Abstract
One in six cancers worldwide is caused by infection and human papillomavirus (HPV) is one of the main culprits. To better understand the dynamics of HPV integration and its effect on both the viral and host methylomes, we conducted whole-genome DNA methylation analysis using MeDIP-seq of HPV+ and HPV- head and neck squamous cell carcinoma (HNSCC). We determined the viral subtype to be HPV-16 in all cases and show that HPV-16 integrates into the host genome at multiple random sites and that this process predominantly involves the transcriptional repressor gene (E2) in the viral genome. Comparative analysis identified 453 (FDR ≤ 0.01) differentially methylated regions (DMRs) in the HPV+ host methylome. Bioinformatics characterization of these DMRs confirmed the previously reported cadherin genes to be affected but also revealed new targets for HPV-mediated methylation changes at regions not covered by array-based platforms, including the recently identified super-enhancers.
Introduction
One in six cancers worldwide is believed to be caused by infection, with human papillomavirus (HPV) being one of the leading culprits.Citation1 HPV is known to represent a major independent risk factor for Head and Neck Squamous Cell Carcinoma (HNSCC).Citation2,Citation3 It is particularly associated with oropharyngeal carcinoma, of which 20–50% test positive for the HPV-16 subtype, showing expression of the E6 and E7 viral oncogenes.Citation2–Citation4 HPV infected and uninfected (hereafter referred to as HPV+ and HPV-) HNSCCs display differences in clinical behavior: Although HPV+ cancer shows an increased propensity to metastasize, HPV infection is generally associated with a more favorable clinical outcome; patients with HPV+ HNSCC respond better to chemotherapy and radiotherapy (82% vs. 55% response rate for HPV- tumors) and have higher overall survival rates (95% vs. 62% at two years after diagnosis).Citation5 The cause for this difference between HPV+ and HPV- tumors remains unknown.
Upon persistent infection, HPV can integrate into the host cell genome, where it becomes subject to epigenetic modification.Citation6,Citation7 The HPV genome does not encode for any DNA methyltransferases (DNMT) and thus it is believed that the viral genome is methylated by DNMTs of the human host cell.Citation6 This may be directed by the virus in order to regulate intrinsic cell-differentiation dependent and temporal viral gene expression through cross-talk between viral proteins and DNMTs. Alternatively, methylation of viral DNA may represent a potential host defense mechanism, with the host aiming to alter viral gene expression once the virus has integrated into the host genome.
In cervical epithelium, HPV-DNA hypermethylation is more closely associated with cervical carcinoma than with asymptomatic infection or with dysplasia.Citation7,Citation8 Methylation of the L1 gene of both HPV-16 and HPV-18 has been reported in cervical cancer and in penile cancer.Citation8–Citation11 Methylation changes within HPV-16 in HNSCC have been demonstrated by Balderas-Loaeza et al.Citation12 They reported methylation of the viral genome near the 3′ end of the L1 ORF (3 CpGs) and in the Long Control Region (LCR), which regulates the expression of the viral oncoproteins E6 and E7 (5 CpGs). The most extensive study of methylation of the HPV-16 genome in HNSCC to date used bisulfite-sequencing (BS-Seq) to determine the methylation status of all 110 CpG sites within the viral genome in advanced stage HPV+ HNSCC. Contrary to previous findings, methylation within the viral LCR was not observed in most cases, and methylation in regions other than the LCR was detected to varying degrees.Citation13
In this study, we show the potential for identifying both the presence of HPV and the localization of HPV integration sites from MeDIP-seq data. Additionally, we utilize the MeDIP-seq methylomes to identify and characterize regions of differential methylation between HPV+ and HPV- cohorts.
Results
HPV type 16 was confirmed in all HPV+ samples and methylation within the viral genome was demonstrated
MeDIP-seq data from 3 fresh-frozen (FF) HPV- (HN29, HN32, and HN96) and 3 FF HPV+ (HN39, HN105, HN125) HNSCCs were analyzed using the MeDUSA computational pipelineCitation14 (Table S1). We attempted to utilize the MeDIP-seq data set to confirm the presence of HPV in the HPV+ samples and the absence of HPV in the HPV- samples.
For each sample, those sequence reads that failed to align to the human reference genome were aligned against a viral “metagenome” containing 1776 viral genomes obtained from NCBI Genomes, including 29 human papillomavirus strains (Table S2). All HPV+ HNSCC samples were found to contain sequence that aligned to HPV type 16 but not other HPV strains. As expected, there were no matches to any HPV type in HPV- samples.
To aid comparison between samples, reads that aligned to the viral genome were normalized according to average HPV copy number per cell (Sample HN39 = 96.94 copies per cell, HN105 = 110.44 copies per cell, HN125 = 0.66 copies per cell) and further normalized to reads per million (RPM). It should be noted that the measured viral load does not account for potential differences in the ratio of integrated to episomal viral DNA between samples.
