Abstract
The E2f6 transcriptional repressor is an E2F-family member essential for the silencing of a group of meiosis-specific genes in somatic tissues. Although E2f6 has been shown to associate with both polycomb repressive complexes (PRC) and the methyltransferase Dnmt3b, the cross-talk between these repressive machineries during E2f6-mediated gene silencing has not been clearly demonstrated yet. In particular, it remains largely undetermined when and how E2f6 establishes repression of meiotic genes during embryonic development. We demonstrate here that the inactivation of a group of E2f6 targeted genes, including Stag3 and Smc1β, first occurs at the transition from mouse embryonic stem cells (ESCs) to epiblast stem cells (EpiSCs), which represent pre- and post-implantation stages, respectively. This process was accompanied by de novo methylation of their promoters. Of interest, despite a clear difference in DNA methylation status, E2f6 was similarly bound to the proximal promoter regions both in ESCs and EpiSCs. Neither E2f6 nor Dnmt3b overexpression in ESCs decreased meiotic gene expression or increased DNA methylation, indicating that additional factors are required for E2f6-mediated repression during the transition. When the SET domain of Ezh2, a core subunit of the PRC2 complex, was deleted, however, repression of Stag3 and Smc1β during embryoid body differentiation was largely impaired, indicating that the event required the enzymatic activity of Ezh2. In addition, repression of Stag3 and Smc1β occurred in the absence of Dnmt3b. The data presented here suggest a primary role of PRC2 in E2f6-mediated gene silencing of the meiotic genes.
Introduction
Cells of developing mammalian embryos gradually become restricted in their potency as they transition from a pluripotent to a terminally differentiated state. This occurs during cell lineage commitment following cell fate choices, which can be prompted by a variety of cues.Citation1 The molecular mechanisms underlying cell fate determination are diverse, but they ultimately lead to the establishment of cell type-specific gene expression programs. This ensures that gene products essential for a particular cell function will be expressed in the appropriate lineages only and silenced in all others. The same is also true for germline-specific genes, which must be permanently repressed in somatic tissues. Failure to faithfully carry out this process and the aberrant activation of these genes can have severe consequences for the cell.Citation2
The E2F-family of transcription factors are very well studied and recognized for their role in cell cycle regulation, a function they exert through binding to the retinoblastoma (Rb) tumor suppressor and the related pocket proteins. E2f6 was discovered over a decade ago as an addition to the E2F family, but investigators quickly realized that it differs from other E2Fs.Citation3,Citation4 Although it still forms a heterodimer with DP1/2 capable of binding to DNA, it does so with a preference for the TCCCGC consensus sequence, which deviates from the canonical E2F site.Citation5 Furthermore, E2f6 lacks an Rb-tumor suppressor binding domain and was therefore classified as a pocket protein-independent transcriptional repressor. In addition, E2f6 is ubiquitously expressed throughout all phases of the cell cycle and can regulate some unusual E2F target genes. Interestingly, these are commonly expressed in germ cells. E2f6-null mouse embryonic fibroblasts (MEFs) have no proliferation defect, but aberrantly reactivate a number of genes, including the subunits of the meiotic cohesin complex Stag3 and Smc1β,Citation6 the testis-specific tubulin isoforms Tuba3a and Tuba7Citation5, and the gonadal ADP/ATP translocator located on the inner mitochondrial membrane, Slc25a31 (Ant4).Citation7 Even more intriguing is the fact that E2f6 knockout mice, although grossly normal, exhibit mild homeotic transformations of the axial skeleton, a hallmark phenotype for polycomb group protein (PcG) deficiencies.Citation8,Citation9 Indeed, E2F6 has been shown to interact with RYBP in the context of a BMI1-containing repressive complex,Citation10 and with EPC1 which itself associates with EZH2.Citation11 In addition, an E2F6 complex has been described that contains RING1 and HP1γ, which binds to its target promoters in quiescent (G0) cells.Citation12 More recently, a PRC1-like 4 (PRC1L4) complex was also described containing RING1, RING2, and L3MBTL2, in addition to E2F6.Citation13,Citation14
Adding to the complexity of E2f6-mediated gene regulation is the fact that this transcription factor is suggested to associate with Dnmt3b,Citation15 one of the de novo DNA methyltransferases. In this study, Velasco et al. utilize a Dnmt3b hypomorphic mutant that partially retains enzymatic activity, and observe the aberrant transcriptional reactivation of germ cell-specific genes in the soma as a result of hypomethylation of their promoters. Some, but not all, of these genes are also E2f6 targets. Complete disruption of Dnmt3b is embryonic lethal and Dnmt3b-null cells reactivate different targets, underscoring the genes’ differential sensitivities to DNA methylation fluctuations.Citation16 The observed discrepancies between genes reactivated as a result of deletion of E2f6 or Dnmt3b suggests that differential regulatory mechanisms may be involved.
Although many observations have been made thus far regarding the role of E2f6 in repression of some germline genes, we still lack a detailed understanding of the mechanisms involved. Up to this point, the precise timing of repression establishment of this group of genes during development has also remained an open question. In addition, a clear demonstration of a murine PcG-containing E2f6 complex that can target germ cell-specific gene promoters in the soma has also been lacking. We propose here that E2f6 might function together with Ezh2, in addition to de novo DNA methylation, to establish stable gene inactivation of Stag3 and Smc1β.
Results
E2f6 disruption results in loss of DNA methylation at Stag3, Smc1β, Tuba3a, and Slc25a31
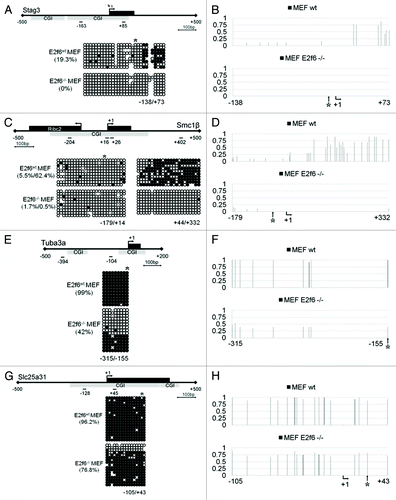
Previous studies demonstrate that E2f6 is required for gene silencing of four meiotic genes (Stag3, Smc1β, Tuba3a, and Slc25a31) in somatic tissues. Indeed, in MEF cells which do not express E2f6 these genes are aberrantly reactivated (see microarray data in ). In order to investigate whether this is accompanied by DNA methylation we performed bisulfite sequencing analysis of their promoter regions comparing wild-type MEFs with MEFs isolated from E2f6 knockout mice. As summarized in , the Stag3, Smc1β, and Slc25a31 genes have promoters with high CpG content and strong CGIs. However, the Tuba3a promoter, although GC-rich, has a lower CpG count, and consequently, a weak CGI. When we examined methylation of CpGs surrounding the E2f6 binding site in wild type MEFs, hypermethylation was found mostly downstream of it in Stag3 and Smc1β (). In contrast, DNA methylation was present at all CpG sites investigated in Tuba3a and Slc25a31. The Stag3 and Smc1β promoters were almost completely devoid of methylation in E2f6-null MEFs. However, deletion of E2f6 only partially affected methylation at the Tuba3a and Slc25a31 promoters (). The overall methylation level of the Tuba3a promoter decreased from 99% in wild-type MEFs to 42% in E2f6−/− MEFs. The change in methylation of the Slc25a31 promoter was even less dramatic, decreasing from 96.2% to 76.8%. This indicates that although E2f6 plays a role in establishing DNA methylation, the levels of its requirement among these genes are different.
