Abstract
DNA methylation can regulate gene expression and has been shown to modulate cancer cell biology and chemotherapy resistance. Therapeutic radiation results in a biological response to counter the subsequent DNA damage and genomic stress in order to avoid cell death. In this study, we analyzed DNA methylation changes at >450,000 loci to determine a potential epigenetic response to ionizing radiation in MDA-MB-231 cells. Cells were irradiated at 2 and 6 Gy and analyzed at 7 time points from 1–72 h. Significantly differentially methylated genes were enriched in gene ontology categories relating to cell cycle, DNA repair, and apoptosis pathways. The degree of differential methylation of these pathways varied with radiation dose and time post-irradiation in a manner consistent with classical biological responses to radiation. A cell cycle arrest was observed 24 h post-irradiation and DNA damage, as measured by γH2AX, resolved at 24 h. In addition, cells showed low levels of apoptosis 2–48 h post-6 Gy and cellular senescence became significant at 72 h post-irradiation. These DNA methylation changes suggest an epigenetic role in the cellular response to radiation.
Introduction
Radiation therapy of cancer elicits a complex and multi-pathway cellular response that results in life or death of the cell. Radiation-induced DNA damage is repaired by non-homologous end joining (NHEJ), homologous recombination (HR) and, to a lesser extent, nucleotide excision repair (NER) pathways. A checkpoint is activated that leads to an arrest of the cell cycle in G2 phase. Following arrest, cells can permanently lose the ability to proliferate or re-enter the cell cycle. The latter activity may result in mitotic catastrophe and cell death or apoptosis. These events are regulated through signaling pathways that link DNA damage, cell cycle, and apoptosis, among other pathways. While a great deal is known about classical responses to radiation, recent biological discoveries have uncovered additional mechanisms cells use to regulate processes. New technologies that have recently been developed now permit us to investigate this biology further.
Epigenetics is becoming more widely studied in cancer biology as a mechanism regulating cancer cell biology. Epigenetics deals with regulation of gene expression primarily through DNA methylation, miRNA, or histone modifications such as acetylation, methylation, phosphorylation, or ubiquitination. DNA methylation is the addition of methyl groups to the cytosine in CpG dinucleotides by DNA methyltransferases. Hypermethylation of gene promoters is usually associated with decreased gene expression while hypomethylation is thought to render a gene permissive for increased expression.
There have been limited studies of the effect of ionizing radiation on site- or gene-specific DNA methylation. Kim et al.Citation1 performed DNA methylation profiling of 27,000 loci in 15,000 genes in two lung cancer cell lines, one of which was radiation resistant and the other radiation sensitive. They found 1,091 differentially methylated genes. This study was not designed to characterize a potential epigenetic response to radiation treatment, as the current study does. DNA methylation of ATM and p16INK4a has been shown to be altered by radiation treatment.Citation2 This resulted in increased transcriptional activity and may alter cellular resistance to radiation. Cisplatin resistance has been shown to have an epigenetic DNA methylation component in ovarian cancer.Citation3 Other evidence suggesting DNA methylation plays a role in radiation resistance have been studies showing treatment with 5-azacitidine to reduce global methylation has a radiosensitizing effect.Citation4,Citation5
DNA methylation profiling technology with the Illumina HumanMethylation450 array allows us to query >450,000 loci within the genome and to cover 99% of RefSeq genes.Citation6 This represents greater than one order of magnitude of genome coverage over that of previous technology and allows a more comprehensive examination of methylation changes occurring under a given set of conditions. In the studies described here, we utilized this technology to test the hypothesis that exposure to ionizing radiation induces an epigenetic DNA methylation response in pathways associated with cell cycle, DNA repair, and apoptosis. We found this to occur with kinetics consistent with that of biological effects to radiation. A differential DNA methylation response was also found between low and high radiation doses.
Results
Radiation induces global changes in DNA methylation
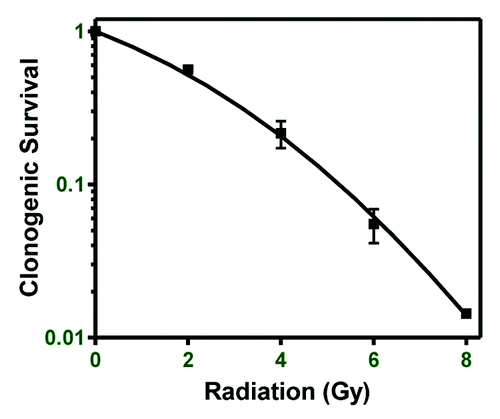
MDA-MB-231 breast cancer cells were chosen as a model for determining DNA methylation changes following ionizing radiation because they display moderate radiation resistance compared with the commonly used breast cancer cell lines MCF-7 and SUM-159 (not shown). Clonogenic survival of MDA-MB-231 cells after exposure to 2, 4, 6, and 8 Gy was 56, 21, 5.5, and 1.4%, respectively (). For DNA methylation profiling, cells were irradiated at 0, 2, or 6 Gy and samples collected at 1, 2, 4, 8, 24, 48, and 72 h post-irradiation. DNA was subjected to whole-genome DNA methylation profiling using an Infinium HumanMethylation450 BeadChip. Each irradiated sample was compared against the time matched 0 Gy sample to determine whether a specific locus showed significantly different DNA methylation following radiation (P < 0.05).