The most pronounced methylation signal was observed at the boundary of the L1 and the L2 gene and within the E1 gene (). Methylation in the boundary region was detected in all samples investigated. The sequence of this part of the viral genome harbors six CpG sites at genomic position 5600, 5606, 5609, 5615, 5707 and 5724. The first four CpG sites are located within a sequence of only 16bp and the remaining two follow 92 bp and 109 bp downstream, respectively.
Figure 1. HPV type 16 methylome in HNSCC. The blue peaks represent regions of the viral genome in which methylation was detected using MeDIP-seq. Methylation was measured as reads per million per HPV copy per cell. Sites of methylation within the viral genome were mainly detected at the boundary of the L1/L2 ORF and within the E1 ORF (created in CircosCitation39)
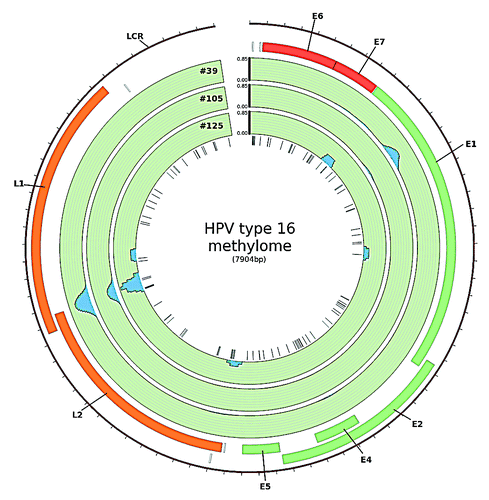
Validation of methylation at the boundary of the L1/L2 ORF of the viral genome in fresh-frozen HNSCC samples and in an independent set of HPV+ HNSCC cell lines by BS-Seq
The results obtained by MeDIP-Seq were validated by applying the targeted BS-Seq technique (see Methods) to a 421 bp region of the viral genome, covering the six CpG sites of interest and two additional CpG sites in genomic positions 5925 and 5961, in the three FF HPV+ HNSCC samples. BS-Seq was also used to test the methylation status of this region in three HPV+ HNSCC cell lines (UPCI:SCC90, UM:SCC47, 93VU-147T). Methylation at all six CpG sites at genomic positions 5600, 5606, 5609, 5615, 5707 and 5724 was observed (Fig. S1).
Integration sites identified within the host genome
Next, we selected for paired-end reads in which one aligned with high confidence to HPV type 16 and the other to human DNA; these constitute potential ‘integration site’ reads (n = 33). While the integration sites across the human genome appear to be random, there is a significant enrichment of potential integration sites within the viral E2 (E4 and E5) region, as illustrated in and .
Figure 2. Potential integration sites of HPV type 16 in the human genome and illustration of location of “integration site reads” in the HPV genome, representing the genomic region of the virus disrupted during the integration process. Links colored dark blue indicate those located in the HPV region most significantly enriched for integration sites. HPV and human genome drawn at different scale (created in CircosCitation39).
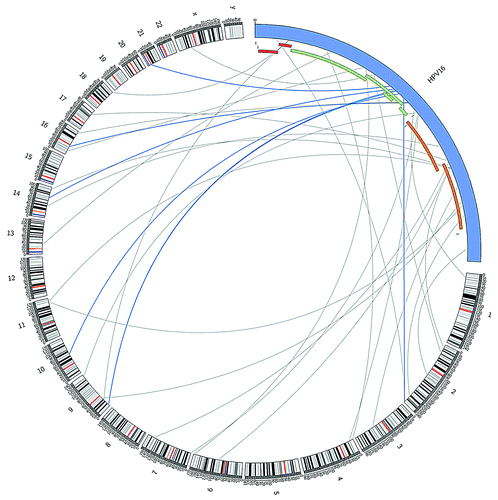
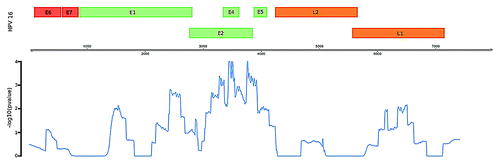
Figure 3. Enrichment of potential integration sites in the HPV genome as measured by –log transformed empirical P-value. Region containing the E3, E4, and E5 genes display the most significant degree of enrichment.
Genome-wide methylome analysis of HPV+ and HPV- HNSCCs
MeDUSA was used to locate differentially methylated regions (DMRs) between the HPV+ and HPV- cohorts. This analysis led to the identification of 1168 DMRs between the HPV+ and HPV- cohort at a false-discovery rate of 0.01. 782 of these DMRs were found to be enriched or hypermethylated in the HPV+ cohort relative to the HPV- cohort. The remaining 386 showed reduced signal in the HPV+ relative to HPV- cohort; we refer to this as hypomethylation. A substantial number of hypermethylated DMRs were found to reside on the X chromosome. This signal was clearly driven by the single sample derived from a female patient (HN125) (Fig. S2). As a result, DMRs on the sex chromosome were removed and recalled using only the five samples derived from males. This resulted in the total DMR count falling to 830 (439 hypermethylated and 391 hypomethylated DMRs).