Table 1. Data from microarray performed on E2f6 wild-type and E2f6 knockout MEFs
Table 2. CGI class definition of Stag3, Smc1β, Tuba3a, and Slc25a31 according to the more stringent criteria proposed by Takai and JonesCitation17
Figure 1. Deletion of E2f6 disrupts somatic cell CGI methylation at E2f6 dependent germline specific gene promoters. (A, C, E, and G) Bisulfite DNA sequencing analysis for CGI methylation of the Stag3, Smc1β, Tuba3a, and Slc25a31 promoters, respectively, in wild-type and E2f6−/− MEFs. Each gene representation includes the first exon in black and the position of the CGI in gray. Short lines with numbers depict the first nucleotide from the forward and reverse primers used for bisulfite sequencing. Percent methylation for the region amplified by the primers is given in parenthesis. (B, D, F, and H) Quantification of bisulfite sequencing data demonstrating the dynamics of methylation at each individual CpG dinucleotide in Stag3, Smc1β, Tuba3a, and Slc25a31 promoters, respectively. A value of one corresponds to 100% methylation. *Designates the position of E2f6 binding; +1 designates the TSS.
E2f6-target genes are first silenced in EpiSCs, a model for the post-implantation epiblast
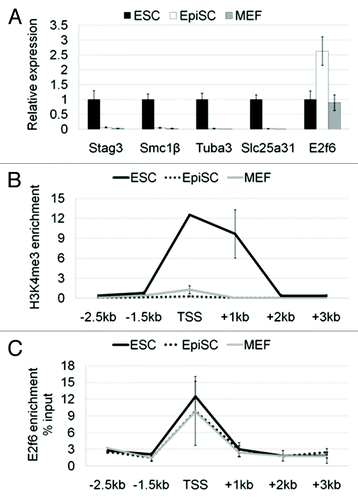
To shed light on E2f6-mediated germ cell-specific gene repression during development we investigated gene expression patterns in two mouse pluripotent stem cell types, ESCs and EpiSCs, and in the terminally differentiated MEFs. We chose to look at ESCs and EpiSCs as representing the 2 different stages in development from which these stem cells are derived, the pre-implantation and post-implantation epiblast, respectively. MEFs represent somatic tissues. To validate the quality and differentiation stages of the cells used in this study, we initially performed a microarray analysis. Markers for the naïve pluripotency state like Zfp42 (Rex1) were highly expressed in ESCs, as was Klf4, which then becomes downregulated in EpiSCs. Pluripotency markers like Pou5f1 (Oct4) and Nanog were highly expressed both in ESCs and EpiSCs, and downregulated in MEFs. Fgf5, a marker for the poised pluripotency state, was upregulated in EpiSCs, similarly to Dnmt3b, which is a known marker for the post-implantation epiblast. As expected, MEFs upregulate Col11a, which is not expressed in ESCs and EpiSCs. Notably, Stag3, Smc1β, and Tuba3a have appreciable expression in ESCs; whereas Slc25a31 is at nearly background levels (see microarray data in ).
Table 3. Data from microarray performed on ESCs, EpiSCs, and MEFs
To confirm the expression patterns of E2f6 and the germline genes we used quantitative RT-PCR. As we expected based on the microarray data, all four genes are expressed albeit to a different extent in naïve ESCs (). However, all four are completely silenced in the primed EpiSCs, coinciding with a 2.5-fold increase of E2f6 mRNA. Using Stag3 as a model for a gene regulated by E2f6 we observed that the Stag3 transcription start site (TSS) in ESCs was enriched for H3K4me3, a histone mark associated with active transcription (). This activation mark was lost in EpiSCs and MEFs. We also found that E2f6 was associated with the Stag3 promoter in all three cell types (). Notably, E2f6 was recruited even in ESCs, while this gene is still being actively transcribed. This suggests that E2f6 occupancy at the Stag3 promoter is not sufficient to silence the gene.
Figure 2. Germline-specific genes regulated by E2f6 are first silenced in primed pluripotent stem cells. (A) RNA expression levels of Stag3, Smc1β, Tuba3a, and Slc25a31 in ESCs, EpiSCs, and MEFs shown by qPCR. The expression of each gene in embryonic stem cells was set to a value of one. (B) ChIP-qPCR analysis for H3K4me3 enrichment spanning 2.5 kb downstream and 3 kb upstream (relative to the TSS) of the Stag3 promoter in ESCs, EpiSCs, and MEFs. Enrichment is shown normalized to the total H3 content at each specific primer pair position. (C) ChIP-qPCR analysis for enrichment of E2f6 at the Stag3 promoter in ESCs, EpiSCs, and MEFs. Enrichment is shown as the percent of Input DNA.