Figure 1. Breast cancer cell line radioresistance. Clonogenic survival of the MDA-MB-231 breast cancer cell line was performed to determine relative radioresistance.
Over 450,000 DNA methylation loci were queried. Depending on the time post-irradiation, 2 Gy resulted in 518–1359 hypermethylated loci, while 419–1661 loci were hypomethylated, compared with 0 Gy, while treatment with 6 Gy resulted in 514–2657 hypermethylated loci, and 789–1408 hypomethylated loci, compared with 0 Gy (). There was no consistent pattern of greater numbers of loci hypermethylated or hypomethylated based on radiation dose or time. Unirradiated control cells showed increased methylation over the culture period (Fig. S1).
Figure 2. Global and specific changes in DNA methylation. Global DNA methylation changes were determined in MDA-MB-231 cells following 2 and 6 Gy. (A) The number of loci hypomethylated or hypermethylated 1–72 h post-irradiation with 2 (left) or 6 Gy (right). (B) The number of genes displaying differential DNA methylation 1–72 h post-irradiation and the number of common differentially methylated genes between the 2 and 6 Gy dose at each time point. (C) Differential DNA methylation and gene expression at 1 h post-2 Gy in 3 chosen genes. (D) Differential DNA methylation in the RB1 gene 1–72 h post-2 Gy correlated with gene expression. (E) Measurement of RB1 gene methylation by meDiP-qPCR 1–72 h post-2 Gy. Values are relative to matched 0 Gy controls.
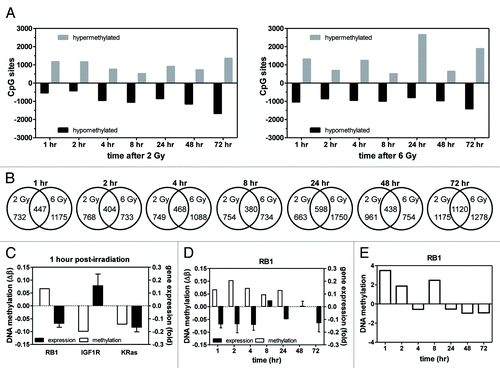
When we analyzed the number of genes showing altered DNA methylation at each time point and radiation dose, there was significant overlap between doses at each time point (P < 0.05; ). There was no consistent increase or decrease in the absolute number of genes showing altered DNA methylation over time, nor was there an apparent dose response. The number of common genes at each time point was consistent except at 72 h post-irradiation, which showed the highest proportion of overlap and the greatest number of altered genes.
Fifteen genes were differentially methylated at all time points post-2 Gy, and 23 genes were differentially methylated at all time points post-6 Gy (). Among all conditions, 7 genes were differentially methylated. In response to the higher radiation dose, IGF1R and KRas, which are involved in the DNA damage response and cell cycle, were differentially methylated. Notably, HDAC4 was differentially methylated under all conditions tested.
Table 1. Common differentially methylated genes
DNA methylation changes can alter gene expression. We first analyzed a panel of 3 genes (RB1, IGF1R, KRas) differentially methylated 1 h after 2 Gy (). Gene expression of RB1 and IGF1R was consistent with changes in DNA methylation; however, KRas expression was not. We then chose to follow up on RB1 more broadly by measuring gene expression over 72 h and comparing it to differential DNA methylation (). We found that an increase in DNA methylation frequently corresponded to a decrease in gene expression across the time period. These data show that changes in DNA methylation induced by radiation can result in changes in gene expression.
We then analyzed RB1 DNA methylation using meDiP-qPCR following 2 Gy as a validation of the profiling data. As shown in and compared with , RB1 methylation changes according to meDiP-qPCR were similar to that of the profiling data.
These data show that treatment with low and high dose radiation results in a large number of site-specific increases and decreases in DNA methylation. These changes occurred as early as 1 h post-irradiation and effects of radiation treatment are observed up to 72 h post-treatment. Gene expression changes followed DNA methylation changes in the RB1 gene.
Changes in DNMT1 expression following irradiation
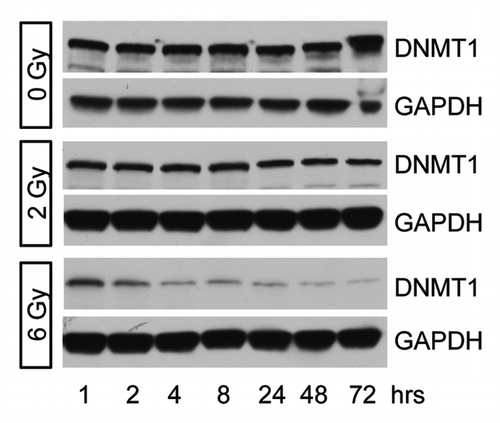
Radiation has been shown to induce global DNA hypomethylation.Citation7 We analyzed DNMT1 protein expression following 0, 2, or 6 Gy as a possible mechanism for differential DNA methylation following irradiation. We found that there was a gradual decrease in DNMT1 expression up to 72 h post-6 Gy (). A much smaller effect was observed following 2 Gy. This suggests that decreased DNMT1 expression could be involved in gene hypomethylation following irradiation.
