Abstract
The term epigenetics refers to the heritable changes in gene expression that do not represent changes in DNA sequence. DNA methylation and histone modification are the best studied epigenetic mechanisms. However, microRNAs, which affect gene expression at the posttranscriptional level, should be considered as members of the epigenetic machinery too. Myelodysplastic syndromes (MDS) are clone disorders of the hematopoietic stem cell with increased risk of leukemic transformation. Over the years, increased number of studies indicates the role of epigenetic mechanisms, including microRNAs, in MDS pathogenesis and prognosis. Indeed, epigenetic therapy with demethylating agents has already been applied to MDS. In this review we summarize current knowledge on the role of epigenetic alterations in MDS pathogenesis and treatment.
Introduction
Epigenetics refer to the heritable changes in gene expression that are not reflected in changes in DNA sequence. The term was first used in the 1940s by Conrad Waddington, who defined epigenetics as “the causal interactions between genes and their products, which bring the phenotype into being.”Citation1 Several definitions of epigenetics have been proposed since then. For example, Holliday R. defined epigenetics as “changes in gene expression that are not due to alteration in DNA sequence.”Citation2 Recently, another definition was proposed by Adrian Bird “the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states.”Citation3 The epigenetic machinery comprises DNA methylation, histone modification and RNA interference. RNA interference refers mostly to the recently discovered microRNAs. microRNAs are small, non-coding RNAs that regulate gene expression at the posttranscriptional level repressing the translation of their mRNA target without affecting the DNA sequence of the targeted gene.
Myelodysplastic syndromes (MDS) are heterogeneous disorders of hematopoietic stem cell associated with ineffective hematopoiesis and increased risk of transformation to acute myeloid leukemia (AML). The precise pathophysiological mechanisms of MDS remain unclear. However, it seems that the pathways involved in the pathogenesis of this disease are genetic alterations, repression of apoptosis and deregulation of the microenvironment.Citation4 Emerging data support the role of the aforementioned epigenetic mechanisms in MDS pathogenesis and prognosis. Indeed, several studies, discussed below, provide evidence that key molecules involved in DNA methylation, histone modification and microRNA regulation are implicated in MDS.
The aim of this review is to summarize the role of epigenetics with respect to pathogenesis, prognosis and treatment of MDS.
DNA Methylation
DNA methylation is the best studied epigenetic mechanism. It involves the addition of a methyl group at the 5′ carbon of a cytosine of a 5′-CpG-3′ dinucleotide by DNA methyltransferases (DNMTs).Citation5 Most of the CpG dinucleotides in mammals are methylated. However, regions in genome with high frequency of CpG dinucleotides, called CpG islands, reside in or near promoters of housekeeping genes and are unmethylated.Citation6,Citation7 DNA methylation is transmitted through mitosis and controls gene function by inhibiting DNA transcription. The latter is achieved by two ways. First, DNA methylation prevents transcription factors from binding to gene promoter and, second, methyl-CpG groups bind proteins, namely methyl-CpG-binding domain proteins (MeCPs), which, in turn, recruit histone-modifying proteins leading to chromatin inactivation.Citation8
Several alterations have been reported in MDS regarding DNA methylation, such as aberrant genome methylation and impaired function of proteins that regulate methylation (). Aberrant methylation of selected genes, such as p15INK4B, has been reported by several groups.Citation9,Citation10 However, it seems that altered methylation in MDS is not restricted to unique genes but affects the whole genome. Widespread promoter hypermethylation in MDS and secondary AML has been reported by the recent study of Figueroa et al. According to this study, bone marrow cells from patients with MDS and secondary AML exhibit an extensive pattern of promoter hypermethylation that differs from normal cells and de novo AML and is not limited to cancer-associated genes but affects almost the whole genome. The extension of this phenomenon suggests that general mechanisms that normally control DNA methylation are deregulated in MDS. Of note, members of WNT and MAPK pathways as well as CDKN2A genes were found to be hypermethylated in this study.Citation11 However, WNT-antagonist genes (sFRP1, sFRP2, sFRP4, sFRP5 and DKK1) are found methylated too in another study. As a result WNT-pathway is activated and WNT downstream genes TCF1 and LEF1 are upregulated.Citation19 A possible explanation of these contradictory phenomena is that the relevant methylation status of WNT-pathway genes and WNT-antagonists determines the level of WNT activation. In accordance to the study of Figueroa et al., another group reported the aberrant methylation of CDKN2A gene in MDS and AML. In this cohort, CDKN2B was hypermethylated too while both genes exhibited higher levels of methylation in AML compared with MDS. Interestingly, aberrant methylation of the aforementioned genes was associated with disease progression.Citation12
Table 1. The effect of gene methylation in MDS
Global methylation analysis of total leukocytes form peripheral blood revealed different methylation status between MDS patients and normal controls. These results were consistent with the ones reported by previous studies in bone marrow cells. Of note, aberrant hypermethylation occurred mainly outside CpG islands. Among the most affected genes was DOCK4 (Dedicator of cytokinesis 4) an activator of G-proteins, whose mutations have been associated with invasion of cancer cells.Citation13 However, the fact that total leukocytes from peripheral blood contain normal and dysplastic cells should always be taken into consideration when evaluating these results.
During the last few years, several studies of gene methylation in MDS have been published. SOX7 (sex determining region Y-box) gene, which codes for an inhibitor of WNT pathway, is found to be methylated in its CpG island in 58.1% of MDS patients and the percentage of patients with hypermethylation of SOX7 gene was higher among patients with advanced disease. Interestingly, the percentage of methylation of CpG island seems to represent a predictive factor of MDS prognosis.Citation14 In addition, methylation of Dab2 (human disabled - 2) gene is associated with MDS pathogenesis, since it is methylated in 50.6% of MDS patients and its hypermethylation is mostly associated with advanced stages of disease.Citation15 Another gene whose methylation is implicated in MDS pathogenesis is the erythroid transcription factor GATA1, whose upregulation is necessary for erythroid and megakaryocytic development. Methylation of GATA1 in MDS patients inhibits upregulation of the gene and leads to ineffective erythropoiesis.Citation20 Finally, the promoter of p73, a tumor suppressor gene and homolog of p53 gene, is hypermethylated in MDS and is negatively associated with patients’ prognosis.Citation21
As previously mentioned, DNA methylation is catalyzed by methyltransferases (DNMTs). There are three DNMTs in mammals, DNMT3A and B that catalyze de novo methylation and DNMT1 that is associated with maintenance of methylation. 5′-methylcytocine is converted to 5′-hydroxymethylcytocine by Ten Eleven Translocation (TET) proteins (TET1, 2 and 3), whose action depends on aKG and Fe2+. 5′-hydroxymethylcytocine (5-hmC) is short lived and is followed by demethylation of cytocine. Of note, isocitrate dehydrogenase 1 (IDH1) converts isocitrate to aKG which is necessary for TET2 function.Citation22 As a result, proper function of DNMTs, TET proteins and IDH1 protein are necessary for normal DNA methylation.