De novo DNA methylation patterns of E2f6-target gene promoters are first set in EpiSCs
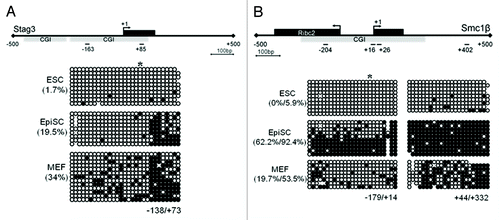
We next demonstrate establishment of DNA methylation during development again using the Stag3 promoter as an example. In agreement with our gene expression data we observed that the Stag3 promoter region (from −138 bp to +73 bp relative to the TSS) is nearly completely unmethylated in ESCs (with 1.7% overall methylation) and increasingly methylated in EpiSCs and MEFs, at 19.5% and 34%, respectively (). This observation was also confirmed for Smc1β, which exhibits 0% methylation at the upstream promoter region (−179 bp/+14 bp) and 5.9% at the downstream exonic region (+44 bp/+332 bp) in ESCs. Methylation of both regions increased in EpiSCs to 62.2% and 92.4%, respectively. In MEFs overall methylation of these regions decreases to 19.7% and 53.5%, respectively, independent of transcriptional silencing (). In addition, we also observe that the Stag3 promoter is unmethylated in E13.5 primordial germ cells (PGCs) and induced pluripotent stem cells (iPSCs) (Fig. S1). Thus, this promoter undergoes DNA demethylation during both in vivo and in vitro reprogramming. Taken together these data indicate that E2f6-mediated DNA methylation and gene repression likely occur during the transition from ground-to-primed state of pluripotency, in a reprogrammable manner.
Figure 3. Differential DNA methylation of the Stag3 and Smc1β promoters in ESCs, EpiSCs, and MEFs. (A) CGI methylation of the Stag3 promoter in ESCs, EpiSCs, and MEFs. (B) CGI methylation of the Smc1β promoter in ESCs, EpiSCs, and MEFs. *Designates the position of E2f6 binding site; +1 designates the TSS.
Overexpression of E2f6 or Dnmt3b in ESCs does not induce premature silencing of E2f6-target genes
Since we observed an increase in E2f6 expression in EpiSCs, the upregulation of this transcription factor protein might underlay silencing of the target genes. To test this hypothesis, we overexpressed E2f6 in ESCs to the level seen in EpiSCs (approximately 2.5-fold increase). However, stable overexpression of E2f6 did not repress any of the genes investigated (). Moreover, we did not observe an increase in DNA methylation at the Stag3 gene promoter (). To ensure that ectopically overexpressed E2f6 can be recruited to DNA we also confirmed that it was enriched at the Stag3 promoter by ChIP (). These data indicate that E2f6 overexpression alone cannot induce premature silencing of E2f6-target genes in ground state ESCs.
Figure 4. The effects of E2f6 and Dnmt3b overexpression on RNA expression and DNA methylation of meiotic genes in ESCs. (A) qPCR analysis of Stag3, Smc1β, and E2f6 RNA expression in ESCs with or without stable overexpression of E2f6. (B) Bisulfite DNA sequencing analysis of the Stag3 promoter in ESCs with or without stable overexpression of E2f6. (C) ChIP-qPCR analysis for E2f6 occupancy at the Stag3 promoter in ESCs ectopically overexpressing FLAG-tagged E2f6. (D) RNA expression levels of Stag3, Smc1β, and Dnmt3b in ESCs with or without stable overexpression of Dnmt3b. (E) Bisulfite DNA sequencing analysis for methylation of the Stag3 promoter in ESCs with or without stable overexpression of Dnmt3b.
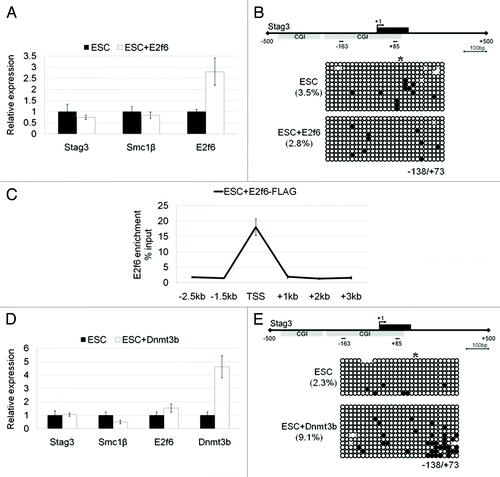
Previously, Dnmt3b has been shown to be essential for repression of E2f6 target genes.Citation15 From our microarray we observed that expression of Dnmt3b is increased 2-fold in EpiSCs relative to ESCs. We used the same rationale as above and utilized a Dnmt3b overexpressing J1 ESC line described previously with an approximately 4.5-fold increase in Dnmt3b expression.Citation18 Again, we did not observe gene inactivation in comparison to the control J1 ESCs (). We note that there was a small increase in Stag3 promoter DNA methylation from 2.3% to 9.7%, but this was not sufficient to silence the gene (). Taken together we conclude that 1) overexpression of E2f6 or Dnmt3b alone cannot establish gene repression in ground state ESCs and 2) additional co-regulator(s) are likely required for initiation and establishment of gene silencing.
Dnmt3b is dispensable for repression of Stag3 and Smc1β
To investigate the requirement of Dnmt3b for the silencing of Stag3 and Smc1β during embryonic development, we used a well-established and routinely employed approach for pluripotent cell differentiation, namely embryoid bodies. Embryoid bodies are complex structures, which faithfully recapitulate the early stages of mammalian embryonic development.
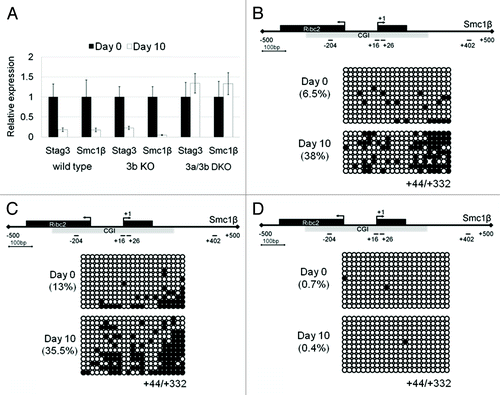
A previous report demonstrated that a hypomorphic mutation of Dnmt3b led to de-repression of meiotic genes in somatic tissues, some of which overlap with the target genes of E2f6.Citation15 To further shed light on the role of Dnmt3b in E2f6-mediated gene repression, here we investigated gene repression and DNA methylation during in vitro differentiation of Dnmt3b knockout (3b KO), Dnmt3a/3b double knockout (3a/3b DKO), and their parental J1 ESCs.Citation19,Citation20 If Dnmt3b-mediated DNA methylation is essential for repression of Stag3 and Smc1β, Dnmt3b−/− ESCs would be deficient in silencing of these genes upon cell differentiation. Surprisingly, both genes were downregulated following embryoid body differentiation (). Importantly, this was accompanied by an increase in overall methylation at the downstream exonic region of Smc1β (), which was similar to what we observed in the control J1 ESCs (). This suggests that Dnmt3b is not indispensable for establishing gene repression and DNA methylation of these meiotic genes. In contrast, the Dnmt3a/3b double knockout cells failed to repress Stag3 and Smc1β () and eventually exhibited no DNA methylation at the Smc1β gene ().