Figure 3. DNMT1 expression following irradiation. DNMT1 expression was determined in MDA-MB-231 cells 1–72 h after treatment with 0, 2, or 6 Gy. Decreased DNMT1 expression is observed following 6 Gy.
Differential DNA methylation of cell cycle pathway genes at low and high dose radiation
Gene ontology analysis was performed on the genes showing significant changes in DNA methylation at specific loci using DAVID Bioinformatics Resources (see Table S1 for gene list and delta-β values).Citation8,Citation9 Among pathways that showed significant enrichment for genes with altered DNA methylation in response to ionizing radiation were pathways involved in cell cycle (; ). We then calculated the average values for DNA methylation (delta-β) for all loci in all genes in the pathway showing these alterations at each dose/time. If this average was positive, it indicates increased DNA methylation and if negative, it indicates decreased DNA methylation, compared with matched controls. We found the direction of DNA methylation at 2 Gy to be different from that at 6 Gy over the time course. At the lower radiation dose, cell cycle pathways indicated a predilection toward hypermethylation in the first 2 h post-irradiation, but hypomethylation at 4–72 h post-irradiation. At the higher radiation dose, the first 8 h post-irradiation were characterized by a tendency toward hypomethylation, but at 24–72 h, pathways were hypermethylated.
Figure 4. DNA methylation of cell cycle pathways. Gene ontology analysis was used to determine that cell cycle-related pathways are enriched for DNA methylated loci and this was compared with biological response. (A) The average DNA methylation state of genes in the indicated pathways at 1–72 h post-irradiation. Blue boxes indicate hypomethylation; red boxes indicate hypermethylation. The absence of a box indicates this pathway was not significantly enriched at that time/dose. (B) Cells were stained with propidium iodide to determine cell cycle. Representative histograms at 3 time points are shown. Numbers shown represent the percentage in G2/M. (C) Time course of the percentage of cells in G2/M following irradiation (n = 3). The percentage of cells in G2 phase approached control levels in 2 Gy-treated cells by 72 h post-irradiation, but a high percentage of G2 cells remained 72 h post-6 Gy.
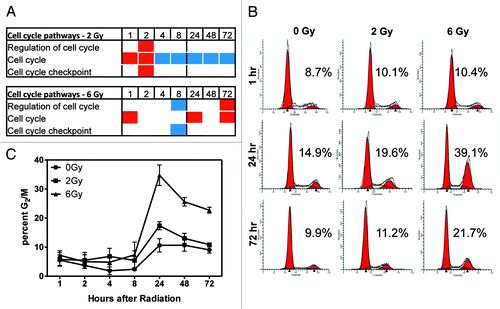
Table 2. DAVID Functional Annotation Chart results
We then investigated the biological changes with respect to cell cycle occurring at the 2 radiation doses. Using PI staining and flow cytometry, we analyzed the proportion of cells in each phase of the cell cycle at 1–72 h post-irradiation (). A notable arrest in the G2 phase of the cell cycle was found at 24 h post-irradiation (). Cells exposed to 6 Gy showed that 35% were arrested in G2/M at 24 h while exposure to 2 Gy resulted in 17% arrested in G2/M (n = 3; ). The arrest with 2 Gy was modestly higher than the control (11%). The small increase in G2 in controls is an artifact of the culturing conditions which results in a slight degree of synchronization.
The switch in cell cycle pathway DNA methylation from predominantly hypomethylation to predominantly hypermethylation between 8 and 24 h after 6 Gy correlates with the observed time course of the G2 arrest. At the 2 Gy dose, however, this switch in methylation occurred earlier, between 2 and 4 h post-radiation and little G2 arrest was found. Cells exposed to 2 Gy show 56% clonogenic survival while cells exposed to 6 Gy show 5.5% clonogenic survival (). This indicates that while 56% of 2 Gy treated cells retained proliferative (clonogenic) capacity, <6% of 6 Gy treated cells are actively proliferating and able to form colonies. These data show a correlation between the kinetics of DNA methylation in cell cycle pathway genes and a prototypical biological response to ionizing radiation such as G2 arrest. There was also a correlation between DNA methylation of cell cycle pathways and radiation doses with differential effects on subsequent cellular proliferation.
DNA methylation changes associated with DNA damage response
A second series of pathways involved in the DNA damage response were found by gene ontology analysis to be enriched for changes in DNA methylation following irradiation at 2 and 6 Gy (; ; Table S1). These pathways include general DNA repair/double-strand break repair as well as specific DNA repair systems such as nucleotide excision repair (NER), recombinational repair (HR), and non-recombinational repair (NHEJ). At both 2 and 6 Gy, hypermethylation predominated at the early time points of 2 and 4 h post-irradiation. At 48 h post-irradiation, these pathways displayed hypomethylation. This was generally consistent at both low and high doses. Cells receiving 6 Gy showed more changes in DNA methylation, particularly in NER, HR, and NHEJ pathways, than 2 Gy.