Mutations in DNMT3A were first reported in AMLCitation23,Citation24 and were recently identified in advanced stages MDS patients (RAEB I and II).Citation16 Additionally, Walter et al. found 13 heterozygous, missence, nonsense and frame shift somatic mutations in DNMT3A in 8% of patients. Four of them occurred in R882 amino acid which residues in methyltransferase domain of DNMT3A and has been associated with impaired function of the enzyme since it reduces its ability to bind DNA. These mutations seem to occur at early stages of MDS course and are associated with worse overall survival (OS) and event free survival (EFS). However, multivariate analysis could not be performed due to the small number of patients.Citation17 Another study confirmed these results and also found that global methylation status did not differ between mutated and wild type patients.Citation25 The result of DNMT3A mutations in DNA methylation and the mechanism by which they affect gene expression remain unclear. Further studies will clarify this subject. Apart from somatic mutations of DNMT3A, overexpression of DNMT1, 3A and B in MDS has also been reported, particularly in refractory anemia (RA) and refractory anemia with excess of blasts (RAEB), which is consistent with the hypermethylation described in MDS.Citation26
Patients with MDS and AML and myeloproliferative neoplasms (MPN) exhibit uniparental disomy (UPD) on chromosome 4q24. The deleted region on chromosome 4q24 contains the aforementioned gene, TET2. Deletions or missence, nonsence and frameshift mutations of TET2 are observed in MDS, MPN and AML. The defects occur at early stages of MDS pathogenesis, which is deduced from the fact that mutations and deletions of TET2 are observed in CD34+ cells.Citation27-Citation30 Homozygous and hemizygous mutations as well as heterozygous and compound heterozygous mutations occur in patients with MDS/MPN and affect conserved regions of TET2 protein and the N-terminal domain.Citation18 The clinical impact of TET2 mutations has not been clarified yet. It seems that mutations are associated with impaired TET2 catalytic activity leading to low levels of 5-hmC. In spite of the fact that the most expected result of such a modification is DNA hypermethylation, low levels of 5-hmC are associated with DNA hypomethylation.Citation31 However, hypermethylation of DNA has also been reported.Citation11 Of note, the effect of TET2 mutations on patients’ prognosis is still a contradictory issue. There are a few studies supporting the favorable effect of TET2 mutations on OS and response rate to azacitidineCitation29,Citation32,Citation33 while Smith et al. did not found any correlation between TET2 mutations and OS.Citation34 As reported by the authors, these contradictory results may be attributed to differences in study design and source of data. Further studies will illuminate this issue.
IDH1 and 2 are enzymes that participate in the citric acid cycle and catalyze the oxidative decarboxylation of isocitrate producing a-ketoglutarate in the cytoplasm and peroxisomes respectively. IDH1 is located in cytoplasm and peroxisomes and IDH2 in mitochondria. IDH1 mutations were first found in gliomas and were then discovered in AML patients by whole genome sequencing.Citation35 Most mutations affect highly conserved arginine residues: IDH1-R132, IDH2-R172 and IDH2-R140. These heterozygous mutations are found to inhibit the catalytic activity of IDH1 in gliomas resulting in loss-of function of IDH1, reduction in aKG and activation of HIF-a pathway.Citation36 However, the fact that only one gene is mutated raised the possibility that these mutations may result in gain of function, not simply in loss of function. This is the reason why Dang et al. performed metabolic studies and showed that IDH1 mutations result in the production of 2-hydroxyglutarate (2-HG), which is an onco-metabolite and its accumulation is associated with progression of gliomas.Citation37 Very little is known about these rare, albeit important, mutations in MDS. Most of the studies published so far agree that IDH1 and 2 mutations are rare in MDS (3–5 and 12% in one study), are associated with normal or good risk karyotype and represent and independent factor of poor prognosis and AML transformation.Citation38-Citation41 Apart from the aforementioned mutations, a new nonsense mutation of IDH1 has been recently discovered; R100X, which results in a truncated molecule, without enzymatic activities.Citation40 It is worth noting that IDH1 and 2 mutations result in DNA hypermethylation in AML and are mutually exclusive with TET2 mutations.Citation42 Thus, IDH1 and 2 mutations may represent a potential prognostic marker as well as therapeutic target in MDS treatment.
Finally, methylation of imprinted genes seems to play a role in MDS. The term genomic imprinting refers to the preferential expression of a gene depending on whether they come from the maternal or paternal DNA. As a result, the inherited genes are unequal expressed and the genetic locus acts as haploid although it is diploid. A mechanism by which this phenomenon takes place is DNA methylation. This is a normal mechanism, however aberrant methylation of imprinted genes have been associated with myeloid malignancies. Thus, the imprinted genes MEG3 (maternally expressed gene 3) and SNRPN (small nuclear ribonucleoprotein polypeptide N) are hypermethylated in MDS and AML. MEG3 methylation is also associated with worse overall survival suggesting that it is a potential prognostic factor.Citation43
Histone Modification
Histones are proteins that organize the DNA of a eukaryotic cell into structural units called nucleosomes. There are two classes of histone proteins: the core histones H2A, H2B, H3 and H4 and the linker histone H1. Each nucleosome is an octamer consisting of two pairs of the core histone proteins and 147 base pairs of DNA wrapped around them. H3 and H4 histones have tails of amino acids that comprise mostly lysine and arginine. Epigenetic modifications of these amino acids (acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ADP-ribosylation, deimination, proline isomerization) are associated with compaction or relaxation of nucleosomes resulting in repression or activation of gene translation, respectively.