Figure 5. Downregulation of Stag3 and Smc1β gene expression during embryoid body (EB) differentiation of wild-type, Dnmt3b knockout (KO), and Dnmt3a/3b double knockout (DKO) J1 ESCs. (A) qPCR analysis of Stag3 and Smc1β RNA expression during EB differentiation of wild type, 3b KO, and 3a/3b DKO J1 ESCs at Day0 and Day 10. (B) CGI methylation of the downstream region of the Smc1β promoter measured using bisulfite sequencing from the sample of Day 0 and Day 10 following EB differentiation of wild-type J1 ESCs, Dnmt3b KO (C), and Dnmt3a/3b DKO (D).
Ezh2 is required for repression of Stag3 and Smc1β
Silencing of developmentally regulated genes by polycomb repressive complexes is often initiated by PRC2, which then modifies the chromatin and provides a binding platform for PRC1. Enhancer of Zeste homolog 2 (Ezh2) is a core subunit of the PRC2 complex, and it is the enzymatic activity of this protein that is responsible for trimethylation of histone H3 at lysine 27 (H3K27me3). In order to investigate whether PRC2 is required for the silencing of our E2f6-target genes we used ground state induced pluripotent stem cells (iPSCs),Citation21 in which we can conditionally delete exons of the SET domain of Ezh2 ().Citation22 These Ezh2fl/fl iPSCs were subjected to embryoid body differentiation. The H3K4me3 mark associated with transcriptional activity was enriched at the Stag3 promoter in Ezh2 fl/fl iPSCs prior to differentiation (at Day0) and is nearly lost by Day 10 (). On the same promoter we also observed enrichment of Ezh2 at Day 0, which also became absent by Day 6 of differentiation (). The Stag3 and Smc1β genes were efficiently silenced after 10 d of differentiation of the Ezh2fl/fl iPSCs (). In contrast, in the Ezh2Δ/Δ iPSCs Stag3 and Smc1β remained transcriptionally active, suggesting that these genes require Ezh2 for their repression ().
Figure 6. Downregulation of Stag3 and Smc1β gene expression during embryoid body (EB) differentiation of wild-type and Ezh2-deficient pluripotent stem cells. (A) Confirmation of Ezh2fl/fl and Ezh2∆/∆ genotype using PCR and RT-PCR, upper and lower panel, respectively. The amplicons for the flox and the Δ allele are 450 bp and 370 bp (for genomic PCR) and 587 bp and 254 bp (for cDNA PCR), respectively. (B) ChIP-qPCR analysis for H3K4me3 enrichment of the Stag3 promoter in Ezh2 fl/fl iPSCs at 3 differentiation time points (Day 0, Day 6, and Day 10). Enrichment is shown normalized to the total H3 content at each specific primer pair position. (C) ChIP-qPCR analysis for Ezh2 occupancy at the Stag3 promoter in Ezh2 fl/fl iPSCs at 3 differentiation time points (Day 0, Day 6, and Day 10). Enrichment is shown normalized to the background from a ChIP with mouse IgG. (D) qPCR analysis of Stag3 and Smc1β RNA expression during EB differentiation of Ezh2fl/fl iPSCs and Ezh2∆/∆ iPSCs at Day 0 and Day 10. The expression of each gene at Day 0 was set to a value of one. (E and F) CGI methylation of the downstream region of the Smc1β promoter measured using bisulfite sequencing from the sample of Day 0 and Day 10 following EB differentiation of Ezh2fl/fl iPSCs (E) and Ezh2∆/∆ iPSCs (F).
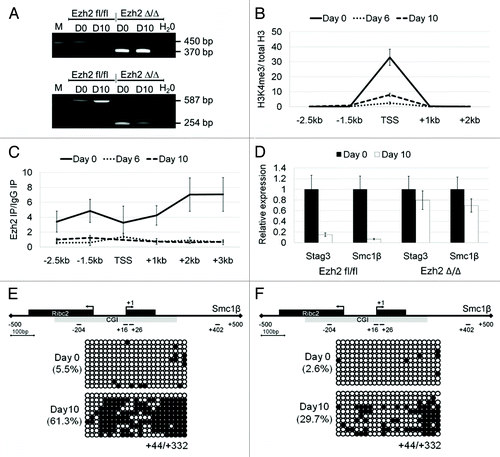
When we examined the downstream exonic Smc1β region for DNA methylation, it was hypomethylated in both the wild type and mutant iPSCs at the onset of differentiation (Day 0), which correlates with transcriptional activity from this promoter. Following 10 d of differentiation, it acquires the 5meC mark in the Ezh2∆/∆ iPSCs but decreased when compared with the Ezh2fl/fl iPSCs. The overall methylation of the downstream exonic Smc1β region increases from 5.5% to 61.3% in Ezh2fl/fl and from 2.6% to 29.7% in Ezh2∆/∆ iPSCs ().
Discussion
Epigenetic mechanisms of gene regulation are fundamental for the establishment and maintenance of the cell type-specific gene expression programs that are first set up during the early stages of mammalian embryonic development. The phenotypic outcomes of chromatin modifications and their inheritance through consecutive cell divisions depend on the concerted efforts of epigenetic “writers, readers, and erasers.” Through their action functional domains like euchromatin, facultative, and constitutive heterochromatin can be established. One potential route for targeting of chromatin modifiers to gene regulatory regions is recruitment through sequence-specific DNA binding molecules, such as transcription factors. The E2f6 transcription factor binds to the proximal promoters of a group of genes, including Stag3, Smc1β, Tuba3a, and Slc25a31, which are restricted to the germline and inevitably get shut down in somatic tissues during development.
We demonstrate here that E2f6-dependent germline gene promoters are first silenced and targeted for de novo methylation in EpiSCs (i.e., the post-implantation epiblast). Temporally, this corresponds to the transition from ground-to-primed state pluripotency. During this transition the open, transcriptionally permissive chromatin of early epiblast cells is progressively compacted. Gene silencing and heterochromatinization can be achieved through the loss of activation-associated histone marks (e.g., H3K4me3), and gradual accumulation of DNA methylation, as we observe for the Stag3 and Smc1β promoters. Strong CpG islands are hypomethylated in mouse ESCs, as are the Stag3 and Smc1β CGIs, and DNA methylation of dense CGIs is often incompatible with transcription from the associated promoters. Our findings here are in agreement with a recent study, which demonstrated that de novo methylation of the E6.5 epiblast tissue is mainly targeted to genes expressed in the germline.Citation23 Importantly, methylation of the Stag3 promoter is reversible during reprogramming in vivo to PGCs and in vitro to iPSCs.
