profile of the MEN1 and CDC73 mutated cases did not reveal any statistically significant differences. This could indicate that the mutational status of these two genes is unlikely to direct the tumors toward a different methylation profile.
Although epigenetic mechanisms do not exclusively constitute DNA methylation, one can assume, based on our results, that aberrant CpG promoter methylation of each of CASR, CDC73, MEN1, P16, PAX1 and VDR is unlikely involved in the pathogenesis parathyroid tumors. On the other hand, inability to detect methylation in these genes does not rule out DNA methylation itself as a regulatory mechanism. DNA methylation of CpG dinucleotides outside promoter CpG islands (CpG shores) has been reported as frequent and probably more commonly encountered sites of differential methylation,Citation47 a phenomenon that cannot be excluded in our case.
Overall, the hypermethylated adenomas for APC 1A, β-catenin and RASSF1A exhibited reduced mRNA expression. This association suggests the silencing effect of the methylation on these genes. However, it is not entirely clear why some methylated samples do not always have reduced mRNA expression. A possible explanation is transcription via an alternative promoter such as via promoter 1B in the case of APC gene, where all the tumors were unmethylated and all expressed APC 1B transcript. A similar finding has been reported in another study of breast and lung cancer, where reduced expression of APC promoter 1A, but not 1B, was reported to be concordant with hypermethylated APC 1A and unmethylated APC 1B, respectively.Citation34 In addition, DNA methylation may require other concurrent epigenetic modifications, such as histone modifications and chromatid remodeling in order to regulate gene function.Citation48 Considering technical issues, one of the limitations of the pyrosequencing technique is the inability to provide information on allele-specific methylation, which per se could be a reason for partial methylation and, therefore, consequently retained gene expression. Incomplete or partial methylation could also be explained by tumor heterogeneity and/or contamination with normal tissues.
The significant correlation between APC 1A methylation status and adenoma weight observed in this study suggest that large parathyroid adenomas could have a different DNA methylation pattern in addition to previously reported specific genetic profile linked to adenoma size.Citation49,Citation50 Furthermore, a statistically significant correlation was observed between the methylation status of RASSF1A with both APC 1A and β-catenin. In our previous study, we have reported a similar correlation between the methylation status of APC 1A and RASSF1A, but not for β-catenin.Citation12 This finding reinforces the concordant aberrant hypermethylation of these genes as of importance in the tumorigenesis of a subgroup of parathyroid adenomas and could suggest the existence of a subgroup of parathyroid tumors with a methylator phenotype including these genes.
The differential hypermethylation of APC 1A, β-catenin and RASSF1A in a subset of the adenomas could mirror the underlying tumor heterogeneity as well as different molecular mechanisms in tumor development. However, the mechanism underlying this differential methylation is still not clearly understood. Furthermore, the question of whether aberrant DNA methylation detected contributes to the initiation of the tumor development or it is just a bystander effect, remain to be answered. Finally, before being able to address the clinical relevance of these findings, the precise mechanism and the functional consequences of such detected aberrations need to be further explored.
One of the limitations of this study is the small sample size of parathyroid carcinomas, atypical adenomas and normal parathyroid samples used as control. However, due to the rarity of these tumor subgroups, identifying additional cases was not feasible for our current study. In addition, a normal parathyroid tissue is of difficult availability for ethical reasons; a known issue in parathyroid research with no currently available alternative solutions.
Another known limitation to pyrosequencing analyses stems from tissue heterogeneity. While primary parathyroid tumors routinely exhibit high tumor cell percentages, we also analyzed two distant metastases of parathyroid carcinoma, raising the possibility of contamination of normal lung tissue. Although tumor representativity was not performed for these cases, previous DNA analyses from these samples using array-comparative genomic hybridization (a-CGH) visualized gross chromosomal alterations in agreement with previous publications regarding parathyroid carcinomas.Citation16 In addition, paraffin-embedded tissue specimens from metastatic cases have shown high tumor cell proportion in comparison to surrounding lung tissue, suggesting a high tumor-to-normal tissue proportion.Citation51
In summary, this study suggests a role of aberrant promoter methylation for APC 1A, RASSF1A and β-catenin genes in the molecular pathogenesis of a subset of parathyroid tumors and may indicate the disruption of multiple pathways. Our results also suggest a role of SFRP1 methylation in parathyroid carcinomas which could contribute to the constitutive activation of Wnt/β-catenin pathway. However, this needs to be validated in a larger cohort.