Histone methylation and acetylation are the best-studied modifications. Acetylation is mediated by histone acetylases (HAT) and results in activation of gene transcription while deacetylation by histone deacetylases (HDAC), prevents transcription. HATs are divided into group A, which are located in the nucleus, and group B, located in the cytoplasm. HDACs are divided into three classes. Class I HDACs are located in the nucleus and participate in transcription. Class II HDACs can be found in the nucleus and in the cytoplasm and mediate protein modifications, and Class III are associated with cell cycle.Citation4
The effect of methylation depends on the residue modified and the degree of modification and could be associated with activation or repression of gene transcription. Trimethylation of lysine 27 of H3 (H3K27me3) by the polycomb complex PRC2 results in transcription inactivation. Similarly, di- and tri- methylation of lysine 9 of H3 (H3K9me2/3) leads to gene silencing. In contrast, trimethylation of lysine 4 (H3K4me3) and lysine 6 of H3 (H3K6me3) are marks of active gene translation. Thus, histone modification determines the “on” or “off” status of DNA, which is necessary for the accurate control of gene transcription. In conclusion, it should be mentioned that the two important epigenetic mechanisms, DNA methylation and histone modification, are related to each other, since histone modification directs DNA methylation in early development and DNA methylation represents a template for the histone modification.Citation44
Alteration of histone modification has been associated with MDS pathogenesis and prognosis (). Genes that regulate histone modification, like Polycomb group (PcG) proteins, are mutated in MDS. PcG acts in two complexes namely PRC1 and 2 that are responsible for histone modification and gene silencing. PRC1 comprises three proteins: RING1A, RING1B and BMI1 while PRC2 contains four core proteins namely EZH2, EED, SUZ12 and RbAp46/48. EZH2, located in 7q36.1, is the catalytic unit of PRC2 and mediates methylation of H3K27 resulting in gene silencing. Missense, nonsense, donor-splice-site and frameshift mutations of EZH2 have been discovered in MDS patients. Hemizygous mutations were found in patients with microdeletions in 7q and homozygous mutations in 7q acquired UPD.Citation45,Citation47,Citation48 These mutations contribute to loss-of-function and decreased H3K27 methylation suggesting that EZH2 acts as tumor suppressor. However, recent studies support the oncogenic role of EZH2 in myeloid malignancies.Citation46,Citation49 Finally, EZH2 mutations are associated with decreased overall survival but more studies are required to clarify the role of these mutations in MDS prognosis.Citation48
Table 2. Histone modification in MDS
SUZ12 (suppressor of zeste 12 homolog), member of PRC2 complex is necessary for cell proliferation in vivo and in vitro and EZH2 activity in vivo.Citation50 Mutation in the SUZ12 gene as well as in the EED gene (also member of PRC2 complex) has been recently discovered in MDS and have been associated with compromised PRC2 function.Citation51
The ASXL1 gene is located on 20q11 chromosome and codes for a nuclear protein that is a member of PRC2 complex and regulates gene expression by repressing transcription of some genes while enhancing the transcription of others. Mutations of ASXL1 have been reported in 11.4–20.7% of MDS patients.Citation52 The vast majority of these mutations are loss-of-function frameshift and nonsense mutations and result in compromised function of PRC2 regarding H3K27 methylation, similarly to the effect of aforementioned EZH2 mutations.Citation53 Finally, combined frameshift and point mutations as well as frameshift mutations of ASXL alone represent an independent prognostic marker in MDS.Citation54
BMI1 protein belongs to PRC1 complex and mediates ubiquitylation of H2AK119 leading to PRC2 recruitment.Citation55 It is responsible for CD34+ cells’ self – renewal and has been associated with poor prognosis MDS (RAEB and RAEB-T) and disease progression to AML.Citation56,Citation57 Finally, overexpression of EZH, RING1 and BMI1in MDS has been recently reported and has been associated with poor prognosis too.Citation58 It is worth noting that combined mutations of the previously mentioned genes: ASLX1, IDH1, IDH2, TET2 and WT1, have been recently reported, highlighting the complexity of the pathways involved in MDS pathogenesis and progression.Citation59
Finally, it should be mentioned that the chromatin remodeling factor ATRX was found to be mutated in α thalassaemia associated MDS (ATMDS). ATRX is a nuclear protein that regulates gene expression through interaction with the HP1 components of heterochromatin. Its normal function is important for α-globin expression, while the hematological phenotype of patients with ATMDS is attributed to ATRX mutations.Citation60
The Role of microRNAs in MDS and Epigenetic Machinery
microRNAs are small non-coding RNAs that regulate gene expression at the posttranscriptional level by inhibiting protein translation or destabilizing target transcripts. They are transcribed as long, capped and polyadenylated primary precursors (pri-microRNA) by RNA polymerase II.Citation61,Citation62 Pri-microRNAs are then cleaved by Drosha (a nuclear RNase III) into a hairpin shaped precursor that is called pre-microRNA and consists of 70–100 nucleotides.Citation63 Drosha creates a complex with the dsRNA-binding protein, DGCR8 (DiGeorge syndrome chromosomal region 8),Citation64-Citation66 which, unlike Drosha, can stably interact with pri-microRNAs.Citation67 In that way, Drosha is stabilized. Pre-microRNAs are exported to the cytoplasm by exportin 5.Citation68 At the cytoplasm, the RNase III Dicer in association with the dsRNA-binding protein TRBP (transactivation-responsive RNA-binding protein) and PACT (in humans), removes the terminal loop of pre-microRNAs and creates a 19–24 nucleotide microRNA duplex.Citation69-Citation71 The protein complex RISC (RNA-induced silencing complex) binds to the aforementioned duplex. The mature microRNA is determined by the strand of the complex that remains stably associated with RISC. Then, the mature microRNA drives RISC to the target mRNA.Citation72,Citation73 microRNAs bind to the 3′-untranslated regions (UTRs) of mRNAs targets by their “seed” region (nucleotides 2 to 8 counted from their 5′ end) through perfect (in plants) or imperfect (in mammals) base-pairing.Citation74
microRNAs have been associated with almost every normal cell function as well as with normal hematopoiesis and hematologic malignancies including MDS.Citation75 These small non-coding RNAs hold a leading role in the epigenetic machinery acting in a bidirectional way: They target key molecules of the epigenetic reactions but are susceptible to epigenetic regulation themselves. However, the available data on this interesting, albeit confusing, subject are still very few. The results of different studies overlap or even oppose to each other suggesting that there is a long way to the understanding of microRNA function in MDS.