That deletion of E2f6 results in aberrant reactivation of a group of germline genes has been demonstrated previously. Here, we can correlate this transcriptional reactivation with a loss of DNA methylation. Indeed, in the absence of E2f6 the Stag3, Smc1β, Tuba3a, and Slc25a31 promoters are unable to undergo proper de novo methylation. This observation is in agreement with a recent report that binding of Dnmt3b to the proximal promoters of Maelstrom, Syce1, and Tex11, in addition to Slc25a31 was lost upon deletion of E2f6 in MEF cells.Citation15 Thus, it is plausible that E2f6 occupancy at germline gene promoters is essential for Dnmt3b-mediated DNA methylation. However, according to our data stable overexpression of Dnmt3b in ESCs is not sufficient to completely silence these genes, irrespective of an increase in Stag3 promoter methylation. Moreover, overexpression of E2f6 and its binding to the Stag3 promoter is not sufficient to induce gene silencing or promoter methylation in ESCs. These data clearly indicate that E2f6 and Dnmt3b alone are not sufficient for initiating gene repression. Moreover, our data indicated that Dnmt3b was not essential for E2f6-mediated repression of the meiosis genes. It appears that Dnmt3a and Dnmt3b have functional redundancy in the methylation. In contrast, double knockout cells failed to either methylate or downregulate the genes, suggesting de novo DNA methylation activity is indeed required for gene repression. It should be noted here, however, that ESCs deleting both Dnmt3a and Dnmt3b are defective for in vitro differentiation ability itself.Citation24
Our data suggest that additional regulators may play a role in the silencing of Stag3 and Smc1β. All DNMTs can interact with the PRC2 complex through associations with EZH2, EED, and perhaps SUZ12.Citation25 The interactions between DNMT and EZH2 are characterized the best and involve the N-terminal portion of EZH2. Binding of EZH2 to its target promoters and the associated H3K27me3 modification occur upstream of DNA methylation because DNMTs dissociate from EZH2 depleted promoters. So, we would expect PRC2 activity to be essential for initiation of repression, while DNA methylation stabilizes the repressed state. In addition, the presence of non-functional PRC2 complexes (e.g., with mutated Ezh2 subunits) should result in a defect in repression initiation. Here we demonstrated that Ezh2 bound to the Stag3 promoter in undifferentiated iPSCs, when the gene is also marked with H3K4me3, an indicator of active transcription. When the cells were differentiated, the H3K4me3 association was lost along with the decrease in gene expression. Interestingly, the Ezh2 association also disappeared swiftly upon differentiation. A bivalent-like status in the ground state pluripotent cells may be crucial for initiation of the repression. When Ezh2 is deleted, this gene repression was mostly eliminated, suggesting that Ezh2 is indeed essential for initiation of the promoter repression. A recent study looking at the interaction between the PRC and DNA methylation repressive machineries found that targeting of a Gal4DBD-EZH2 fusion protein to a Gal4 binding site array in MEL cells can lead to the recruitment of Bmi1 and Suz12, and an increase in H3K27me3.Citation26 However, it only resulted in the recruitment of Dnmt3a, which importantly, was not accompanied by de novo deposition of the 5meC mark. Unfortunately, no definitive results could be obtained for Dnmt3b. Of note, a similar system using GAL4-EZH2 targeted to a GAL4TK-Luciferase construct was capable of silencing the reporter independently of the SET enzymatic activity when a ΔSET mutant was employed.Citation27 The fact that Stag3 and Smc1β continue to be expressed following differentiation of Ezh2∆/∆ iPSCs suggests that H3K27me3 is likely required for initiation of repression of these genes.
We have attempted to describe here, in further detail, the molecular mechanisms of E2f6-mediated germline gene regulation. Surprisingly, we observe that differential mechanisms govern silencing of these genes in the soma. We point out that the methylation changes we observe upon deletion of E2f6 vary depending on gene context, traversing from a severe loss (Stag3 and Smc1β), to a mild (Tuba3a) or subtle decrease (Slc25a31). It is possible that additional mechanisms exist to safeguard Slc25a31 promoter methylation. Slc25a31 is more hypermethylated even in undifferentiated ESCsCitation28 or E2f6 null MEFs (data shown here), compared with other three genes. Accordingly, it was recently described that silencing of this gene is mainly DNA-methylation dependent. This was observed in a study aimed at identifying genes that are permanently upregulated after a prolonged recovery period following 5aza-dC treatment of somatic cells.Citation16 In addition, Ezh2 activity is dispensable for silencing of both Tuba3a and Slc25a31 following differentiation of Ezh2∆/∆ iPSCs (data not shown). The discovery that deletion of E2f6 in somatic cells can deregulate genes with functions in the germline, and that a conserved E2f6 binding element exists in the proximal promoters of murine meiosis-specific genesCitation7 put forth the idea that E2f6 might serve as a common, “master” regulator involved in their coordinated silencing. However, E2f6-mediated regulation of these genes appears to be more nuanced than we initially anticipated. It seems likely that within the larger group of testis-specific, germline genes additional subgroups exist and that the mechanisms of E2f6-mediated repression are complex and dependent upon the gene context. It is probable that genes with similar functions, such as Stag3 and Smc1β which are subunits of the meiotic cohesin complex, are regulated through mechanisms that are more closely related than functionally distant genes.
Materials and Methods
Cells and culture conditions
R1 and J1 ESCs were maintained on dishes coated with 0.1% gelatin (EMD Millipore, ES-006-B) in KO-DMEM (Life Technologies, 10829-018), containing 10% knockout serum replacement (KSR) (Life Technologies, 10828-028), 1% FCS (Atlanta Biologicals, S11550H), 25 mM HEPES (Corning Cellgro, 25-060-Cl), 100 U/ml penicillin, 100 μg/ml streptomycin, 0.3 mg/ml L-glutamine (Corning Cellgro, 30-009-Cl), monothioglycerol (Sigma-Aldrich, M6145), and 1,000 U/ml LIF (EMD Millipore, ESG1107). J1 ESCs constitutively overexpressing Dnmt3b were generated as described previously.Citation18 R1 ESCs constitutively overexpressing E2f6 were generated as described previously.Citation7 Dnmt3b−/− and Dnmt3a/3b double knockout J1 ESCs are a gift from Dr. En Li as we described previously.Citation20 ESCs aggregation was promoted by hanging drop culture. Approximately 2,000 cells/25 μl drops were seeded on the lid of Petri dishes. Embryoid body differentiation was performed in IMDM media (Corning Cellgro, 10-016-CV) with 20% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and monothioglycerol. Two days after embryoid body formation the cell aggregates were collected and transferred to non-adherent Petri dishes. At Day 4 the aggregates were allowed to attach to gelatin-coated culture dishes and grown until Day 10.