Materials and Methods
This study included a panel of 72 (T1-T72) fresh frozen parathyroid tumors and three non-tumorous parathyroid tissue samples (N1, N2 and N3) used as normal references. Sixty-six tumors were diagnosed as adenomas (T1-T66), 3 as atypical adenomas (T67-T69) and 3 as carcinomas (T70-T72). Twenty-one tumors were previously published.Citation52 The mutational status of both MEN1 and CDC73 genes were known for a number of the cases.Citation16,Citation51-Citation54 Five cases had a MEN1 mutation (T57-T61) and 8 cases, including the 3 carcinomas, had a CDC73 mutation (T62-T66 and T70-T72). Apart from the three carcinomas which were obtained from the Tokyo Women's Medical University Hospital in Japan, all the other samples were retrieved from Karolinska University Hospital. Histopathological evaluation of all the tumors was performed according to the World Health Organization guidelines (DeLellis RA, Lloyd RV et al. 2004). All samples were collected with informed oral (Karolinska) or written (Tokyo) consents and local ethical committee approval.
In general, primary parathyroid tumors contain a high proportion of tumor cells and very seldom a significant amount of stromal tissue, making micro-dissection somewhat redundant for this tumor type. All tumor tissue included in this study was specifically handled by an experienced endocrine pathologist following excision in the operation theater. All primary parathyroid tumors from our institution are regularly macrodissected with a sterile scalpel, so that macroscopically normal parathyroid tissue and adipose tissue is separated from the tumor prior to biobank sampling and freezing in liquid nitrogen. As a consequence, all tissue sampled to the biobank for research purposes contain tumor tissue to a very large extent, which previous representativity testing has proven.Citation54
The clinical and biochemical data for a subset of the cases were previously published.Citation16,Citation52Table S2 summarizes the clinical and biochemical details for all the cases.
DNA and RNA extraction
Extraction of genomic DNA was performed using DNeasy Blood and Tissue DNA isolation kit (Qiagen AB) following the manufacturer’s protocol. Total RNA isolation was performed using the protocol recommended with the mirVana miRNA Isolation Kit (Ambion, Life Technologies, Applied Biosystems). Tissues from the 3 normal parathyroid samples and 52 adenomas were available for RNA extractions. Quantification and quality check of the extracted DNA and RNA were performed using NanoDrop A100 (Thermo Scientific).
Bisulfite conversion
For bisulfite conversion, the manufacturer’s protocol was followed as recommended with no modifications. Briefly, 500 ng of genomic DNA was bisulfite treated using EpiTect Bisulfite conversion kit (Qiagen AB). Bisulfite conversion was performed in a thermal cycler using the following cycling conditions: Denaturation at 95°C for 5 min (min), incubation at 60°C for 25 min, denaturation at 95°C for 5 min, incubation at 60°C for 85 min, denaturation at 95°C for 5 min, incubation at 60°C for 175 min and storage at +20°C. Modified DNA was bound to the EpiTect spin columns, washed twice, desulfonated and then eluted in 20 ul volume. The bisulfite converted DNA was either used immediately or stored at -20°C and used within a month of storage.
Pyrosequencing reaction and analysis
PCR amplification of bisulfite converted DNA with specific methylation assay was performed using EpiTect PCR kit (Qiagen). Ready-made assays were purchased from Qiagen (Qiagen) (APC 1A, β-catenin, CASR, CDC73, LINE-1, MEN1, P16, PAX1, RASSF1A, SFRP1 and VDR). APC1B was designed using PyroMark assay design software version 2.0 (Qiagen) and ordered from biomers.net. All primers were located within CpG islands in the promoter region of the target gene. Regions overlapping with known single nucleotide polymorphisms (SNPs) were avoided. Bisulfite treated PCR products were run in 2% agarose gel for quality Table S3.
For pyrosequencing reactions, 15–20 ul of the bisulfite converted amplified DNA was added to a total reaction volume of 85 ul containing binding buffer and sepharose streptavidine. This reaction mix was agitated for 10 min at room temperature in order to facilitate the attachment of the beads to the biotinylated strands. The double stranded DNA was separated using the filter pump and this allowed having only single stranded (biotinylated) DNA as a template. This was released to the pyromark plate well each containing sequencing primer with annealing buffer in a reaction volume of 25 ul. The mixture was denatured for 2 min at 80°C and re-annealed at room temperature for at least 5 min. The plate was put in the PyroMark Q24 machine and run with relevant run file.