Expression of microRNAs in MDS
As shown in and , many studies using different techniques have tried to shed light on the role of microRNAs in MDS pathogenesis and prognosis, the exact pathways are not yet understood.Citation82 Interestingly, the expression profile of microRNAs was found different between MDS patients and normal controls. Indeed, microarray and RT-PCR analysis of microRNA expression in mononuclear cells (MNC) of patients suffering from low risk MDS identified 13 microRNAs differentially expressed between patients and controls.Citation76 This was also true when Pons et al. compared the expression of microRNAs in MNC from PB and BM of patients and controls using RT-PCR. Twelve microRNAs were differentially expressed between these two groups.Citation80 Furthermore, expression analysis of microRNAs with Taqman arrays and RT-PCR showed differential expression between patients with 5q- syndrome and controls.Citation77 A recent study identified another microRNA whose expression differs between patients and controls. MiR-21 is overexpressed in bone marrow samples of MDS patients compared with normal controls and directly targets SMAD-7, by binding to its 3′-UTR. SMAD-7 is a negative regulator of TGF-β receptor-I kinase. Thus, SMAD-7 inhibition by miR-21 leads to the activation of TGF-β pathway and ineffective hematopoiesis and especially anemia and dysplasia. Indeed, miR-21 inhibition resulted in SMAD elevation in vivo and stimulated erythropoiesis in vitro.Citation81 Finally, miR-150 has been found upregulated in patients with del (5q) MDS compared with controls and miR-221 was found decreased in sAML compared with other subgroups of MDS.Citation78
Table 3. Downregulated microRNAs in MDS patients
Table 4. Upregulated microRNAs in MDS
In addition, microRNA expression differs between MDS subgroups and is associated with disease prognosis. Hussein et al. used low-density RT–PCR based array and found different microRNA signature between patients with normal karyotype and those with chromosomal alteration.Citation83 Furthermore, Merkerova et al. used illumina array platform and demonstrated that 22 microRNAs distinguished between patients and controls and MDS subtypes.Citation79 Our group found that mir-17–5p and mir-20a were under expressed in high risk MDS, compared with low risk MDS patients and that let-7a was under expressed in patients with intermediate or high-risk karyotype. Additionally, mir-17–5p and mir-20a expression was associated with increased overall survival of MDS patients suggesting that these two microRNAs are favorable prognostic factors.Citation84 Finally, Sokol et al. focused on the prognostic value of microRNA expression in MDS and found 10 microRNAs that were associated with International Prognostic Scoring System (IPSS) and that patients with high mir-181 expression had worse overall survival than patients with lower expression.Citation85
microRNAs control epigenetic machinery in MDS
microRNAs that affect the epigenetic machinery are called epi-microRNAs. The first study reporting the existence of epi-microRNAs was performed by Fabbri et al., who found that mir-29 family downregulates DNMT3A and B levels in lung cancer cells and its enforced expression results in DNA hypomethylation (through DNMT3A and B inhibition) and tumor-suppressor genes re-expression.Citation86 The same group confirmed the role of mir-29b in DNMT1, DNMT3A and B downregulation in AML cells. They illustrated that mir-29b downregulates DNMT3A and B by binding to the 3′- UTR of their mRNA and DNMT1 by inhibiting the transcription factor Sp1 (specific protein 1). In addition, enforced expression of mir-29b reduced global DNA methylation while its overexpression resulted in upregulation of the tumor suppressor genes ESR1 and p15INK4b.Citation87 These studies illuminated the role of mir-29 family in lung cancer and AML and paved the way for the possible therapeutic use of mir-29. Despite the paucity of data for the role of mir-29 family in MDS, we can hypothesize that, similarly to AML, overexpression of DNMTs and global promoter hypermethylation (including p15INK4b promoter) occurring in MDS could be associated with mir-29 function. Therefore, studies of the role of mir-29 in MDS pathogenesis and prognosis would be of great interest.
Besides their role in posttranscriptional regulation of gene expression, microRNAs seem to act as regulators of gene translation and hematopoietic stem cell differentiation too. Mir-223, which regulates human granulopoiesis by targeting the nuclear factor I-A (NFA-I)Citation88 localizes in the nucleus, forms a complex with PcG proteins and binds NFA-I promoter. This results in NFA-I silencing, which favors granulopoiesis. On the other hand, NFA-I activation leads to erythroid differentiation.Citation89 This is a novel pathway of direct control of hematopoietic stem cell differentiation by a microRNA suggesting that deregulation of mir-223 could contribute to impaired granulopoiesis and possibly disease pathogenesis. Indeed, in AML, AML-ETO fusion gene binds to mir-223 promoter, recruits a complex consisting of DNMTs, HDAC and MCPS and results in its silencing. This modification is associated with impaired myeloid differentiation, which characterizes AML blast cells.Citation90
Epigenetic regulation of microRNA expression in MDS
Apart from their role in regulation of gene expression, microRNAs are susceptible to epigenetic control themselves. microRNA expression depends on their genomic location (microRNAs associated with cancer are located in genomic fragile regions), transcription regulation (i.e., promoter hyper/hypomethylation, transcription factors, etc.) and posttranscriptional events including maturation process.
A recent study demonstrated that Dicer and Drosha, two ribonucleases necessary for microRNA maturation, are found downregulated in mesenchymal stromal cells (MSC) from BM of MDS patients compared with normal controls and MNC from MDS patients. The same study found global microRNA downregulation (including the hematopoiesis related mir-155, 181 and 222), which shows that the maturation mechanism of microRNAs in these cells is impaired. Additionally, the authors note that mir-155, 181 and 222 target Dicer and Drosha, which highlights the complexity of the pathways involved in MDS pathogenesis.Citation91 To our knowledge, this is the first study that brings evidence of decreased levels of Dicer and Drosha in BM MSC from MDS patients and addresses the role of the microenvironment in MDS.
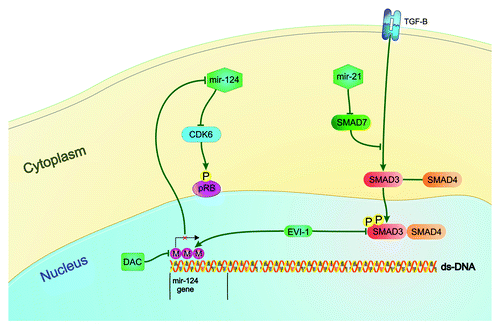
Another microRNA affected by the epigenetic machinery in MDS is miR-124. It is transcribed in three regions of the genome as miR-124a-1 (8p), miR-124a-2 (8q) and miR-124a-3 (20q). In the study of Castoro et al., miR-124a-1 was methylated in the majority of MDS (78%) and AML (95%) patients and was associated with worse overall survival while miR-124a-3 was methylated in 47% of MDS and 65% of AML patients. Treatment with the DNA methylation inhibitor decitabine (DAC) resulted in a significant decrease in methylation status of miR-124a-1 and 3 in patients who responded to treatment. On the other hand, non-responders had no significant de-methylation of this microRNA. Methylation levels were measured at day 5, 12 and 30 after DAC treatment. In addition, patients who responded to DAC showed a significant increase in miR-124 expression at day 5 while patients who did not respond to treatment had no significant increase in miR-124 expression. Finally, miR-124 re-expression was associated with the repression of its target mRNA, CDK6, at day 5.Citation92 This study provides a representative example of the effects of DNA methylation and epigenetic therapies on microRNA expression. Of interest, these results were confirmed by a recent study according to which, miR-124 is downregulated in MDS patients and is related with the level of its promoter methylation.Citation93
The role of the aforementioned miR-124 in MDS pathogenesis and its relationship with its regulator EVI1 was later studied by another group. EVI1 is a transcription factor that regulates the expression of many genes responsible for cell cycle, including SMAD3 (a member of TGF-β pathway) and generates a model of MDS when expressed in murine bone marrow. Its activation in MDS patients is associated with megakaryocytic and erythroid dysplasia, refractory anemia and bone marrow failure. Dickstein at al created an MDS model by expressing EVI1 in murine bone marrow and studied the role of this transcription factor in MDS pathogenesis. One of the major pathways resulting in MDS phenotype in this study was miR-124 silencing by CpG methylation of its promoter.Citation94 These data, which are summarized in , demonstrate an example of epigenetic alteration of microRNAs in MDS pathogenesis.