Copy number analysis was performed on the six samples using the Illumina HumanOmni1-Quad BeadChip genotyping array. These data revealed that potential copy number variable (CNV) regions from the six samples cover 64% of the genome. “False-enriched CNV regions” were defined as regions that show a gain in copy number, defined as a score of > 2.3, in the HPV+ cohort or a loss of copy number, defined as a score of < 1.7, in the HPV- cohort. Conversely, “false-reduced CNV regions” were defined as showing a loss in copy number, defined as a score of < 1.7, in the HPV+ cohort, or a gain in copy number, defined as a score of > 2.3, in the HPV- cohort. Significant association of hypermethylated DMRs with false-enriched CNV regions (p ≤ 0.001, permutation test) but not with false-reduced CNV regions (p = 0.976, permutation test) was observed. Likewise, hypomethylated DMRs were associated with false-reduced CNV regions (p ≤ 0.001, permutation test) but not with false-enriched CNV regions (p = 0.996, permutation test). Therefore, hypermethylated DMRs falling into “false-enriched CNV regions” and hypomethylated DMRs falling into “false-reduced CNV regions” were removed, yielding a final set of 209 hypermethylated and 244 hypomethylated DMRs.
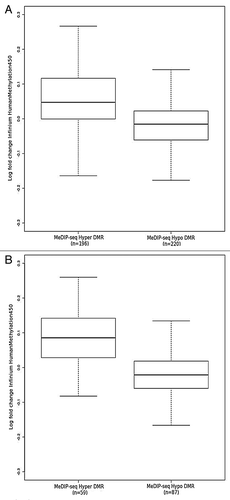
These DMRs were compared with previously published data from Lechner et al.Citation15 This data set was comprised of 20 HPV+ and 20 HPV- samples derived from formalin-fixed paraffin-embedded (FFPE) tissue, with the analysis performed on the Illumina Infinium 450K platform. A significant trend (p = < 0.001, K-S test) is found whereby the fold change in probes located within a DMR between HPV+ and HPV- is in concordance with the directionality of the MeDIP-seq DMR (). This trend becomes stronger if only DMRs that contain ≥ 5 probes are included (). In reality, the majority of the MeDIP-seq DMRs (71% (149/209) hypermethylated and 64% (155/244) hypomethylated) actually contain fewer than 5 Infinium probes. Of these DMRs, however, 98% contained ≥ 5 CpGs. This suggests that there are potentially interesting methylation variable regions, containing numerous CpG sites, absent from the Infinium array. Such regions can currently only be detected by genome-wide techniques such as MeDIP-seq.
Figure 4. Log fold change obtained from comparison of FFPE HPV+ (n = 20) and FFPE HPV- (n = 20) probes on Illumina Infinium 450K arrayCitation15 located within (A) all MeDIP-seq DMR regions and (B) MeDIP-seq DMR regions containing ≥ 5 probes.
Utilizing our DMR set, we sought to determine significant associations with different genomic features. Both hyper- and hypo-methylated DMRs were strongly associated (p-value ≤ 0.001, permutation test) with CpG islands, CpG island shores, promoter regions and genes (Fig. S3A). Within genes, hypomethylated DMRs (p = 0.025 FDR-adjusted, 1-tailed Fisher’s Exact Test), but not hypermethylated DMRs (p = 0.83), showed a preference for coding genes (Fig. S3C, left panel). Additionally, there was strong association with exons (p = 2.2 × 10-28 and p = 1.6 × 10-59 for hyper- and hypomethylated DMRs respectively, FDR-adjusted, 1-tailed Fisher’s Exact test) but not with introns (p = 1) (Fig. S3C, right panel). Within CpG islands, we observed a strong association with promoter CpG islands (p = 1.59 × 10-9 for hypermethylated DMRs, p = 6.13 × 10-15 for hypomethylated DMRs, both FDR-adjusted, 1-tailed Fisher’s Exact test) but not 3′-, intergenic, or intragenic CpG islands (Fig. 3SB).
To determine whether the DMRs were associated with particular genomic regulatory features, the Ensembl ENCODE regulatory segmentationCitation16 was utilized. Using the output from the programs chromHMMCitation17 and Segway,Citation18 the segmentation classifies the genome into regions of similar signal over 14 assays to obtain a summary of the functional architecture of the human genome. Of the six cell lines for which this segmentation data are available, we selected the HepG2 cell line as being the most similar to oropharyngeal HNSCC. As expected, we discovered significant association with predicted promoter regions in both hyper- and hypomethylated DMRs (p < 0.001 Fisher’s Exact test, FDR-adjusted). Additionally, there was a highly significant association (p < 0.001 Fisher’s Exact test, FDR adjusted) with predicted enhancer regions. This was supported by significant association with the ENCODE ChIP-seq data for the histone mark H3K4me1 (HepG2) (p < 0.001 Fisher’s Exact test, FDR-adjusted) and H3K27ac (HepG2) (p < 0.001 Fisher’s Exact test, FDR-adjusted) known to be present at enhancer regions.Citation19 Furthermore, utilizing annotations from previously identified multiple myeloma super-enhancers,Citation20 we observe overlaps in 28 of our DMRs.