All sequencing primers contained at least one nucleotide (C not followed by G) to control for bisulfite conversion efficiency. A methylation standard (0%, 50 and 100% methylated) (EpiTect control set, Qiagen, Sweden, AB) and water were used as positive and negative controls, respectively. Data analysis was performed using the PyroMark Q24 software (Qiagen, Sweden, AB). The program evaluates the target CpG sites and converts pyrograms to numerical values for peak heights and calculates the proportion of methylation at each base as a C/T ratio. For each samples and assay a methylation index (MetI) was calculated as the mean methylation of all CpG dinucleotides covered. Tumor methylation status was defined based on comparison to the methylation status of the normal reference samples. A difference of more than 10% in MetI for a particular assay between parathyroid tumor and normal references was regarded as significant difference. All experiments were repeated twice to exclude any technical errors.
Quantitative reverse transcription real-time PCR (qRT-PCR)
qRT-PCR analysis was applied on 52 parathyroid adenomas and 3 normal parathyroid tissues. For TaqMan gene expression analysis, cDNA was synthesized from 100 ng total RNA using High Capacity cDNA Reverse Transcription Kit with RNase Inhibitor following manufacturer’s protocol (Applied Biosystems). Briefly, 2× RT master mix was prepared in a 10 ul reaction volume containing: 2 ul of 10X RT Buffer, 0.8 ul of 25 X dNTP Mix (100 mM), 2 ul of 10 X RT random primers, 1 ul of reverse transcriptase (50 U/ul), 1 ul of RNase inhibitor and 3.2 ul of nuclease free water. Ten ul of 2X RT master mix was added to each well and to which a total of 10 ul RNA (10 ng/ul) was pipetted on a 96 well plate. The reactions were set up on ice. Reverse transcription was performed in a StepOnePlus real-time PCR machine (Applied Biosystems) using the following cycling conditions: 25 °C for 10 min, 37 °C for 120 min and then at 85 °C for 5 min. The quality and concentration of cDNA was assessed by spectrophotometry and samples were stored at 4 °C when used in less than 24hrs or at -20 °C for longer storage.
For the qRT-PCR, a 20 ul of PCR reaction mix containing 1 ul of 20 × TaqMan gene expression assay, 10 ul of 2 × TaqMan gene expression master mix, 100 ng of cDNA template and 5 ul of nuclease free water. The analyzed assays included TaqMan gene expression probes (Applied Biosystems) targeting total APC mRNA, the APC 1B-specific transcript, RASSF1A and CTNNB1 transcripts. For assay details see Table S3. RPLP0 (ribosomal protein, large, P0) was used as an endogenous control and amplified in the same plate with each target assay. In a previous study in our group (unpublished data), the endogenous control RPLP0 (also known as 36B4) along with three others commonly used endogenous controls namely the glyceraldehydes-3-phosphate dehydrogenase (GAPDH), β-actin and 18S rRNA (rRNA) were tested in a set of parathyroid tumors using the same target gene expression assays analyzed in this study and found the expression of RPLP0 to be the most stable and ubiquitously expressed gene. The RPLP0 has also been used in previous publications by the authors and was selected as an endogenous reference gene known to demonstrate little variation in parathyroid tissues.Citation54 Samples were run along with the endogenous control and non-template control in triplicates in 96-well plates in a StepOnePlus real-time PCR machine (Applied Biosystems) using standard amplification method and the following thermal cycling conditions: hold at 50 °C for 2 min, hold for 95 °C for 10 min, 1 cycle at 95 °C for 15 sec followed by 40 cycles at 60 °C for 1 min. Results were analyzed with SDS software v2.4 and the expression value was expressed as fold changes calculated as 2-∆∆ Ct value for each assay using DataAssit v3.0 software (Applied Biosystems).
Statistics
Spearman’s rank correlation test was used to analyze correlations between the differentially methylated genes and the clinical/biochemical parameters and gene expression. The same test was also used to assess the correlations among the differentially methylated genes. For testing statistically significant differences in the methylation level between the tumor groups and the normal parathyroids we used Mann-Whitney Rank Sum Test. A P value of < 0.05 was regarded as statistically significant. The statistical analyses were performed using the SPSS software (Statistical Software Package for Windows, V.20, V.20, SPPS Inc.).