Figure 1. Epigenetic alteration of microRNAs and MDS pathogenesis Mir-124, which is hypermethylated and under-expressed in MDS patients, participates in MDS pathogenesis. It down-regulates the cyclin dependent kinase 6 (CDK6), which in turn phosphorylates and inactivates the tumor-suppressor protein Rb. EVI1 represses miR-124 expression by promoter methylation. It also down-regulates the expression of SMAD3, a member of TGF-β pathway that regulates cell differentiation, proliferation and apoptosis. Mir-124 activity can be restored by the treatment with the DNA methylation inhibitor, Decitabine. The TGF-β pathway is also regulated by mir-21 which down-regulates the expression of SMAD7, which in turn is a negative regulator of TGF-β receptor.
MiR-10a expression is induced by the transcription factor TWIST-1, which is upregulated in CD34+ cells of MDS, interacts with the DNA-binding domain of p53 and leads to resistance in apoptosis. In addition, TWIST-1 binds to the promoter of miR-10a/b and induces its expression. A recent study found that mir-10a/b is overexpressed in CD34+ cells from BM of MDS patients compared with healthy controls correlating with TWIST-1 levels. Furthermore, miR-10a/b, TWIST-1, NF-kB and p53 form a pathway which control sensitivity to apoptosis in MDS patients.Citation95 In summary, transcriptional regulation of mir-10a/b seems to be important for MDS pathogenesis and constitutes a candidate pathway for target therapy.
Epigenetic Therapy of MDS
The term epigenetic therapy refers to treatment of a disease by targeting epigenetic pathways. As illustrated above, the role of epigenetic mechanisms in MDS pathogenesis and prognosis has received very much attention. The observation that DNA methylation, histone modification and microRNA interference play a critical role in MDS raised hope that key molecules of these pathways could represent new therapeutic targets or even therapeutic agents. The first epigenetic drugs for MDS treatment, 5-azacytidine (azacitidine) and 5-aza-2’-deoxycytidine (decitabine), have already been used in MDS patients with promising results.
Targeting DNA methylation
The central epigenetic pathway (DNA methylation) has already been targeted by the aforementioned hypomethylating agents azacitidine and decitabine. Both drugs were synthesized in the 1960s and represent cytocine analogs in which the carbon at position 5 of the ring is replaced by N. Azacitidine incorporates first into RNA and then into DNA and binds irreversibly DNMTs causing its degradation. As a result, when DNA replicates, the newly synthesized DNA strands remain unmethylated. The anti-neoplastic effect of azacitidine is a dose-dependent: At low doses it inhibits DNMTs while in high doses is cytotoxic. It was established in the treatment of high-risk MDS with a randomized, open-label phase III study of azacitidine compared with conventional regimens, which proved the survival benefit of patients receiving azacitidine. Nowadays, azacitidine is used for the treatment of intermediate II and high risk IPSS MDS.Citation96 Decitabine, on the other hand, incorporates directly into DNA and inhibits DNMTs action in a similar way as azacitidine. It is also indicated for MDS and AML treatment in the US
Although azacitidine has proved its demethylating effect in vivo and vitro, questions were raised about the exact mechanism of action in MDS. According to many scientists the belief that demethylation itself is sufficient for the anti-tumor effect of azacitidine and decitabine is just simplicity of what really takes place into the cell. Indeed, there are still no data proving a direct relationship between DNA hypomethylation and disease response. On the other hand, azacitidine is administered in low doses in which the drug exhibits hypomethylating function.Citation97 Whether DNA hypomethylation is the only mechanism of action or a part of a complicated pathway still remains an open question. Finally, it should be mentioned that several clinical trials that study the combination of azacitidine with other agents like lenalidomide, valproic acid etc or even oral administration of azacitidine have been already launched aiming to change the current treatment approach of MDS.
Targeting histone modification
Targeting histone modification currently means targeting histone deacetylases. As discussed above, histone modifications are involved in MDS pathogenesis so the use of histone deacetylace inhibitors (HDACi) in MDS treatment seems rational. HDACi induce differentiation, cell cycle arrest and expression of p21cip/waf1.Citation98,Citation99 However, the exact mechanism of their action has not been clarified yet. The application of HDACi as therapeutic agents of hematological malignancies is still under clinical research. A phase II study of belinostat (PXD101) for the treatment of MDS stopped at the first stage of enrollment.Citation100 However, there are several phase I and II clinical trials of HDACi with or without azacitidine and chemotherapy regiments for the treatment of MDS and AML. In addition, it has been showed that valproic acid targets HDAC and in combination with all-trans retinoic acid can induce differentiation of malignant cells.Citation101,Citation102 Thus, the possibility that new therapeutic agents will change the current treatment of MDS cannot be excluded.
Conclusions and Future Directions
In this review we have summarized the current knowledge in epigenetic alterations, including microRNAs, in MDS. Growing evidence supports the role of aberrant DNA methylation, histone modification and microRNA regulation in MDS pathogenesis and prognosis and helps decipher the complicated pathways that are involved in this disease. The greatest offer of these data so far is the development of epigenetic therapies. The most illustrative example is azacitidine, which is the only agent associated with increased overall survival in MDS patients. However, the development of drugs that target histone modification is expected to change thoroughly the therapeutic approach in MDS.
In conclusion, the current knowledge on epigenetic alterations in MDS seems to be only the tip of the iceberg since many important issues remain unclear. Further studies are required that will elucidate the exact role of epigenetic alterations in MDS as well as the role of microRNAs in epigenetic machinery.
Acknowledgments
The authors acknowledge the help of Dr. A. Pouliakis who designed .