Putative transcription factor binding sites in regions where DMRs overlap with CpG islands were identified by both affinity prediction using TRAPCitation21 and association with published ChIP-seq data from ENCODE. Interestingly, given their role in HPV associated cancers,Citation22 E2F1 and E2F6 binding sites are significantly enriched in targets of both enriched and reduced DMRs (Table S3). Importantly, the sets of target genes of hypermethylated and hypomethylated DMRs are non-overlapping (Table S4 and S5). E2F1 and E2F6 themselves do not appear to be differentially methylated in HPV+ with respect to HPV- HNSCC. Association of methylation variable positions (MVPs) with E2Fs has also been detected in the analysis of 450K array data, thus reinforcing that the key findings by Lechner et al.Citation15 can be replicated with a small data set.
Gene enrichment analysis using PANTHERCitation23 revealed that promoter-associated hypermethylated DMRs affect genes associated with the biological processes cell-cell adhesion (p = 5.83 × 10-3, Bonferroni corrected) and ion transport (p = 0.0387, Bonferroni corrected). Hypomethylated DMRs found in promoter regions are associated with cation transport (p = 3.61 × 10-2, Bonferroni corrected). Target genes associated with cell-cell adhesion include members of the cadherin family (CDH2, CDH4, CDH6, and CDH11). This family was also identified as significantly associated with MVPs in the analysis of a much larger data set using the 450K array technology.Citation15
Discussion
We have analyzed MeDIP-seq data on 3 HPV- and 3 HPV+ HNSCCs to characterize both the viral and host methylomes. Given the small sample size, we first attempted to validate previous findingsCitation15 using our independent data set obtained with a different method. Additionally, unlike the previous array-based analysis, we were able to utilize the same MeDIP-seq data to interrogate the viral methylome and potential sites of viral integration.
Analysis of HPV-induced changes in the tumor methylome identified 453 regions of differential methylation with high confidence, given a 1% false-discovery rate for DMR identification and removal of DMRs falling into relevant copy-number variable regions. CpG Island regions that are targeted by differential methylation were found to harbor binding sites for a range of transcription factors, notably E2F1 and E2F6. While the gene sets targeted by hypermethylated and hypomethylated DMRs are non-redundant, there are target genes within each class that harbor both E2F1, E2F6 and other transcription factor binding sites within the methylated region. Within the E2F family, E2F1 is known to act as an activator transcription factor to promote cell proliferation.Citation24 E2F6, on the other hand, lacks the Cdk- and Rb-interaction domains found in other members of the E2F family.Citation24 Given the resolution limit of the MeDIP-seq technique (~200 bp), it is not possible to definitively state which binding site is affected by methylation in these particular cases. On a global scale, however, it is interesting to note that many genes targeted by differential methylation are involved in cell-cell adhesion processes as supported by gene enrichment analysis. This includes members of the cadherin family as well as NCAM2. Given the known role of these proteins in the epithelial-to-mesenchymal transition and metastasis,Citation25 it is tempting to speculate that epigenetic changes induced by HPV infection might underlie the difference in metastatic potential between HPV+ and HPV- HNSCC observed in the clinic. We conclude that we can replicate some of the key findings from previous analysis of the methylomes of HPV+ and HPV- HNSCC with MeDIP-seq and a small data set.
One key advantage of MeDIP-seq over 450K in this context is that it allows us to interrogate the viral methylome. In line with previous reports showing that HPV type 16 is found in almost all cases of HPV+ HNSCC,Citation26 all FF HPV+ HNSCC samples tested in this study had integrated copies of HPV type 16. Specifically, methylation within one region of the HPV type 16 genome was detected consistently across all samples. This 124 bp region lies at the boundary of the L1 and the L2 gene and harbors a total of 6 CpG sites.