MEFs were maintained in DMEM media, containing 15% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin. For the purpose of EpiSC culture MEF feeder cells were prepared by treating sub-confluent MEF cultures with 10 μg/ml Mitomycin C (Roche Diagnostics, 107409) for 3 h at 37 °C. EpiSC cell clumps were transferred on to MEF feeders plated at 5 × 104 cells/cm2 and maintained in KO-DMEM with 20% KSR, 5 ng/ml FGF2 (Life Technologies, PHG0021) 0.1 mM β-mercaptoethanol (Life Technologies, 21985-023), 2mM L-glutamine (Life Technologies, 25030-081), and non-essential amino acids (Life Technologies, 11140-050). Colonies were passaged every 3 d using gentle dissociation with 1.5 mg/ml Collagenase IV (Life Technologies, 17104-019).
Ezh2fl/fl iPSCsCitation21 were generated by reprogramming of ROSA26:CreER MEF cells carrying a loxP flanked SET domain of Ezh2, and generously provided to us by Dr Manuel Serrano. They were maintained in the ESC culture conditions described above. To obtain Ezh2∆/∆ these iPSCs were treated with 1 μM final concentration of 4-hydroxytamoxifen (Sigma-Aldrich, H-7904) for 4 d. Three Ezh2∆/∆ clones were expanded and used for experimentation.
Genotyping PCR for Ezh2fl/fl and Ezh2∆/∆ iPSCs
Deletion of the SET domain of Ezh2 was confirmed by genotyping PCR. Genotyping PCR was performed with 1.5 U of Taq polymerase (5 PRIME, 2200010), with 320 μM dNTPs (Life Technologies, 10297-018), and 500nM oligonucleotides (Integrated DNA Technologies) using 200 ng gDNA template. The following conditions were used: 15 min initial denaturation at 95 °C, followed by 40 cycles of 45 s at 95 °C denaturation, annealing for 30 s (Ta = 58.7 °C for the wild type allele; Ta = 62.6 °C for the mutant allele), extension for 1.5 min at 72 °C, and final extension for 10 min. Deletion of the SET domain at the mRNA level was confirmed by RT-PCR with 15 ng cDNA and the following conditions: 1 min initial denaturation at 94 °C, followed by 30 cycles for 5 s at 94 °C, annealing for 5 s at Ta = 55 °C, and final extension for 10 min at 72 °C. The oligonucleotides used for genotyping have been described previously.Citation22 For primer sequences see Table S1.
Microarray
Total RNA was extracted from ESCs, EpiSCs, and MEFs using RiboPure RNA Isolation Kit (Life Technologies, AM1924). Gene expression profiling was performed by Genus Biosystems. Total RNA samples were quantified by UV spectrophotometry (OD260/280). Quality and quantity of total RNA was assessed using an Agilent Bioanalyzer (Agilent Technologies). First and second strand cDNA was prepared from the total RNA samples. cDNA was fragmented to uniform size and hybridized with Agilent Whole Genome 4×44K arrays. Slides were washed and scanned on an Agilent G2565 Microarray Scanner. Data was analyzed with Agilent Feature Extraction and GeneSpring GX v7.3.1 software packages. Intensity values were normalized to the median value of the array.
Real time RT-PCR
Total mRNA for gene expression analysis was extracted using the RNAqueous Kit (Life Technologies, AM1912). First strand cDNA was synthesized using High CapacitycDNAReverse Transcription Kit (Life Technologies, 4368814) and the supplied random primers. The qPCR was performed in 20 μl reactions/well on a 96-well plate (Bio-Rad, MLL9601) using 2 μM of each respective primer pair (Integrated DNA Technologies), 2.5 ng cDNA, and 2× Power SYBR Green PCR Master mix (Life Technologies, 4367659). We used a DNA Engine Opticon 2 Thermo cycler (MJ Research Inc.), and the data was analyzed with MJ Opticon Monitor analysis software 3.1 (Bio-Rad). The expression level of all genes was normalized to β-Actin. Relative quantification was performed following the 2−∆∆Ct Livak method. Primer sequences are listed in Table S1.
Bisulfite DNA sequencing
Genomic DNA used for bisulfite conversion was extracted using the Wizard Genomic DNA Purification Kit (Promega, PRA1125) according to the manufacturer’s protocol. All DNA sequences for primer design were obtained from the UCSC genome browser (http://genome.ucsc.edu). Primer design and CpG island (CGI) prediction was performed using the MethPrimer softwareCitation29 (http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi). This software scores a DNA sequence as a CGI when it fulfills the following criteria: at least a 200-bp sequence, with a G+C content >50%, and an Obs/Exp CpG ratio >0.6. Bisulfite conversion was done using the EZ DNA Methylation Gold Kit (Zymo Research, D5005). Up to 2 μg of genomic DNA was converted using the C1000 Thermal Cycler (Bio-Rad) at 98 °C for 10 min, followed by 64 °C for 2.5 h.
PCR products from bisulfite converted templates were gel purified using QIAquick Gel Extraction Kit (Qiagen, 28704) and cloned in the pCR2.1 vector using TOPO TA Cloning Kit (Life Technologies, K4520-01) according to the manufacturer’s protocol. For blue-white screening 10–20 white colonies were chosen for expansion in Luria Broth (Research Products International Corp., L24041-1000) liquid culture overnight containing 100 μg/ml ampicillin (Fisher Scientific, BP1760) at 37 °C and 250 rpm. Plasmid DNA was isolated using Qiaprep Spin Miniprep Kit (Qiagen, 27106). Clones containing the insert of interest were subjected to automated DNA sequencing (DNA Sequencing facilities, Center for Mammalian Genetics, University of Florida) using M13 primers supplied with the TOPO TA Cloning Kit.