Figure 5. Gene ontology analysis of DNA repair pathways and the time course of DNA repair. (A) Gene ontology analysis showed enrichment of differentially methylated genes in DNA repair pathways. Red boxes indicate the average delta-β of genes differentially methylated at that condition and represent hypermethylation. Blue boxes indicate hypomethylation. The absence of a box indicates that pathway was not enriched at that condition. (B) Analysis of DNA damage and repair by γH2AX immunofluorescence. γH2AX is in green and blue is Dapi staining of nuclei. Shown are three representative time points. (C) Quantitation of γH2AX foci across the full time range (n = 3). (D) western blot showing γH2AX staining following irradiation (representative of n = 3). DNA damage was greatest 1 h post-irradiation and decreased to basal levels in 2 Gy samples. Some staining remained >24 h post-6 Gy.
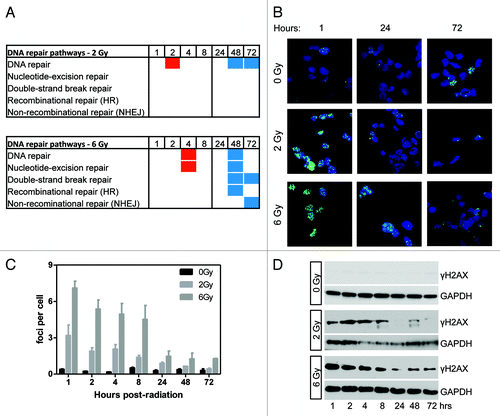
We then determined the kinetics of DNA repair signaling by analyzing γH2AX foci formation and resolution by immunofluorescence (; quantified in ). γH2AX foci number were highest 1 h post-irradiation, with roughly twice as many foci/cell being observed after 6 Gy than after 2 Gy, indicating greater DNA damage (). Foci number approached control levels over time at both doses. Similar results were obtained by γH2AX western blotting (). Staining was highest at 2 h post-2 Gy and 1 h post-6 Gy. At 2 Gy, γH2AX approached control levels at 24–72 h post-irradiation. In 6 Gy irradiated samples, γH2AX staining decreased until 24 h post-irradiation. Low levels were detected up to 72 h post-irradiation. These data show that DNA repair pathways are enriched for differentially-methylated genes following irradiation. Multiple specific DNA repair pathways were affected following a high radiation dose, compared with a low radiation dose.
DNA methylation of apoptosis pathway associated genes
Gene ontology analysis of genes which showed changes in DNA methylation after irradiation were found to be enriched in pathways involved in apoptosis (; ; Table S1). When we calculated the average delta-β for genes in enriched pathways at 2 Gy, we found those pathways to be, on average, hypomethylated and 40–44% of included genes had negative delta-β values. In contrast, 56–83% of apoptosis pathway genes in cells treated with 6 Gy showed positive delta-β values, signifying hypermethylation.
Figure 6. Gene ontology analysis shows enrichment of apoptosis pathways following irradiation. (A) Differentially methylated genes were enriched in apoptosis pathways following 2 and 6 Gy. Red and blue boxes indicate genes in the pathway were hypermethylated or hypomethylated, respectively. (B) Analysis of cellular senescence 8–72 h post-irradiation. Significant senescence was observed at 72 h post-irradiation (P < 0.05, n = 3). (C) Determination of apoptosis by western blotting for cleavage of caspase 3 (representative of n = 3). Low levels of cleaved caspase 3 were seen 4–48 h post-6 Gy.
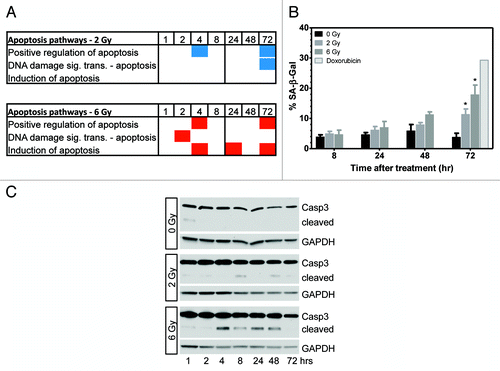
Irradiated cells may undergo apoptosis or survive in a senescence-like state.Citation10 To determine the proportion of cells displaying this senescence phenotype, we used senescence-associated β-galactosidase (SA-β-gal) staining. The senescence phenotype trended higher from 8–72 h post-irradiation and reached significance vs. untreated cultures at 72 h. Cultures treated with 2 and 6 Gy displayed 11.3% and 17.9% SA-β-gal+ cells, respectively, while untreated cultures showed 3.8% SA-β-gal+ cells 72 h post-irradiation (P < 0.05; ). Analysis at 5 d post-irradiation did not show a further in SA-β-gal+ cells (not shown).
Since radiation-induced cell death can occur through apoptosis, we investigated apoptosis in irradiated cells by examining cleavage of caspase 3 by western blotting. Cleaved caspase 3 levels in cells exposed to 2 Gy were low to undetectable (). We found cells treated with 6 Gy showed low levels of cleaved caspase 3 at 4–48 h post-irradiation. No indication of apoptosis was found at 72 h post-irradiation, which is notably when the proportion of senescent cells became significantly higher than untreated cultures (; P < 0.05). Apoptosis can also be quantified as cells showing sub-G1 DNA content after staining with PI and performing flow cytometry as in . Levels of apoptosis detected by quantitation of the proportion of sub-G1 cells was less than 0.5% under all conditions tested. This suggests that the cleaved caspase 3 signal on the western blot represents a small proportion of cells.
These data show that ionizing radiation dose affects DNA methylation of apoptosis pathways. Higher radiation dose results in hypermethylation of apoptosis pathways in conjunction with an increased senescence-like phenotype and modest induction of apoptosis in a small population of cells.