Additional material
Download Zip (1.3 MB)Acknowledgments
The study was financially supported by Swedish Cancer Society, Swedish Research Council (No. 5210–2010–3518), Gustav V Jubilee Foundation, Karolinska Institutet and Stockholm County Council. L.S. is supported by a research stipend donated from Mr. Ahmad Ismail Nanakaly. The authors wish to express their gratitude to Lisa Ånfalk at the Department of Oncology-Pathology, Karolinska University Hospital Solna, for samples collection and preparation. We also thank the medical genetics group at our department for their valuable comments. L.S., C.C.J., I.LN., O.F., C.F. and J.H. conceived and designed the study; L.S. performed the experiments and the analyses and wrote the manuscript; O Fotouhi performed qRT-PCR. I.L.R., C.C.J. and C.L. contributed to the tumor and patient characterization. All authors contributed to interpretation of results and have revised and approved the final version of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- DeLellis RA, Lloyd RV, Heitz PU, C E. Pathology and Genetics of the Tumours of Endocrine Organs, WHO Classification of Tumours. Lyon: IARC Press, 2004.
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997; 276:404 - 7; http://dx.doi.org/10.1126/science.276.5311.404; PMID: 9103196
- Lemmens I, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet 1997; 6:1177 - 83; http://dx.doi.org/10.1093/hmg/6.7.1177; PMID: 9215690
- Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet 2002; 32:676 - 80; http://dx.doi.org/10.1038/ng1048; PMID: 12434154
- Carling T, Rastad J, Szabó E, Westin G, Akerström G. Reduced parathyroid vitamin D receptor messenger ribonucleic acid levels in primary and secondary hyperparathyroidism. J Clin Endocrinol Metab 2000; 85:2000 - 3; http://dx.doi.org/10.1210/jc.85.5.2000; PMID: 10843188
- Gogusev J, Duchambon P, Hory B, Giovannini M, Goureau Y, Sarfati E, et al. Depressed expression of calcium receptor in parathyroid gland tissue of patients with hyperparathyroidism. Kidney Int 1997; 51:328 - 36; http://dx.doi.org/10.1038/ki.1997.41; PMID: 8995751
- Carling T, Szabo E, Bai M, Ridefelt P, Westin G, Gustavsson P, et al. Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. J Clin Endocrinol Metab 2000; 85:2042 - 7; http://dx.doi.org/10.1210/jc.85.5.2042; PMID: 10843194
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429:457 - 63; http://dx.doi.org/10.1038/nature02625; PMID: 15164071
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415 - 28; PMID: 12042769
- Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet 2000; 16:168 - 74; http://dx.doi.org/10.1016/S0168-9525(99)01971-X; PMID: 10729832
- Das PM, Singal R. DNA methylation and cancer. J Clin Oncol 2004; 22:4632 - 42; http://dx.doi.org/10.1200/JCO.2004.07.151; PMID: 15542813
- Juhlin CC, Kiss NB, Villablanca A, Haglund F, Nordenström J, Höög A, et al. Frequent promoter hypermethylation of the APC and RASSF1A tumour suppressors in parathyroid tumours. PLoS One 2010; 5:e9472; http://dx.doi.org/10.1371/journal.pone.0009472; PMID: 20208994
- Starker LF, Svedlund J, Udelsman R, Dralle H, Akerström G, Westin G, et al. The DNA methylome of benign and malignant parathyroid tumors. Genes Chromosomes Cancer 2011; 50:735 - 45; http://dx.doi.org/10.1002/gcc.20895; PMID: 21638518
- Hewitt KM, Sharma PK, Samowitz W, Hobbs M. Aberrant methylation of the HRPT2 gene in parathyroid carcinoma. Ann Otol Rhinol Laryngol 2007; 116:928 - 33; PMID: 18217513
- Hahn MA, Howell VM, Gill AJ, Clarkson A, Weaire-Buchanan G, Robinson BG, et al. CDC73/HRPT2 CpG island hypermethylation and mutation of 5′-untranslated sequence are uncommon mechanisms of silencing parafibromin in parathyroid tumors. Endocr Relat Cancer 2010; 17:273 - 82; http://dx.doi.org/10.1677/ERC-09-0291; PMID: 20026646