Methylation in this location has previously been shown in cervical cancer.Citation7,Citation8 Fernandez et al.Citation7 showed that methylated CpG sites within this region of both the HPV type 16 and HPV type 18 genome is present in the majority of cervical cancer samples tested, rather than in premalignant cervical lesions and in carriers. As part of their comprehensive HPV type 16 methylome analysis Park et al.Citation13 assessed the methylation status of this region in advanced stage III/IV HNSCC patients. Although not specifically mentioned by the authors, they found consistent methylation of CpGs within this region across every sample tested. The main result presented by Park et al.Citation13 was that large parts of the HPV type 16 genome, in particular the LCR, which controls transcription of the E6 and E7 oncogenes,Citation27 was found to be unmethylated in the majority of examined cases. In line with the findings by Park et al.Citation13 the LCR region is found unmethylated in all FF HPV+ HNSCC samples tested in this study, suggesting that methylation in this genomic location may be a rare event in HNSCC. The LCR harbors multiple binding sites for E2. Methylation within the LCR is known to prevent the binding of E2, thereby increasing the expression of E6 and E7, the two key oncogenes.Citation6 Thus, one would expect to find methylated CpG sites within this region. One explanation may be that, as the E2 gene is often disrupted or lost upon viral integration, the selective pressure to methylate this genomic segment is lost in the absence of the transcriptional repressor E2.Citation13
Based on the findings by Fernandez et al.,Citation7 it is conceivable that methylation of the viral genome plays an important role in HPV-induced carcinogenesis, rather than a bystander role only. Moreover, the boundary of the L1 and L2 gene is reported more frequently methylated in cervical cancer samples than in premalignant and normal tissue.Citation8 Irrespective of the potential effects of methylation in this specific location, it may serve as a diagnostic biomarker for HPV+ HNSCC. Park et al.Citation13 employed methylation-specific PCR (MSP) to test for LCR methylation status in serum and saliva samples of HPV+ HNSCC patients. As no control group was included in that study, it can be regarded as a successful feasibility trial. However, based on the findings discussed in this section and the results by Brandsma et al.Citation8 and Fernandez et al.,Citation7 testing the methylation status of this region at the boundary of L1/L2 in serum and saliva samples may represent a far more powerful diagnostic biomarker.
Apart from methylation at the L1/2 boundary region of the viral genome, methylation within the E1 gene was detected to varying degrees in the FF HPV+ HNSCC tissue data set. The significance of these findings remains to be investigated.
A further advantage of having employed MeDIP-Seq is that potential integration sites can be detected by selecting for paired-end reads that align both to the viral and host genome (“integration site reads”). Our data show that integration sites across the human genome appear to be random. However, there was a significant enrichment of potential “integration site reads” within the viral E2 (E4 and E5) region, suggesting that this is a common location of disruption of the viral sequence upon integration into the host genome in HNSCC. This is in line with integration events observed for cervical HPV infection.Citation28 It is noteworthy that the disruption of E2 function (an inhibitor of E6 and E7 viral gene expression) leads to increased expression of the E6 and E7 viral oncogenes and, thus, may represent a key step in viral carcinogenesis.
In conclusion, we show that integrated virus-host methylome analysis using MeDIP-seq complements array-based studies for the identification of novel targets for differential DNA methylation in HPV+ HNSCC.
Materials and Methods
Human Tissue Samples and Cell lines used for the experiments
Ethical approval for the study of human tissue samples was granted by the UCL/UCLH Ethics Committee (Reference number 04/Q0505/59). Three fresh-frozen (FF) HPV+ and three FF HPV– HNSCC samples (Table S6) were obtained from the UCLH Head and Neck Tumour Bank and were used for MeDIP-Seq analysis. HNSCC cell lines UPCI:SCC090 (HPV+), UPCI:SCC003 (HPV–), UPCI:SCC036 (HPV–) and PCI-30 (HPV–) were generous gifts from Dr Susanne Gollin and Dr Theresa Whiteside (University of Pittsburgh Cancer Institute, US). 93VU-147T (HPV+) was a generous gift from Dr Hans Joenje (VU Medical Center, Netherlands) and UM:SCC047 (HPV+) was from Dr Thomas Carey (University of Michigan, US). All cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. The main features of these cell lines are described in Table S7. HPV status was confirmed by E6 qPCR from both FF and cell line samples as described previously.Citation29
DNA extraction
DNA from FF tumor and cell line samples was extracted using the QIAamp DNA Blood Mini Kit (QIAGEN). This DNA purification method yields DNA sized from 200 bp to 50 kb. Thus, it is possible that episomal viral DNA was co-purified together with the genomic DNA that included integrated viral DNA.
MeDIP-Seq data analysis of the HPV genome
DNAs from 3 HPV+ and 3 HPV- fresh-frozen HNSCC samples were subjected to AutoMeDIP-seq as previously described,Citation30 using the NEBNext DNA Sample Prep Mastermix (New England Biolabs) for the library preparation and the MagMeDIP kit (Diagenode) for immunoprecipitation. Adequate enrichment of the methylated DNA fraction (compared with input) was quality controlled using qPCR. Following adaptor-mediated PCR, the library was subjected to size selection (300–350 bp) from low melting-point agarose gels. The excised fraction was quality controlled by qPCR. Cluster generation and 36 base paired-end sequencing was performed by the UCL Genomics Core Facility according to the manufacturer’s recommendation, using an Illumina GAIIX platform.