Discussion
Here we show that treatment of cancer cells with ionizing radiation results in extensive changes in the DNA methylation state of genes. This result was observed at a dose level (2 Gy) commonly used clinically as one fraction of a multi-fraction regimen, as well as at a high dose (6 Gy) that is highly toxic to cells. Gene ontology analysis showed that genes with altered DNA methylation were enriched in pathways such as cell cycle, DNA repair, and apoptosis—pathways classically associated with the radiation response. The pattern of hypermethylation and hypomethylation was associated with the characterized biological responses and suggests that this DNA methylation response to radiation was non-random.
Previous studies of DNA methylation changes occurring after ionizing radiation treatment have investigated global differences or fewer loci than we have done here. Decreased overall genomic DNA methylation was observed at 24–72 h post-irradiation in cell lines and this was reported to be due to decreased levels of DNA methyltransferase.Citation7 A decrease in global DNA methylation coupled with decreased histone H4K20 trimethylation was observed in the thymus of irradiated mice.Citation11 This was also associated with decreased expression of DNA methyltransferase. Chaudhry et al.Citation12 show global hypomethylated DNA after irradiation which was time-dependent and displayed different profiles in radioresistant and radiosensitive cells. In our study of loci-specific changes in DNA methylation in response to radiation, we find global hypomethylation occurring at some time points, but not all. This may be partially explained by noting that additional changes in DNA methylation were measured in our experiment which, while they did not reach significance at specific loci, together may show a trend if global methylation was measured non-specifically. Also, in spite of our array experiment providing a great deal of genomic coverage, not all potential DNA methylation sites are queried with this technology. Global hypomethylation may require DNA replication to be fully realized and we limited our studies to 72 h, which may be insufficient time for this to occur under radiation-treated conditions. We did observe decreased DNMT1 expression over 72 h post-6 Gy, which may be a mechanism that results in global hypomethylation at later time points when DNA replication resumes.
MCF7 cells that regrew 2–14 d after fractionated radiation have been investigated using a 255k DNA methylation microarray.Citation13 A small number of differentially-methylated genes were found based on stringent criteria. This study differs from ours in that much later effects were examined—up to 14 d post-irradiation in order to investigate changes during regrowth of the cells, gene ontology analysis could not be performed, and fractionated radiation was used. We sought to characterize DNA methylation changes occurring as an acute consequence of radiation by analyzing cells 1–72 h after a single radiation dose, which is within the expected acute biological effects of radiation. Our data thus complements and extends that of Kuhmann et al.Citation13
DNA methylation profiling to determine responses to ionizing radiation has not been well described. One study performed DNA methylation profiling comparing a radioresistant lung cancer cell line (H1299) to that of a radiosensitive lung cancer cell line (H460).Citation1 Using Illumina’s BeadChip to query 27,578 CpG sites, they found that 1091 genes were differentially methylated. While this work demonstrates that DNA methylation may be important in radioresistance, the study was not designed to show a DNA methylation response following radiation treatment as we do here. A limitation of the Kim et al.Citation1 study was also that two completely different cell lines were compared such that many DNA methylation differences may not have been related to their radiation sensitivity phenotype. Indeed, H460 cells express wild type p53 while H1299 cells do not.Citation14 Other gene mutation differences may have had a greater effect on the radioresistant phenotype. In our study, comparisons were made between doses and time points in the same cell line and each condition was normalized to a matched control to minimize variations based on factors such as cell density, media conditions, etc.
DNA methylation profiling studies following chemotherapy treatment are few. In one study, methyl-capture sequencing was used to compare differences between 2 isogenic ovarian cancer cell lines, one of which has been rendered more resistant to cisplatin.Citation3 They found 2,440 differentially methylated regions that mapped to several cell signaling pathways, among others.
Characterization of epigenetic changes that result from treatment with radiation or chemotherapy may lead to new therapies that alter the DNA methylation response or reveal new targets for traditional therapy. Epigenetic therapies targeting DNA methylation are under study and have been shown to sensitize cells to radiation.Citation4,Citation15-Citation17 Treatment of cells with the DNA methyltransferase inhibitors psammaplin A or zebularine partially abrogated the G2 phase arrest, increased apoptosis, and reduced the ability to repair DNA double strand breaks in cells exposed to 6 Gy radiation.Citation4
The DNA damage response is complex and involves changes to the cell cycle, activation of DNA repair pathways, and induction of apoptosis or, alternatively, senescence. Although it has been previously shown that irradiation affects global changes in DNA methylation, it was not known whether these changes were enriched in pathways directly involved in the radiation response. We show here enrichment of differentially methylated genes in classical radioresponse pathways; however, other pathways also affected were related to signal transduction, transcription, invasion, among others. The pathways described here are thought to play a major role in radiation resistance.
Hypermethylation of cell cycle pathway genes starting at 24 h post-irradiation in 6 Gy irradiated cells is consistent with the fact that 94% of the cells have lost clonogenic capacity and may be on a trajectory toward senescence or death. Meanwhile, most 2 Gy treated cells maintain their clonogenic potential and this is reflected in hypomethylation of pathways associated with cell cycle. The difference in gene ontology results and the trend in differential methylation of DNA repair pathway genes suggest that the higher radiation dose activated a more robust response than cells treated with the lower radiation dose. However, this does not translate into increased clonogenic survival. We show that the gene RB1 is hypermethylated, together with decrease gene expression, following 2 Gy, which would be expected to be associated with continued proliferation. This is in agreement with the high clonogenic survival at this dose.