- Sulaiman L, Haglund F, Hashemi J, Obara T, Nordenström J, Larsson C, et al. Genome-wide and locus specific alterations in CDC73/HRPT2-mutated parathyroid tumors. PLoS One 2012; 7:e46325; http://dx.doi.org/10.1371/journal.pone.0046325; PMID: 23029479
- Chan AO, Kim SG, Bedeir A, Issa JP, Hamilton SR, Rashid A. CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene 2003; 22:924 - 34; http://dx.doi.org/10.1038/sj.onc.1206123; PMID: 12584572
- Di Vinci A, Perdelli L, Banelli B, Salvi S, Casciano I, Gelvi I, et al. p16(INK4a) promoter methylation and protein expression in breast fibroadenoma and carcinoma. Int J Cancer 2005; 114:414 - 21; http://dx.doi.org/10.1002/ijc.20771; PMID: 15578730
- Lai HC, Lin YW, Huang TH, Yan P, Huang RL, Wang HC, et al. Identification of novel DNA methylation markers in cervical cancer. Int J Cancer 2008; 123:161 - 7; http://dx.doi.org/10.1002/ijc.23519; PMID: 18398837
- Dong SM, Sun DI, Benoit NE, Kuzmin I, Lerman MI, Sidransky D. Epigenetic inactivation of RASSF1A in head and neck cancer. Clin Cancer Res 2003; 9:3635 - 40; PMID: 14506151
- Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Fong K, Gao B, et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst 2001; 93:691 - 9; http://dx.doi.org/10.1093/jnci/93.9.691; PMID: 11333291
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev 2007; 17:45 - 51; http://dx.doi.org/10.1016/j.gde.2006.12.007; PMID: 17208432
- Juhlin CC, Haglund F, Villablanca A, Forsberg L, Sandelin K, Bränström R, et al. Loss of expression for the Wnt pathway components adenomatous polyposis coli and glycogen synthase kinase 3-beta in parathyroid carcinomas. Int J Oncol 2009; 34:481 - 92; PMID: 19148484
- Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 2004; 36:417 - 22; http://dx.doi.org/10.1038/ng1330; PMID: 15034581
- Hoque MO, Prencipe M, Poeta ML, Barbano R, Valori VM, Copetti M, et al. Changes in CpG islands promoter methylation patterns during ductal breast carcinoma progression. Cancer Epidemiol Biomarkers Prev 2009; 18:2694 - 700; http://dx.doi.org/10.1158/1055-9965.EPI-08-0821; PMID: 19789364
- Costa VL, Henrique R, Ribeiro FR, Carvalho JR, Oliveira J, Lobo F, et al. Epigenetic regulation of Wnt signaling pathway in urological cancer. Epigenetics 2010; 5:343 - 51; http://dx.doi.org/10.4161/epi.5.4.11749; PMID: 20421722
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983; 301:89 - 92; http://dx.doi.org/10.1038/301089a0; PMID: 6185846
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol 2005; 2:Suppl 1 S4 - 11; http://dx.doi.org/10.1038/ncponc0354; PMID: 16341240
- Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol 2005; 45:629 - 56; http://dx.doi.org/10.1146/annurev.pharmtox.45.120403.095832; PMID: 15822191
- Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev 2012; 22:50 - 5; http://dx.doi.org/10.1016/j.gde.2012.02.008; PMID: 22402447
- Gavert N, Ben-Ze’ev A. beta-Catenin signaling in biological control and cancer. J Cell Biochem 2007; 102:820 - 8; http://dx.doi.org/10.1002/jcb.21505; PMID: 17854061
- Zeki K, Spambalg D, Sharifi N, Gonsky R, Fagin JA. Mutations of the adenomatous polyposis coli gene in sporadic thyroid neoplasms. J Clin Endocrinol Metab 1994; 79:1317 - 21; http://dx.doi.org/10.1210/jc.79.5.1317; PMID: 7962323
- Hayes MJ, Thomas D, Emmons A, Giordano TJ, Kleer CG. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin Cancer Res 2008; 14:4038 - 44; http://dx.doi.org/10.1158/1078-0432.CCR-07-4379; PMID: 18593979
- Virmani AK, Rathi A, Sathyanarayana UG, Padar A, Huang CX, Cunnigham HT, et al. Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin Cancer Res 2001; 7:1998 - 2004; PMID: 11448917
- Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res 2000; 60:4366 - 71; PMID: 10969779
- Björklund P, Akerström G, Westin G. Accumulation of nonphosphorylated beta-catenin and c-myc in primary and uremic secondary hyperparathyroid tumors. J Clin Endocrinol Metab 2007; 92:338 - 44; http://dx.doi.org/10.1210/jc.2006-1197; PMID: 17047023