The data were analyzed using the MeDUSA pipeline.Citation14 First, paired-end sequencing reads were aligned to the human reference genome (hg19) using BWA (v0.5.8),Citation31 with default parameters. Next, SAMToolsCitation32 and custom scripts were used to remove (1) incorrectly mapped pairs, (2) pairs in which neither read obtained a Phred score of alignment quality greater than 10, and (3) duplicate fragments (i.e fragments of exactly the same coordinates), which likely represent artifacts of PCR amplification. Read counts are illustrated in Table S1. Quality control was performed using FastQC (v0.9.4, http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/) and MEDIPS (v1.0.0).Citation33 Next, the MultipleReplicaScanSeqs (MRSS) and EnrichedRegionMaker functions of the USeq software suite (v6.8)Citation34 were used to identify DMRs between the HPV+ and HPV- cohorts. MRSS processes point data for use in the BioConductor package DESeq.Citation35 Window size was set to 500. Only regions containing a minimum of 10 reads summed from the two cohorts were included for DMR analysis. DESeq’s variance outlier filter function was bypassed.
DMRs were further filtered to remove those located on the sex chromosomes due to the presence of a single female sample in the HPV+ cohort. Additionally, hypermethylated DMRs falling into “false-enriched CNV regions” and reduced DMRs falling into “false-reduced CNV regions” were removed. “False-enriched CNV regions” were defined as regions that show a gain in copy number, defined as a score of > 2.3, in the HPV+ cohort or a loss of copy number, defined as a score of < 1.7, in the HPV- cohort. Conversely, “false-reduced CNV regions” were defined as showing a loss in copy number, defined as a score of < 1.7, in the HPV+ cohort, or a gain in copy number, defined as a score of > 2.3, in the HPV- cohort.
CNVs were determined using the HumanOmni1-Quad bead chip (Illumina). 1ug of DNA was utilized and processed according to the manufacturers instructions. Copy number alterations were identified using Illumina Genome Studio. The average log ratio of the X chromosome in male compared with female was used to determine the value of a single copy alteration as 0.3, hence a gain was defined as > 2.3 and a loss as < 1.7.
DMR annotation
DMRs identified by the MeDUSA pipeline at a false-discovery rate of 0.01 were annotated using BEDTools.Citation36 Feature annotation was obtained from Ensembl (Build 70).Citation16 A DMR was annotated with a feature if a minimum 51% overlap in one direction, i.e., 51% of the DMR overlapping the feature or 51% of the feature overlapping the DMR, was observed. Statistical testing was performed using either Fisher’s exact test or a permutation test. For both these tests, all regions containing a minimum of 10 reads summed from the two cohorts were used as genomic background thus accounting for biases in the genomic distribution of MeDIP-seq reads.
Gene enrichment analysis
Gene enrichment analysis was performed on the subset of DMRs falling into promoter regions (minimum 51% overlap) against the whole-genome background using the web tool PANTHER (v8.0).Citation23
Identification of putative transcription factor binding sites
Putative transcription factor binding sites in regions where DMRs overlap with CpG islands were identified by two methods: (1) regions were submitted to the online tool TRAPCitation21 for affinity-based prediction of TF binding sites. (2) Overlap between DMRs and Encode ChIP-seq data for various DNA binding factors was determined (minimum 51% overlap) and the significance of association determined using Fisher’s Exact Test.
Characterization of the HPV methylome
Reads that failed to align to the human genome were mapped, using BWA,Citation31 to 1776 viral genomes, including 29 human papillomaviruses (Table S2), obtained from NCBI Genomes (http://www.ncbi.nlm.nih.gov/genomes/GenomesHome.cgi?taxid = 10239) (accessed 28/03/2011).
Filtering was performed using SAMtools (v0.1.9)Citation32 to remove erroneously mapped and low quality (score of < 10) reads. In order to compare viral aligned reads between samples, the read counts were normalized for viral load by dividing the number of reads in 10bp windows by the viral load as estimated by E6 qPCR. Further normalization to account for total library size led to read counts being transformed to reads per million (RPM) as described in Chavez et al. 2010.Citation33
Additionally, the MeDIP-seq data were analyzed to identify potential viral integration sites. For this, reads that failed to form a correctly aligned pair in the human genome were stored. For each HPV+ sample, these stored reads were aligned to the HPV-16 (NC_001526) genome using BWA. Pairs in which one read mapped to the viral genome with an alignment score ≥ 10 and the corresponding mate failed to map, were identified (n = 182). These reads were compared with the relevant human alignment to find pairs. In order to be classed as a pair, the human read also required an alignment score ≥ 10 and to have an unmapped mate (n = 660428). Therefore the resulting pair comprises of one read aligning with high confidence to HPV-16 and the other to human, indicating that a potential integration site is located within the fragment (n = 33).