Chromatin immunoprecipitation (ChIP) -qPCR
Approximately 5 × 106 cells were used per ChIP assay. Cells were cross-linked using 1% formaldehyde (Sigma-Aldrich, F8775) for 10 min at room temperature. The formaldehyde was quenched using 250 mM glycine (Fisher Scientific, BP381-1) for 5 min. The cells were washed with 1× ice cold PBS, containing 137 mM NaCl (Fisher Scientific, BP3581), 2.7 mM KCl (Sigma-Aldrich, P-9333), 10 mM Na2HPO4 (Sigma-Aldrich, S-7907), and 2 mM KH2PO4 (Sigma-Aldrich, P-9791) and collected by scraping in 1 ml ice cold PBS with protease inhibitor cocktail (PIC) (Sigma-Aldrich, P-8340) followed by centrifugation. The cell pellets were resuspended in lysis buffer containing: 5 mM PIPES pH8.0 (Sigma-Aldrich, P-6757), 85 mM KCl, and 0.5% NP-40 (USB Corporation, 19628) and incubated on ice for 10 min. The cell nuclei were pelleted at 5,000 rpm for 5 min at 4 °C and resuspended in nuclei lysis buffer containing 50 mM TRIS-HCl pH8.1 (Fisher scientific, BP152-1), 10mM EDTA (Fisher Scientific, BP120), and 1% SDS (Sigma-Aldrich, L4509). The chromatin was sheared using a Sonic Dismembrator 100 (Fisher scientific) at power setting 4. Each burst was 10 s, followed by a 1 min rest on ice. The number of bursts required to generate average chromatin fragments between 200–700 bp was optimized for each cell type. The chromatin was diluted in buffer containing: 167 mM NaCl, 16.7 mM TRIS-HCl pH 8.2, 1.2 mM EDTA, 1.1% Triton X-100 (Sigma Aldrich, T-9284), 0.01% SDS. The diluted chromatin was then pre-cleared with blocked recombinant Protein G Sepharose 4B conjugate beads (Life Technologies, 10-1241) for 20 min at 4 °C. Some of this chromatin was set aside and used later as Input. The chromatin equivalent of around 5 × 106 cells was then incubated overnight with 2 μg of each antibody. The antibodies used in this study are α-E2f6 polyclonal rabbit antibody (a generous gift from Dr Stefan Gaubatz), α-H3K4me3 (EMD Millipore, 07-473), and α-H3 (Abcam, ab1791). In the case of the ChIP for Ezh2 we used 4 μg of α-Ezh2 antibody (EMD Millipore, 17-662), and mouse IgG was used as a control. On the next day antigen-antibody complexes were pulled down with Protein G Sepharose beads for 1 h at 4 °C. The beads were collected and washed 7 times with wash buffer containing 250 mM LiCl (Sigma-Aldrich, L-4408), 10 mM TRIS-HCl pH 8.0, 1 mM EDTA, 0.5% DOC (Fisher Scientific, BP349), 0.5% NP-40. The chromatin was eluted from the beads using elution buffer containing 50 mM TRIS-HCl pH 8.0, 10 mM EDTA, 1% SDS. The covalent cross-linking was reversed by incubating the chromatin with 200 mM NaCl overnight at 65 °C. The DNA was then extracted using phenol/chloroform/isoamylalcohol (Sigma-Aldrich, P-2069) and purified using QIAquick PCR Purification Kit (Qiagen, 28106). This DNA was then subjected to qPCR. For qPCR the Input DNA was diluted 1:50 and the IP DNA samples 1:5, which was accounted for during quantification. Six primer pairs (Integrated DNA Technologies) spanning 5 kb of the Stag3 gene (from −2.5 kb to +3 kb around the transcription start site) were designed using Primer3 primer design software (http://frodo.wi.mit.edu/; also see Table S1).
| Abbreviations: | ||
| 4-OHT | = | 4-hydroxytamoxifen |
| bp | = | base pair |
| CGI | = | CpG island |
| ChIP | = | chromatin immunoprecipitation |
| EB | = | embryoid body |
| ESC | = | embryonic stem cell |
| EpiSC | = | epiblast stem cell |
| FCS | = | fetal calf serum |
| FGF | = | fibroblast growth factor |
| iPSC | = | induced pluripotent stem cell |
| KSR | = | knockout serum replacement |
| LIF | = | leukemia inhibitory factor |
| MEF | = | mouse embryonic fibroblast |
| PGC | = | primordial germ cell |
| PRC | = | polycomb repressive complex |
| RT-PCR | = | reverse transcription polymerase chain reaction |
| TSS | = | transcription start site |
Additional material
Download Zip (130.1 KB)Acknowledgments
The authors wish to thank Dr Stefan Gaubatz (University of Würzburg) for his generosity in providing us with E2f6−/− MEFs and the α-E2f6 antibody used in this study. We would also like to thank Dr Manuel Serrano (Spanish National Cancer Research Center) for sharing the Ezh2fl/fl iPSCs generated by his group. In addition, we are grateful to Dr Paul Tesar (Case Western Reserve University) for supplying us with the EpiSCs used in this study and to Dr En Li (Harvard Medical School) for the Dnmt3b−/− and Dnmt3a/3b double knockout J1 ESCs. We also thank Drs Jörg Bungert, Paul Oh, James Resnick, and Thomas Yang (University of Florida) for helpful discussion and critical reading of the manuscript. This research was supported in part by National Institutes of Health Grants, HD060474 and GM091238 to NT. ML was a recipient of the International Fulbright science and technology award scholarship.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here:
http://www.landesbioscience.com/journals/epigenetics/article/25522
References
- Stephenson RO, Rossant J, Tam PP. Intercellular interactions, position, and polarity in establishing blastocyst cell lineages and embryonic axes. Cold Spring Harb Perspect Biol 2012; 4:4; http://dx.doi.org/10.1101/cshperspect.a008235; PMID: 23125013
- Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 2005; 5:615 - 25; http://dx.doi.org/10.1038/nrc1669; PMID: 16034368
- Cartwright P, Müller H, Wagener C, Holm K, Helin K. E2F-6: a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene 1998; 17:611 - 23; http://dx.doi.org/10.1038/sj.onc.1201975; PMID: 9704927
- Kherrouche Z, Begue A, Stehelin D, Monté D. Molecular cloning and characterization of the mouse E2F6 gene. Biochem Biophys Res Commun 2001; 288:22 - 33; http://dx.doi.org/10.1006/bbrc.2001.5718; PMID: 11594747