We found low levels of cleaved caspase 3 indicating apoptosis after 6 Gy and a significant increase in senescent cells (). Apoptotic pathways were generally hypermethylated in 6 Gy treated cells and it has been shown that senescent cells are more resistant to radiation.Citation18 A potential mechanism of this affect may be downregulation of apoptotic pathways through DNA hypermethylation.
It is our hypothesis that ionizing radiation/DNA damage elicits a cellular response which includes changes in DNA methylation, specifically the pathways most associated with the DNA damage response. While it is possible that the increased DNA repair due to greater DNA damage at 6 Gy contributes to hypomethylation of the genome, our data does not support this conclusion. Specifically, does not show a trend in hypomethylation of CpG sites with increasing radiation dose or incubation time. Likewise, in , the apoptosis pathways analyzed were hypermethylated at 6 Gy but hypomethylated at 2 Gy, an observation that would be expected to be reversed if DNA repair processes led to general global hypomethylation. DNMT1 has been shown to be recruited to sites of DNA damage, suggesting a link between DNA methylation and DNA repair.Citation19 However, the changes in DNA methylation we observe do not appear to be related to physical DNA damage or the repair process itself, for reasons outlined above.
In summary, we have shown that treatment of breast cancer cells with ionizing radiation results in DNA methylation changes which correlate with biological responses to radiation. DNA methylation changes were enriched in pathways involved in cell cycle, DNA repair, and apoptosis. Cell cycle pathway genes were generally hypomethylated at 2 Gy at 4–72 h but hypermethylated at 24–72 h post-irradiation. DNA repair pathways showed a high degree of hypomethylation 48 h after 6 Gy, while 2 Gy resulted in a less robust response. Apoptosis pathways were hypomethylated, on average, after 2 Gy, but hypermethylated after 6 Gy. These studies show that a DNA methylation response to radiation runs in parallel to classical biological responses to radiation and suggests a possible link between radiation resistance and epigenetics in the form of DNA methylation.
Methods and Materials
Cell culture and irradiation
MDA-MB-231 breast cancer cells were obtained from American Type Culture Collection (ATCC) and grown in DMEM (Thermo Scientific Hyclone) with 10% FBS (Omega Scientific), 2 mM glutamine (Thermo Scientific Hyclone) and without antibiotics. Cells were incubated at 37 °C in 5% CO2.
The day before irradiation, 3.2 × 106 cells were plated in T-75 flasks. Irradiation with X-rays (2 and 6 Gy) was performed with a Pantak 320 kV orthovoltage unit at 0.86 Gy/min. and 10 mA. At 1, 2, 4, 8, 24, 48, and 72 h post-irradiation, cells were trypsinized, washed in cold complete culture media, and cell pellets stored at −20 °C. Untreated time-matched samples were processed in parallel with irradiated samples.
Methylation profiling
DNA methylation profiling was performed by the Applied Genomics Technology Center at Wayne State University. DNA from cell pellets was isolated on a Qiagen EZ1 Advanced (Qiagen) and subjected to bisulfate conversion using a Zymo EZ DNA Methylation kit (Zymo Research). The bisulfite conversion efficiency was determined by sample-dependent controls on the chip and is displayed in the quality control panel in the software. All samples passed quality control measurements. Samples were run on an Infinium HumanMethylation450 BeadChip (Illumina) and scanned on an Illumina iScan instrument according to the manufacturer’s instructions.
The Illumina Infinium 450K methylation files obtained from the Illumina iScan scanner were uploaded to GenomeStudio (V2011.1) using the Methylation module (v1.9.0). Data were normalized using the Controls Normalization method. Differentially methylated probes were identified using the Illumina Custom Error Model with Benjamini-Hochberg False Discovery Rate correction. A P-value is associated with every probe (Detection P-value) and probes were discarded if this Detection pvalue was more than 0.05. Furthermore, probes with a P-value more than 0.05 for the differential call were filtered out.
Gene ontology analysis
Gene ontology analysis was performed using DAVID Bioinformatics Resources 6.7.Citation8,Citation9 Differentially methylated genes were uploaded and analyzed using the Functional Annotation Chart tool and GOTERM_BP_FAT category. GOTERM IDs are as follows: regulation of cell cycle (GO:0051726), cell cycle (GO:0007049), cell cycle checkpoint (GO:0000075), DNA repair (GO:0006281), double-strand break repair (GO:0006302), recombinational repair (GO:0000725), nucleotide-excision repair (GO:0006289), non-recombinational repair (GO:0000726), positive regulation of apoptosis (GO:0043065), induction of apoptosis (GO:0006917), and DNA damage response, signal transduction resulting in induction of apoptosis (GO:0008630). A p-value cutoff of 0.05 was used to call a pathway significantly enriched. The delta-β for all probes in the genes enriched in each pathway was averaged, and if the results were positive or negative, the pathway was noted as being hypermethylated or hypomethylated, respectively. DAVID output and delta-β values of differentially-methylated genes in enriched pathways are contained in Table S1.