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Waddington CH. Preliminary Notes on the Development of the Wings in Normal and Mutant Strains of Drosophila. Proc Natl Acad Sci U S A 1939; 25:299 - 307; http://dx.doi.org/10.1073/pnas.25.7.299; PMID: 16577903
- Holliday R. The inheritance of epigenetic defects. Science 1987; 238:163 - 70; http://dx.doi.org/10.1126/science.3310230; PMID: 3310230
- Bird A. Perceptions of epigenetics. Nature 2007; 447:396 - 8; http://dx.doi.org/10.1038/nature05913; PMID: 17522671
- Stintzing S, Kemmerling R, Kiesslich T, Alinger B, Ocker M, Neureiter D. Myelodysplastic syndrome and histone deacetylase inhibitors: “to be or not to be acetylated”?. J Biomed Biotechnol 2011; 2011:214143; http://dx.doi.org/10.1155/2011/214143; PMID: 21629744
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell 2007; 128:669 - 81; http://dx.doi.org/10.1016/j.cell.2007.01.033; PMID: 17320505
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003; 349:2042 - 54; http://dx.doi.org/10.1056/NEJMra023075; PMID: 14627790
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457 - 66; http://dx.doi.org/10.1038/ng1990; PMID: 17334365
- Ballestar E, Esteller M. Methyl-CpG-binding proteins in cancer: blaming the DNA methylation messenger. Biochem Cell Biol 2005; 83:374 - 84; http://dx.doi.org/10.1139/o05-035; PMID: 15959563
- Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P. Promoter hypermethylation of p15INK4B, HIC1, CDH1, and ER is frequent in myelodysplastic syndrome and predicts poor prognosis in early-stage patients. Eur J Haematol 2006; 76:23 - 32; http://dx.doi.org/10.1111/j.1600-0609.2005.00559.x; PMID: 16343268
- Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Methylation of p15INK4B is common, is associated with deletion of genes on chromosome arm 7q and predicts a poor prognosis in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2003; 17:1813 - 9; http://dx.doi.org/10.1038/sj.leu.2403054
- Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 2009; 114:3448 - 58; http://dx.doi.org/10.1182/blood-2009-01-200519; PMID: 19652201
- Cechova H, Lassuthova P, Novakova L, Belickova M, Stemberkova R, Jencik J, et al. Monitoring of methylation changes in 9p21 region in patients with myelodysplastic syndromes and acute myeloid leukemia. Neoplasma 2012; 59:168 - 74; http://dx.doi.org/10.4149/neo_2012_022; PMID: 22248274
- Zhou L, Opalinska J, Sohal D, Yu Y, Mo Y, Bhagat T, et al. Aberrant epigenetic and genetic marks are seen in myelodysplastic leukocytes and reveal Dock4 as a candidate pathogenic gene on chromosome 7q. J Biol Chem 2011; 286:25211 - 23; http://dx.doi.org/10.1074/jbc.M111.235028; PMID: 21532034
- Fan R, Zhang LY, Wang H, Yang B, Han T, Zhao XL, et al. Methylation of the CpG island near SOX7 gene promoter is correlated with the poor prognosis of patients with myelodysplastic syndrome. Tohoku J Exp Med 2012; 227:119 - 28; http://dx.doi.org/10.1620/tjem.227.119; PMID: 22706399
- Yang Y, Zhang Q, Xu F, Chang C, Li X. Aberrant promoter methylation of Dab2 gene in myelodysplastic syndrome. Eur J Haematol 2012; 89:469 - 77; http://dx.doi.org/10.1111/ejh.12014; PMID: 23005040
- Ewalt M, Galili NG, Mumtaz M, Churchill M, Rivera S, Borot F, et al. DNMT3a mutations in high-risk myelodysplastic syndrome parallel those found in acute myeloid leukemia. Blood Cancer J 2011; 1:e9; http://dx.doi.org/10.1038/bcj.2011.7; PMID: 22829128
- Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2011; 25:1153 - 8; http://dx.doi.org/10.1038/leu.2011.44
- Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009; 113:6403 - 10; http://dx.doi.org/10.1182/blood-2009-02-205690; PMID: 19372255
- Masala E, Valencia A, Buchi F, Nosi D, Spinelli E, Gozzini A, et al. Hypermethylation of Wnt antagonist gene promoters and activation of Wnt pathway in myelodysplastic marrow cells. Leuk Res 2012; 36:1290 - 5; http://dx.doi.org/10.1016/j.leukres.2012.05.023; PMID: 22742816
- Hopfer O, Nolte F, Mossner M, Komor M, Kmetsch A, Benslasfer O, et al. Epigenetic dysregulation of GATA1 is involved in myelodysplastic syndromes dyserythropoiesis. Eur J Haematol 2012; 88:144 - 53; http://dx.doi.org/10.1111/j.1600-0609.2011.01715.x; PMID: 21967505
- Zhao Y, Gu S, Guo J, Zhang Z, Zhang X, Li X, et al. Aberration of p73 promoter methylation in de novo myelodysplastic syndrome. Hematology 2012; 17:275 - 82; http://dx.doi.org/10.1179/1607845412Y.0000000018; PMID: 22971533
- Graubert T, Walter MJ. Genetics of myelodysplastic syndromes: new insights. Hematology Am Soc Hematol Educ Program 2011; 2011:543 - 9; http://dx.doi.org/10.1182/asheducation-2011.1.543; PMID: 22160087
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010; 363:2424 - 33; http://dx.doi.org/10.1056/NEJMoa1005143; PMID: 21067377
- Yamashita Y, Yuan J, Suetake I, Suzuki H, Ishikawa Y, Choi YL, et al. Array-based genomic resequencing of human leukemia. Oncogene 2010; 29:3723 - 31; http://dx.doi.org/10.1038/onc.2010.117; PMID: 20400977
- Thol F, Winschel C, Lüdeking A, Yun H, Friesen I, Damm F, et al. Rare occurrence of DNMT3A mutations in myelodysplastic syndromes. Haematologica 2011; 96:1870 - 3; http://dx.doi.org/10.3324/haematol.2011.045559; PMID: 21880636
- Länger F, Dingemann J, Kreipe H, Lehmann U. Up-regulation of DNA methyltransferases DNMT1, 3A, and 3B in myelodysplastic syndrome. Leuk Res 2005; 29:325 - 9; http://dx.doi.org/10.1016/j.leukres.2004.08.004; PMID: 15661269
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009; 360:2289 - 301; http://dx.doi.org/10.1056/NEJMoa0810069; PMID: 19474426
- Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 2009; 41:838 - 42; http://dx.doi.org/10.1038/ng.391; PMID: 19483684
- Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, et al, Groupe Francophone des Myélodysplasies. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood 2009; 114:3285 - 91; http://dx.doi.org/10.1182/blood-2009-04-215814; PMID: 19666869
- Mohamedali AM, Smith AE, Gaken J, Lea NC, Mian SA, Westwood NB, et al. Novel TET2 mutations associated with UPD4q24 in myelodysplastic syndrome. J Clin Oncol 2009; 27:4002 - 6; http://dx.doi.org/10.1200/JCO.2009.22.6985; PMID: 19528370
- Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010; 468:839 - 43; http://dx.doi.org/10.1038/nature09586; PMID: 21057493
- Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2011; 25:1147 - 52; http://dx.doi.org/10.1038/leu.2011.71
- Wang J, Ai X, Gale RP, Xu Z, Qin T, Fang L, et al. TET2, ASXL1 and EZH2 mutations in Chinese with myelodysplastic syndromes. Leuk Res 2013; 37:305 - 11; PMID: 23099237
- Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gäken J, Lea NC, et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood 2010; 116:3923 - 32; http://dx.doi.org/10.1182/blood-2010-03-274704; PMID: 20693430
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361:1058 - 66; http://dx.doi.org/10.1056/NEJMoa0903840; PMID: 19657110
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009; 324:261 - 5; http://dx.doi.org/10.1126/science.1170944; PMID: 19359588
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462:739 - 44; http://dx.doi.org/10.1038/nature08617; PMID: 19935646
- Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2010; 24:1094 - 6; http://dx.doi.org/10.1038/leu.2010.52
- Thol F, Weissinger EM, Krauter J, Wagner K, Damm F, Wichmann M, et al. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica 2010; 95:1668 - 74; http://dx.doi.org/10.3324/haematol.2010.025494; PMID: 20494930
- Yoshida K, Sanada M, Kato M, Kawahata R, Matsubara A, Takita J, et al. A nonsense mutation of IDH1 in myelodysplastic syndromes and related disorders. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2011; 25:184 - 6; http://dx.doi.org/10.1038/leu.2010.241
- Patnaik MM, Hanson CA, Hodnefield JM, Lasho TL, Finke CM, Knudson RA, et al. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic study of 277 patients. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2012; 26:101 - 5; http://dx.doi.org/10.1038/leu.2011.298
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18:553 - 67; http://dx.doi.org/10.1016/j.ccr.2010.11.015; PMID: 21130701
- Benetatos L, Hatzimichael E, Dasoula A, Dranitsaris G, Tsiara S, Syrrou M, et al. CpG methylation analysis of the MEG3 and SNRPN imprinted genes in acute myeloid leukemia and myelodysplastic syndromes. Leuk Res 2010; 34:148 - 53; http://dx.doi.org/10.1016/j.leukres.2009.06.019; PMID: 19595458
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10:295 - 304; http://dx.doi.org/10.1038/nrg2540; PMID: 19308066
- Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010; 42:722 - 6; http://dx.doi.org/10.1038/ng.621; PMID: 20601953
- Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 2012; 120:1107 - 17; http://dx.doi.org/10.1182/blood-2011-11-394932; PMID: 22677129
- Makishima H, Jankowska AM, Tiu RV, Szpurka H, Sugimoto Y, Hu Z, et al. Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2010; 24:1799 - 804; http://dx.doi.org/10.1038/leu.2010.167
- Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tönnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 2010; 42:665 - 7; http://dx.doi.org/10.1038/ng.620; PMID: 20601954
- Herrera-Merchan A, Arranz L, Ligos JM, de Molina A, Dominguez O, Gonzalez S. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat Commun 2012; 3:623; http://dx.doi.org/10.1038/ncomms1623; PMID: 22233633
- Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 2004; 23:4061 - 71; http://dx.doi.org/10.1038/sj.emboj.7600402; PMID: 15385962
- Score J, Hidalgo-Curtis C, Jones AV, Winkelmann N, Skinner A, Ward D, et al. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 2012; 119:1208 - 13; http://dx.doi.org/10.1182/blood-2011-07-367243; PMID: 22053108
- Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol 2012; 5:12; http://dx.doi.org/10.1186/1756-8722-5-12; PMID: 22436456
- Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012; 22:180 - 93; http://dx.doi.org/10.1016/j.ccr.2012.06.032; PMID: 22897849
- Thol F, Friesen I, Damm F, Yun H, Weissinger EM, Krauter J, et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol 2011; 29:2499 - 506; http://dx.doi.org/10.1200/JCO.2010.33.4938; PMID: 21576631
- Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 2012; 12:599 - 612; http://dx.doi.org/10.1038/nrc3343; PMID: 22898539
- Mihara K, Chowdhury M, Nakaju N, Hidani S, Ihara A, Hyodo H, et al. Bmi-1 is useful as a novel molecular marker for predicting progression of myelodysplastic syndrome and patient prognosis. Blood 2006; 107:305 - 8; http://dx.doi.org/10.1182/blood-2005-06-2393; PMID: 16160010
- Xu F, Yang R, Wu L, He Q, Zhang Z, Zhang Q, et al. Overexpression of BMI1 confers clonal cells resistance to apoptosis and contributes to adverse prognosis in myelodysplastic syndrome. Cancer Lett 2012; 317:33 - 40; http://dx.doi.org/10.1016/j.canlet.2011.11.012; PMID: 22120066
- Xu F, Li X, Wu L, Zhang Q, Yang R, Yang Y, et al. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: relation to adverse epigenetic alteration and poor prognostic scoring. Ann Hematol 2011; 90:643 - 53; http://dx.doi.org/10.1007/s00277-010-1128-5; PMID: 21125401
- Rocquain J, Carbuccia N, Trouplin V, Raynaud S, Murati A, Nezri M, et al. Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer 2010; 10:401; http://dx.doi.org/10.1186/1471-2407-10-401; PMID: 20678218
- Gibbons RJ, Pellagatti A, Garrick D, Wood WG, Malik N, Ayyub H, et al. Identification of acquired somatic mutations in the gene encoding chromatin-remodeling factor ATRX in the alpha-thalassemia myelodysplasia syndrome (ATMDS). Nat Genet 2003; 34:446 - 9; http://dx.doi.org/10.1038/ng1213; PMID: 12858175
- Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004; 10:1957 - 66; http://dx.doi.org/10.1261/rna.7135204; PMID: 15525708
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004; 23:4051 - 60; http://dx.doi.org/10.1038/sj.emboj.7600385; PMID: 15372072
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003; 425:415 - 9; http://dx.doi.org/10.1038/nature01957; PMID: 14508493
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature 2004; 432:231 - 5; http://dx.doi.org/10.1038/nature03049; PMID: 15531879
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004; 432:235 - 40; http://dx.doi.org/10.1038/nature03120; PMID: 15531877
- Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 2004; 18:3016 - 27; http://dx.doi.org/10.1101/gad.1262504; PMID: 15574589
- Yeom KH, Lee Y, Han J, Suh MR, Kim VN. Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing. Nucleic Acids Res 2006; 34:4622 - 9; http://dx.doi.org/10.1093/nar/gkl458; PMID: 16963499
- Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 2003; 17:3011 - 6; http://dx.doi.org/10.1101/gad.1158803; PMID: 14681208
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281 - 97; http://dx.doi.org/10.1016/S0092-8674(04)00045-5; PMID: 14744438
- Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. The role of PACT in the RNA silencing pathway. EMBO J 2006; 25:522 - 32; http://dx.doi.org/10.1038/sj.emboj.7600942; PMID: 16424907