To determine if there was a significant enrichment of these reads within a specific location on the viral genome, counts of the aligned ‘integration site’ reads were obtained for 500bp windows with 10bp slide. Given the low number of ‘integration site’ reads, permutation analysis was performed (n = 10000) to indicate whether the read counts obtained in the 500bp windows were different to that expected by chance. An additional factor to take account of was that, by chance, we were more likely to find an “integration site” read in regions of the viral genome that had more MeDIP reads aligning. Thus the probability of an “integration site” read aligning to a specific genomic region was weighted according to the total number of reads aligning in that window. Permutations were performed, utilizing the probabilities determined for each window, by placing 33 simulated reads across the viral genome. The number falling within each window was counted and compared with the real data. This was repeated (n = 10000) to obtain an empirical p-value. The resulting empirical p-values were log transformed (-log10) and plotted along the HPV16 genome.
Validation of obtained results by bisulfite sequencing (BS-Seq)
Generation of methylated and unmethylated control samples
In vitro methylation of HPV+ HNSCC DNA samples was performed using CpG Methyltransferase (M.Sssi) (New England Biolabs Inc.), according to the protocol of the supplier, in order to obtain a positive control for subsequent BS-Seq analysis.
Whole-genome amplification was performed using the REPLI-g Mini Kit (QIAGEN), according to the protocol of the supplier, in order to obtain a negative control for subsequent BS-Seq analysis.
Bisulfite modification
One microgram of genomic DNA from 3 FF HPV+ HNSCC tissue samples (analyzed by MeDIP-Seq previously) and from 3 HPV+ HNSCC cell lines was bisulfite converted using the Zymo EZ DNA Methylation kit (Zymo Research Corp, cat No. D5001). The protocol was modified to improve bisulfite conversion efficiency by inclusion of a cyclic denaturation step as described previously.Citation37 Bisulfite conversion efficiency was confirmed by methylation-specific PCR (MSP) for a methylated and unmethylated DNA fragment, as described.Citation37
Targeted HPV methylome analysis
In order to obtain the DNA segment of interest (the region at the boundary of the L1 and L2 gene within the HPV-16 genome), the targeted modified DNA fragment was amplified using touchdown PCR with 1× Reaction Buffer, 0.5 mM dNTPs, 2.0–3.0 mM MgCl2, 0.4 μM of forward and reverse primers respectively and 1 unit of HotStar Taq (Qiagen), in a total volume of 20 μl (Qiagen). Reagents were denatured at 95°C for 10 min, followed by 20 cycles of 95°C for 45 sec, 60°C with a decrease of half a degree per cycle for 45 sec, 68°C for 60 sec and 30 cycles of 95°C for 45 sec, 50°C for 45 sec, 68°C for 60 sec and ending with a seven-minute extension at 68°C. One in vitro methylated control sample (positive control) and one unmethylated (whole-genome amplified) control sample (negative control) were included in each PCR, as previously described.Citation38 PCR primers used for BS-Seq, covering the 6 CpG sites of interest in a region at the boundary of the L1 and L2 gene within the HPV-16 genome, were used to validate the viral methylome analysis employing MeDIP-Seq. Sequences of these primers were obtained fromCitation13 and are summarized in Table S8. A product of 421 bp (see Fig. S4) was generated by PCR using the PCR program outlined above.
The specificity of the reaction products was inspected using 1.5% agarose gel electrophoresis before being purified using the Illustra ExoStar 1-Step kit (GE Healthcare Life Sciences).
Bisulfite sequencing
Methylation analysis was performed with bisulfite genomic sequencing using the bisulfite modified and PCR amplified and purified DNA segment of interest (421 bp segment at the border of the L1 and L2 gene in the HPV-16 genome, harboring the 6 CpG sites of interest in genomic positions 5600, 5606, 5609, 5615, 5707, and 5724 (and two additional CpG sites). The same primers (Table S8) were used for BS-Seq.
After bisulfite treatment and PCR amplification, a subcloning step is frequently included before sequencing. This is done to avoid a molecular weight shift, detected during the sequencing reaction, when CpG rich regions are analyzed. This shift is a result of the mixture of methylated (+ CH3) and unmethylated (- CH3) CpG sites within the segment of interest in a heterogeneous DNA population. In this case however, the segment of interest only contained 8 CpG sites (within a 421 bp viral DNA segment). Thus, no significant molecular weight shift was expected. In order to cause a molecular weight shift of one base, the DNA segment would have to contain 22 CpG sites that are differentially methylated in the cell population. Hence, a subcloning step was not required. Although Sanger sequencing is not a quantitative method, the results obtained from the positive (in vitro methylated) and the negative (whole-genome amplified) control, confirms the methylation status of this locus in the HPV type 16 genome.
BS-Seq was performed by the UCL Genomics Core Facility according to the manufacturer’s recommendation using Applied Biosystems Bigdye3.1 chemistry. Sequencing reactions were cleaned up using in-house-made sephadex filtration plates and then run on Applied Biosystems 3730XL Genetic Analyzer.