Western blotting
Cell extracts were prepared with sodium dodecyl sulfate (SDS) lysis buffer (0.004% NaF, 0.045% Na2PO7, 2%, SDS, 1% Phosphatase Inhibitor Cocktails 2 and 3 [Sigma-Aldrich]), and 1% Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Pierce BCA Protein Assay Kit (Pierce Protein Research Products) was used to determine protein concentration. Following SDS-PAGE and Western transfer, membranes were blocked with 5% non-fat milk in 1× TBST (25 mM Tris, 150 mM NaCl, 2 mM KCl, pH 7.4; 0.1% Tween-20). Primary antibodies used were anti-phospho-histone H2AX (Ser139), Clone JBW301 (Millipore), Caspase-3 (3G2), DNMT1, and GAPDH (14C10) (Cell Signaling Technology). The blots were incubated overnight at 4 °C, washed, incubated with peroxidase-conjugated secondary antibodies (Cell Signaling Technology), and signal detected with SuperSignal West Pico® (Pierce Protein Research Products). Western blot images were scanned and brightness/contrast of the whole image adjusted for clarity. At least 3 independent experiments were performed.
Senescence assay
Cells treated or untreated with X-ray radiation or doxorubicin (50 μg/ml; positive control) were grown on coverslips placed in 6-well plates. Samples were processed 8–72 h post-irradiation using a Senescence β-Galactosidase Staining Kit (Cell Signaling Technology) according to the manufacturer’s instructions. Coverslips were then mounted using Fluorsave Reagent (Calbiochem). Percentages were calculated based on β-galactosidase positive cells and negative cells counted in 3 different fields of view using light microscopy.
Immunofluorescence
The levels of γH2AX foci were monitored before and after ionizing radiation. Cells (1.5 × 105) were seeded on coverslips in 12-well plates in triplicate. The following day, cells were treated with 0, 2, or 6 Gy. Cells were then formalin fixed and washed with cold PBS containing 0.5% BSA and 0.2% Tween-20. Cells were blocked in PBS, 0.5% BSA, 2.5% donkey serum, and 0.05% Triton X-100 and incubated with anti-phospho-H2AX antibody. The coverslips were washed and then incubated in Alexa Fluor® 488 labeled donkey anti-mouse secondary antibodies (Invitrogen). Coverslips were mounted using Prolong Gold antifade Reagent with Dapi (Invitrogen). Slides were viewed with a Leica TCS SP5 laser scanning confocal microscope. The brightness and contrast of whole images was adjusted for clarity.
Cell cycle
Cell cycle analysis was done by propidium iodide (PI) staining as described.Citation20 Briefly, cells were collected, fixed with 70% ethanol, treated with RNase A, and stained with PI before being run on a BD LSRII flow cytometer. Quantification of cells in different phases of the cell cycle was conducted using ModFit software (Variety Software House).
Clonogenic assay
To determine clonogenic survival, 5 × 105 cells were seeded in T-25 flasks and the next day treated with radiation (0, 2, 4, 6, or 8 Gy) and then plated for colonies. Resulting colonies were fixed and stained with crystal violet. Colonies with ≥50 cells were scored. Data was fit to a linear quadratic model.
Methylated DNA immunoprecipitation – quantitative PCR (meDiP-qPCR)
Methylated DNA was immunoprecipitated using an EpiQuik Methylated DNA Immunoprecipitation kit (Epigentek) according to the manufacturer’s instructions. Quantitative real time PCR was performed using RB1 gene-specific primers (forward: 5′- TCCGCTCATCTTCACCCAAA-3′, reverse: 5′- ATGGCTTGATGAGCCACACA-3′) on an Applied Biosystems 7500 Fast Real-time PCR System. PCR was done in duplicate using SYBR Premix Ex TaqII (TaKaRa). Relative quantities were determined using the relative standard curve method. Immunoprecipitated DNA was normalized to the quantity of input DNA. Values for irradiated samples were divided by values for time-matched controls.
qRT-PCR
Cells were cultured under identical conditions to that of the DNA methylation profiling experiment and irradiated with 0 or 2 Gy X-rays. Cells were harvested and RNA extracted using a Qiagen (Valencia, CA) RNeasy kit. Real time RT-PCR was performed using Taqman Gene Expression Assays (Life Technologies) for KRas (Hs00364284_g1), IGF1R (Hs00609566_m1), and RB1 (Hs01078066_m1) on an Applied Biosystems 7500 Fast Real-time PCR System. The comparative CT method was used for quantitation using MRPL-19 (Hs00608519) as a control. MRPL-19 did not show any differential DNA methylation following irradiation.