- Pohlers M, Truss M, Frede U, Scholz A, Strehle M, Kuban RJ, et al. A role for E2F6 in the restriction of male-germ-cell-specific gene expression. Curr Biol 2005; 15:1051 - 7; http://dx.doi.org/10.1016/j.cub.2005.04.060; PMID: 15936277
- Storre J, Schäfer A, Reichert N, Barbero JL, Hauser S, Eilers M, et al. Silencing of the meiotic genes SMC1beta and STAG3 in somatic cells by E2F6. J Biol Chem 2005; 280:41380 - 6; http://dx.doi.org/10.1074/jbc.M506797200; PMID: 16236716
- Kehoe SM, Oka M, Hankowski KE, Reichert N, Garcia S, McCarrey JR, et al. A conserved E2F6-binding element in murine meiosis-specific gene promoters. Biol Reprod 2008; 79:921 - 30; http://dx.doi.org/10.1095/biolreprod.108.067645; PMID: 18667754
- Storre J, Elsässer HP, Fuchs M, Ullmann D, Livingston DM, Gaubatz S. Homeotic transformations of the axial skeleton that accompany a targeted deletion of E2f6. EMBO Rep 2002; 3:695 - 700; http://dx.doi.org/10.1093/embo-reports/kvf141; PMID: 12101104
- Courel M, Friesenhahn L, Lees JA. E2f6 and Bmi1 cooperate in axial skeletal development. Dev Dyn 2008; 237:1232 - 42; http://dx.doi.org/10.1002/dvdy.21516; PMID: 18366140
- Trimarchi JM, Fairchild B, Wen J, Lees JA. The E2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proc Natl Acad Sci U S A 2001; 98:1519 - 24; http://dx.doi.org/10.1073/pnas.98.4.1519; PMID: 11171983
- Attwooll C, Oddi S, Cartwright P, Prosperini E, Agger K, Steensgaard P, et al. A novel repressive E2F6 complex containing the polycomb group protein, EPC1, that interacts with EZH2 in a proliferation-specific manner. J Biol Chem 2005; 280:1199 - 208; http://dx.doi.org/10.1074/jbc.M412509200; PMID: 15536069
- Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 2002; 296:1132 - 6; http://dx.doi.org/10.1126/science.1069861; PMID: 12004135
- Trojer P, Cao AR, Gao Z, Li Y, Zhang J, Xu X, et al. L3MBTL2 protein acts in concert with PcG protein-mediated monoubiquitination of H2A to establish a repressive chromatin structure. Mol Cell 2011; 42:438 - 50; http://dx.doi.org/10.1016/j.molcel.2011.04.004; PMID: 21596310
- Qin J, Whyte WA, Anderssen E, Apostolou E, Chen HH, Akbarian S, et al. The polycomb group protein L3mbtl2 assembles an atypical PRC1-family complex that is essential in pluripotent stem cells and early development. Cell Stem Cell 2012; 11:319 - 32; http://dx.doi.org/10.1016/j.stem.2012.06.002; PMID: 22770845
- Velasco G, Hubé F, Rollin J, Neuillet D, Philippe C, Bouzinba-Segard H, et al. Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc Natl Acad Sci U S A 2010; 107:9281 - 6; http://dx.doi.org/10.1073/pnas.1000473107; PMID: 20439742
- Hackett JA, Reddington JP, Nestor CE, Dunican DS, Branco MR, Reichmann J, et al. Promoter DNA methylation couples genome-defence mechanisms to epigenetic reprogramming in the mouse germline. Development 2012; 139:3623 - 32; http://dx.doi.org/10.1242/dev.081661; PMID: 22949617
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A 2002; 99:3740 - 5; http://dx.doi.org/10.1073/pnas.052410099; PMID: 11891299
- Oka M, Rodić N, Graddy J, Chang LJ, Terada N. CpG sites preferentially methylated by Dnmt3a in vivo. J Biol Chem 2006; 281:9901 - 8; http://dx.doi.org/10.1074/jbc.M511100200; PMID: 16439359
- Rodić N, Oka M, Hamazaki T, Murawski MR, Jorgensen M, Maatouk DM, et al. DNA methylation is required for silencing of ant4, an adenine nucleotide translocase selectively expressed in mouse embryonic stem cells and germ cells. Stem Cells 2005; 23:1314 - 23; http://dx.doi.org/10.1634/stemcells.2005-0119; PMID: 16051982
- Oka M, Meacham AM, Hamazaki T, Rodić N, Chang LJ, Terada N. De novo DNA methyltransferases Dnmt3a and Dnmt3b primarily mediate the cytotoxic effect of 5-aza-2′-deoxycytidine. Oncogene 2005; 24:3091 - 9; http://dx.doi.org/10.1038/sj.onc.1208540; PMID: 15735669
- Villasante A, Piazzolla D, Li H, Gomez-Lopez G, Djabali M, Serrano M. Epigenetic regulation of Nanog expression by Ezh2 in pluripotent stem cells. Cell Cycle 2011; 10:1488 - 98; http://dx.doi.org/10.4161/cc.10.9.15658; PMID: 21490431
- Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 2003; 4:124 - 31; http://dx.doi.org/10.1038/ni876; PMID: 12496962
- Borgel J, Guibert S, Li Y, Chiba H, Schübeler D, Sasaki H, et al. Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet 2010; 42:1093 - 100; http://dx.doi.org/10.1038/ng.708; PMID: 21057502
- Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J, et al. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol 2004; 24:8862 - 71; http://dx.doi.org/10.1128/MCB.24.20.8862-8871.2004; PMID: 15456861
- Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006; 439:871 - 4; http://dx.doi.org/10.1038/nature04431; PMID: 16357870
- Rush M, Appanah R, Lee S, Lam LL, Goyal P, Lorincz MC. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of Dnmt3a but not de novo DNA methylation. Epigenetics 2009; 4:404 - 14; http://dx.doi.org/10.4161/epi.4.6.9392; PMID: 19717977
- Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, et al. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 2008; 10:1291 - 300; http://dx.doi.org/10.1038/ncb1787; PMID: 18931660
- Suzuki M, Sato S, Arai Y, Shinohara T, Tanaka S, Greally JM, et al. A new class of tissue-specifically methylated regions involving entire CpG islands in the mouse. Genes Cells 2007; 12:1305 - 14; http://dx.doi.org/10.1111/j.1365-2443.2007.01136.x; PMID: 18076568
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002; 18:1427 - 31; http://dx.doi.org/10.1093/bioinformatics/18.11.1427; PMID: 12424112