- Saito K, Ishizuka A, Siomi H, Siomi MC. Processing of pre-microRNAs by the Dicer-1-Loquacious complex in Drosophila cells. PLoS Biol 2005; 3:e235; http://dx.doi.org/10.1371/journal.pbio.0030235; PMID: 15918769
- Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003; 115:209 - 16; http://dx.doi.org/10.1016/S0092-8674(03)00801-8; PMID: 14567918
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003; 115:199 - 208; http://dx.doi.org/10.1016/S0092-8674(03)00759-1; PMID: 14567917
- Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc Natl Acad Sci U S A 2007; 104:9667 - 72; http://dx.doi.org/10.1073/pnas.0703820104; PMID: 17535905
- Vasilatou D, Papageorgiou S, Pappa V, Papageorgiou E, Dervenoulas J. The role of microRNAs in normal and malignant hematopoiesis. Eur J Haematol 2010; 84:1 - 16; http://dx.doi.org/10.1111/j.1600-0609.2009.01348.x; PMID: 19744129
- Erdogan B, Facey C, Qualtieri J, Tedesco J, Rinker E, Isett RB, et al. Diagnostic microRNAs in myelodysplastic syndrome. Exp Hematol 2011; 39:915 - 26, e2; http://dx.doi.org/10.1016/j.exphem.2011.06.002; PMID: 21703983
- Votavova H, Grmanova M, Dostalova Merkerova M, Belickova M, Vasikova A, Neuwirtova R, et al. Differential expression of microRNAs in CD34+ cells of 5q- syndrome. J Hematol Oncol 2011; 4:1; http://dx.doi.org/10.1186/1756-8722-4-1; PMID: 21211043
- Hussein K, Theophile K, Büsche G, Schlegelberger B, Göhring G, Kreipe H, et al. Significant inverse correlation of microRNA-150/MYB and microRNA-222/p27 in myelodysplastic syndrome. Leuk Res 2010; 34:328 - 34; http://dx.doi.org/10.1016/j.leukres.2009.06.014; PMID: 19615744
- Dostalova Merkerova M, Krejcik Z, Votavova H, Belickova M, Vasikova A, Cermak J. Distinctive microRNA expression profiles in CD34+ bone marrow cells from patients with myelodysplastic syndrome. Eur J Hum Genet 2011; 19:313 - 9; http://dx.doi.org/10.1038/ejhg.2010.209; PMID: 21150891
- Pons A, Nomdedeu B, Navarro A, Gaya A, Gel B, Diaz T, et al. Hematopoiesis-related microRNA expression in myelodysplastic syndromes. Leuk Lymphoma 2009; 50:1854 - 9; http://dx.doi.org/10.3109/10428190903147645; PMID: 19883312
- Bhagat TD, Zhou L, Sokol L, Kessel R, Caceres G, Gundabolu K, et al. miR-21 mediates hematopoietic suppression in MDS by activating TGF-β signaling. Blood 2013; 121:2875 - 81; http://dx.doi.org/10.1182/blood-2011-12-397067; PMID: 23390194
- Rhyasen GW, Starczynowski DT. Deregulation of microRNAs in myelodysplastic syndrome. Leukemia 2012; 26:13 - 22; PMID: 21852786
- Hussein K, Theophile K, Büsche G, Schlegelberger B, Göhring G, Kreipe H, et al. Aberrant microRNA expression pattern in myelodysplastic bone marrow cells. Leuk Res 2010; 34:1169 - 74; http://dx.doi.org/10.1016/j.leukres.2010.04.012; PMID: 20627384
- Vasilatou D, Papageorgiou SG, Kontsioti F, Kontos CK, Tsiotra P, Mpakou V, et al. Expression analysis of mir-17-5p, mir-20a and let-7a microRNAs and their target proteins in CD34+ bone marrow cells of patients with myelodysplastic syndromes. Leuk Res 2013; 37:251 - 8; PMID: 23246221
- Sokol L, Caceres G, Volinia S, Alder H, Nuovo GJ, Liu CG, et al. Identification of a risk dependent microRNA expression signature in myelodysplastic syndromes. Br J Haematol 2011; 153:24 - 32; http://dx.doi.org/10.1111/j.1365-2141.2011.08581.x; PMID: 21332710
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A 2007; 104:15805 - 10; http://dx.doi.org/10.1073/pnas.0707628104; PMID: 17890317
- Garzon R, Heaphy CE, Havelange V, Fabbri M, Volinia S, Tsao T, et al. MicroRNA 29b functions in acute myeloid leukemia. Blood 2009; 114:5331 - 41; http://dx.doi.org/10.1182/blood-2009-03-211938; PMID: 19850741
- Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005; 123:819 - 31; http://dx.doi.org/10.1016/j.cell.2005.09.023; PMID: 16325577
- Zardo G, Ciolfi A, Vian L, Starnes LM, Billi M, Racanicchi S, et al. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood 2012; 119:4034 - 46; http://dx.doi.org/10.1182/blood-2011-08-371344; PMID: 22327224
- Fazi F, Racanicchi S, Zardo G, Starnes LM, Mancini M, Travaglini L, et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 2007; 12:457 - 66; http://dx.doi.org/10.1016/j.ccr.2007.09.020; PMID: 17996649
- Santamaría C, Muntión S, Rosón B, Blanco B, López-Villar O, Carrancio S, et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica 2012; 97:1218 - 24; http://dx.doi.org/10.3324/haematol.2011.054437; PMID: 22371183
- Castoro RJ, Saraf AJ, Watanabe Y, Chung W, Adhab SE, Jelinek J, et al. Microrna 124 and Its Role in Response to Epigenetic Therapy in Patients with Acute Myelogenous Leukemia and Myelodysplastic Syndrome. blood(ASH Annual Meeting Abstracts)2008; 112.
- Xia Q, Hu J, Meng YS. [Abnormal expression of microRNA-124 in patients with leukemia or myelodysplastic syndrome and its significance]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2012; 20:358 - 61; PMID: 22541098
- Dickstein J, Senyuk V, Premanand K, Laricchia-Robbio L, Xu P, Cattaneo F, et al. Methylation and silencing of miRNA-124 by EVI1 and self-renewal exhaustion of hematopoietic stem cells in murine myelodysplastic syndrome. Proc Natl Acad Sci U S A 2010; 107:9783 - 8; http://dx.doi.org/10.1073/pnas.1004297107; PMID: 20448201
- Li X, Xu F, Chang C, Byon J, Papayannopoulou T, Deeg HJ, et al. Transcriptional regulation of miR-10a/b by TWIST-1 in myelodysplastic syndromes. Haematologica 2013; 98:414 - 9; PMID: 22983574
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al, International Vidaza High-Risk MDS Survival Study Group. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009; 10:223 - 32; http://dx.doi.org/10.1016/S1470-2045(09)70003-8; PMID: 19230772
- Tuma RS. Epigenetic therapies move into new territory, but how exactly do they work?. J Natl Cancer Inst 2009; 101:1300 - 1; http://dx.doi.org/10.1093/jnci/djp342; PMID: 19755677
- Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A 2004; 101:1241 - 6; http://dx.doi.org/10.1073/pnas.0307708100; PMID: 14734806
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A 2000; 97:10014 - 9; http://dx.doi.org/10.1073/pnas.180316197; PMID: 10954755
- Cashen A, Juckett M, Jumonville A, Litzow M, Flynn PJ, Eckardt J, et al. Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Ann Hematol 2012; 91:33 - 8; http://dx.doi.org/10.1007/s00277-011-1240-1; PMID: 21538061
- Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001; 20:6969 - 78; http://dx.doi.org/10.1093/emboj/20.24.6969; PMID: 11742974
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 2001; 276:36734 - 41; http://dx.doi.org/10.1074/jbc.M101287200; PMID: 11473107