| Abbreviations: | ||
| NHEJ | = | non-homologous end joining |
| HR | = | homologous recombination |
| NER | = | nucleotide excision repair |
| PI | = | propidium iodide |
| SA-β-gal | = | senescence-associated β-galactosidase |
Additional material
Download Zip (357.7 KB)Acknowledgments
We would like to thank Adele Kruger (WSU Applied Genomics Technology Center) for bioinformatics assistance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplementary materials may be found here:
http://www.landesbioscience.com/journals/epigenetics/article/25498
References
- Kim EH, Park AK, Dong SM, Ahn JH, Park WY. Global analysis of CpG methylation reveals epigenetic control of the radiosensitivity in lung cancer cell lines. Oncogene 2010; 29:4725 - 31; http://dx.doi.org/10.1038/onc.2010.223; PMID: 20531302
- Kumar A, Rai PS, Upadhya R, Vishwanatha, Prasada KS, Rao BS, et al. γ-radiation induces cellular sensitivity and aberrant methylation in human tumor cell lines. Int J Radiat Biol 2011; 87:1086 - 96; http://dx.doi.org/10.3109/09553002.2011.605417; PMID: 21815748
- Yu W, Jin C, Lou X, Han X, Li L, He Y, et al. Global analysis of DNA methylation by Methyl-Capture sequencing reveals epigenetic control of cisplatin resistance in ovarian cancer cell. PLoS One 2011; 6:e29450; http://dx.doi.org/10.1371/journal.pone.0029450; PMID: 22216282
- Kim HJ, Kim JH, Chie EK, Young PD, Kim IA, Kim IH. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat Oncol 2012; 7:39; http://dx.doi.org/10.1186/1748-717X-7-39; PMID: 22429326
- Hofstetter B, Niemierko A, Forrer C, Benhattar J, Albertini V, Pruschy M, et al. Impact of genomic methylation on radiation sensitivity of colorectal carcinoma. Int J Radiat Oncol Biol Phys 2010; 76:1512 - 9; http://dx.doi.org/10.1016/j.ijrobp.2009.10.037; PMID: 20338477
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288 - 95; http://dx.doi.org/10.1016/j.ygeno.2011.07.007; PMID: 21839163
- Kalinich JF, Catravas GN, Snyder SL. The effect of gamma radiation on DNA methylation. Radiat Res 1989; 117:185 - 97; http://dx.doi.org/10.2307/3577319; PMID: 2922465
- Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44 - 57; http://dx.doi.org/10.1038/nprot.2008.211; PMID: 19131956
- Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1 - 13; http://dx.doi.org/10.1093/nar/gkn923; PMID: 19033363
- Sabin RJ, Anderson RM. Cellular Senescence - its role in cancer and the response to ionizing radiation. Genome Integr 2011; 2:7; http://dx.doi.org/10.1186/2041-9414-2-7; PMID: 21834983
- Pogribny I, Koturbash I, Tryndyak V, Hudson D, Stevenson SM, Sedelnikova O, et al. Fractionated low-dose radiation exposure leads to accumulation of DNA damage and profound alterations in DNA and histone methylation in the murine thymus. Mol Cancer Res 2005; 3:553 - 61; http://dx.doi.org/10.1158/1541-7786.MCR-05-0074; PMID: 16254189
- Chaudhry MA, Omaruddin RA. Differential DNA methylation alterations in radiation-sensitive and -resistant cells. DNA Cell Biol 2012; 31:908 - 16; http://dx.doi.org/10.1089/dna.2011.1509; PMID: 22185261
- Kuhmann C, Weichenhan D, Rehli M, Plass C, Schmezer P, Popanda O. DNA methylation changes in cells regrowing after fractioned ionizing radiation. Radiother Oncol 2011; 101:116 - 21; http://dx.doi.org/10.1016/j.radonc.2011.05.048; PMID: 21704414
- Zou Z, Gao C, Nagaich AK, Connell T, Saito S, Moul JW, et al. p53 regulates the expression of the tumor suppressor gene maspin. J Biol Chem 2000; 275:6051 - 4; http://dx.doi.org/10.1074/jbc.275.9.6051; PMID: 10692390
- Luzhna L, Kovalchuk O. Modulation of DNA methylation levels sensitizes doxorubicin-resistant breast adenocarcinoma cells to radiation-induced apoptosis. Biochem Biophys Res Commun 2010; 392:113 - 7; http://dx.doi.org/10.1016/j.bbrc.2009.12.093; PMID: 20034463
- Meador JA, Su Y, Ravanat JL, Balajee AS. DNA-dependent protein kinase (DNA-PK)-deficient human glioblastoma cells are preferentially sensitized by Zebularine. Carcinogenesis 2010; 31:184 - 91; http://dx.doi.org/10.1093/carcin/bgp284; PMID: 19933707
- Brieger J, Mann SA, Pongsapich W, Koutsimpelas D, Fruth K, Mann WJ. Pharmacological genome demethylation increases radiosensitivity of head and neck squamous carcinoma cells. Int J Mol Med 2012; 29:505 - 9; PMID: 22109647
- Chaturvedi V, Qin JZ, Denning MF, Choubey D, Diaz MO, Nickoloff BJ. Apoptosis in proliferating, senescent, and immortalized keratinocytes. J Biol Chem 1999; 274:23358 - 67; http://dx.doi.org/10.1074/jbc.274.33.23358; PMID: 10438513
- Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc Natl Acad Sci U S A 2005; 102:8905 - 9; http://dx.doi.org/10.1073/pnas.0501034102; PMID: 15956212
- Zielske SP, Gerson SL. Cytokines, including stem cell factor alone, enhance lentiviral transduction in nondividing human LTCIC and NOD/SCID repopulating cells. Mol Ther 2003; 7:325 - 33; http://dx.doi.org/10.1016/S1525-0016(03)00005-4; PMID: 12668128