- Cetani F, Pardi E, Banti C, Collecchi P, Viacava P, Borsari S, et al. Beta-catenin activation is not involved in sporadic parathyroid carcinomas and adenomas. Endocr Relat Cancer 2010; 17:1 - 6; http://dx.doi.org/10.1677/ERC-09-0147; PMID: 19755524
- Greenwald BD, Harpaz N, Yin J, Huang Y, Tong Y, Brown VL, et al. Loss of heterozygosity affecting the p53, Rb, and mcc/apc tumor suppressor gene loci in dysplastic and cancerous ulcerative colitis. Cancer Res 1992; 52:741 - 5; PMID: 1346256
- Zhang HY, Rumilla KM, Jin L, Nakamura N, Stilling GA, Ruebel KH, et al. Association of DNA methylation and epigenetic inactivation of RASSF1A and beta-catenin with metastasis in small bowel carcinoid tumors. Endocrine 2006; 30:299 - 306; http://dx.doi.org/10.1007/s12020-006-0008-1; PMID: 17526942
- Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res 2005; 65:3497 - 508; http://dx.doi.org/10.1158/0008-5472.CAN-04-4088; PMID: 15867337
- Pfeifer GP, Yoon JH, Liu L, Tommasi S, Wilczynski SP, Dammann R. Methylation of the RASSF1A gene in human cancers. Biol Chem 2002; 383:907 - 14; http://dx.doi.org/10.1515/BC.2002.097; PMID: 12222680
- Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 2000; 25:315 - 9; http://dx.doi.org/10.1038/77083; PMID: 10888881
- Agathanggelou A, Honorio S, Macartney DP, Martinez A, Dallol A, Rader J, et al. Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 2001; 20:1509 - 18; http://dx.doi.org/10.1038/sj.onc.1204175; PMID: 11313894
- Li J, El-Naggar A, Mao L. Promoter methylation of p16INK4a, RASSF1A, and DAPK is frequent in salivary adenoid cystic carcinoma. Cancer 2005; 104:771 - 6; http://dx.doi.org/10.1002/cncr.21215; PMID: 15959912
- Dahl E, Wiesmann F, Woenckhaus M, Stoehr R, Wild PJ, Veeck J, et al. Frequent loss of SFRP1 expression in multiple human solid tumours: association with aberrant promoter methylation in renal cell carcinoma. Oncogene 2007; 26:5680 - 91; http://dx.doi.org/10.1038/sj.onc.1210345; PMID: 17353908
- Veeck J, Niederacher D, An H, Klopocki E, Wiesmann F, Betz B, et al. Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis. Oncogene 2006; 25:3479 - 88; http://dx.doi.org/10.1038/sj.onc.1209386; PMID: 16449975
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 86; http://dx.doi.org/10.1038/ng.298; PMID: 19151715
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 2007; 8:286 - 98; http://dx.doi.org/10.1038/nrg2005; PMID: 17339880
- Sulaiman L, Nilsson IL, Juhlin CC, Haglund F, Höög A, Larsson C, et al. Genetic characterization of large parathyroid adenomas. Endocr Relat Cancer 2012; 19:389 - 407; http://dx.doi.org/10.1530/ERC-11-0140; PMID: 22454399
- Rosen JE, Costouros NG, Lorang D, Burns AL, Alexander HR, Skarulis MC, et al. Gland size is associated with changes in gene expression profiles in sporadic parathyroid adenomas. Ann Surg Oncol 2005; 12:412 - 6; http://dx.doi.org/10.1245/ASO.2005.03.103; PMID: 15915376
- Juhlin CC, Villablanca A, Sandelin K, Haglund F, Nordenström J, Forsberg L, et al. Parafibromin immunoreactivity: its use as an additional diagnostic marker for parathyroid tumor classification. Endocr Relat Cancer 2007; 14:501 - 12; http://dx.doi.org/10.1677/ERC-07-0021; PMID: 17639063
- Sulaiman L, Nilsson IL, Juhlin CC, Haglund F, Höög A, Larsson C, et al. Genetic characterization of large parathyroid adenomas. Endocr Relat Cancer 2012; 19:389 - 407; http://dx.doi.org/10.1530/ERC-11-0140; PMID: 22454399
- Shattuck TM, Välimäki S, Obara T, Gaz RD, Clark OH, Shoback D, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med 2003; 349:1722 - 9; http://dx.doi.org/10.1056/NEJMoa031237; PMID: 14585940
- Juhlin C, Larsson C, Yakoleva T, Leibiger I, Leibiger B, Alimov A, et al. Loss of parafibromin expression in a subset of parathyroid adenomas. Endocr Relat Cancer 2006; 13:509 - 23; http://dx.doi.org/10.1677/erc.1.01058; PMID: 16728578