Additional material
Download Zip (745.6 KB)Acknowledgments
We would like to thank Dr Susanne Gollin and Dr. Theresa Whiteside (University of Pittsburgh Cancer Institute, US), Dr Hans Joenje (VU Medical Center, Netherlands) and Dr Thomas Carey (University of Michigan, US) for the provision of HNSCC cell lines. We would also like to thank the teams at the Head and Neck Centre and the Department of Histopathology at the University College London Hospital (UCLH), UCL Advanced Diagnostics and UCL Genomics for their support. This study was supported in part by the UCLH Comprehensive Biomedical Research Centre (CBRC). The UCLH Head and Neck Tumour Bank was supported by UCLH/UCL NIHR CBRC. HC was supported by the Swedish Research Council and the Wenner-Gren foundation. ML was supported by a Wellcome Trust Fellowship (WT093855MA) and the Austrian Science Fund (J2856). AK was supported by a Cancer Research UK PhD Fellowship. Research in the Boshoff lab was supported by Cancer Research UK. Research in the Beck lab was supported by the Wellcome Trust (084071), Royal Society Wolfson Research Merit Award (WM100023) and IMI-JU OncoTrack (115234).
Disclosure of Potential Conflicts of Interests
No potential conflicts of interest were disclosed.
Note
MeDIP-Seq data were submitted to GEO (Gene Expression Omnibus, NCBI) according to the instructions provided (GEO accession number: GSE38263).
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/25614
References
- de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol 2012; 13:607 - 15; http://dx.doi.org/10.1016/S1470-2045(12)70137-7; PMID: 22575588
- D’Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med 2007; 356:1944 - 56; http://dx.doi.org/10.1056/NEJMoa065497; PMID: 17494927
- Tran N, Rose BR, O’Brien CJ. Role of human papillomavirus in the etiology of head and neck cancer. Head Neck 2007; 29:64 - 70; http://dx.doi.org/10.1002/hed.20460; PMID: 16823878
- Mendenhall WM, Logan HL. Human papillomavirus and head and neck cancer. Am J Clin Oncol 2009; 32:535 - 9; http://dx.doi.org/10.1097/COC.0b013e31818b8fee; PMID: 19652580
- Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst 2008; 100:261 - 9; http://dx.doi.org/10.1093/jnci/djn011; PMID: 18270337
- Fernandez AF, Esteller M. Viral epigenomes in human tumorigenesis. Oncogene 2010; 29:1405 - 20; http://dx.doi.org/10.1038/onc.2009.517; PMID: 20101211
- Fernandez AF, Rosales C, Lopez-Nieva P, Graña O, Ballestar E, Ropero S, et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res 2008; 19:438 - 51; http://dx.doi.org/10.1101/gr.083550.108; PMID: 19208682
- Brandsma JL, Sun Y, Lizardi PM, Tuck DP, Zelterman D, Haines GK 3rd, et al. Distinct human papillomavirus type 16 methylomes in cervical cells at different stages of premalignancy. Virology 2009; 389:100 - 7; http://dx.doi.org/10.1016/j.virol.2009.03.029; PMID: 19443004
- Kalantari M, Villa LL, Calleja-Macias IE, Bernard HU. Human papillomavirus-16 and -18 in penile carcinomas: DNA methylation, chromosomal recombination and genomic variation. Int J Cancer 2008; 123:1832 - 40; http://dx.doi.org/10.1002/ijc.23707; PMID: 18688866
- Turan T, Kalantari M, Calleja-Macias IE, Cubie HA, Cuschieri K, Villa LL, et al. Methylation of the human papillomavirus-18 L1 gene: a biomarker of neoplastic progression?. Virology 2006; 349:175 - 83; http://dx.doi.org/10.1016/j.virol.2005.12.033; PMID: 16472835
- Turan T, Kalantari M, Cuschieri K, Cubie HA, Skomedal H, Bernard HU. High-throughput detection of human papillomavirus-18 L1 gene methylation, a candidate biomarker for the progression of cervical neoplasia. Virology 2007; 361:185 - 93; http://dx.doi.org/10.1016/j.virol.2006.11.010; PMID: 17175003
- Balderas-Loaeza A, Anaya-Saavedra G, Ramirez-Amador VA, Guido-Jimenez MC, Kalantari M, Calleja-Macias IE, et al. Human papillomavirus-16 DNA methylation patterns support a causal association of the virus with oral squamous cell carcinomas. Int J Cancer 2007; 120:2165 - 9; http://dx.doi.org/10.1002/ijc.22563; PMID: 17278110
- Park IS, Chang X, Loyo M, Wu G, Chuang A, Kim MS, et al. Characterization of the methylation patterns in human papillomavirus type 16 viral DNA in head and neck cancers. Cancer Prev Res (Phila) 2011; 4:207 - 17; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0147; PMID: 21292634
- Wilson GA, Dhami P, Feber A, Cortázar D, Suzuki Y, Schulz R, et al. Resources for methylome analysis suitable for gene knockout studies of potential epigenome modifiers. Gigascience 2012; 1:3; http://dx.doi.org/10.1186/2047-217X-1-3; PMID: 23587164
- Lechner M, Fenton T, West J, Wilson G, Feber A, Henderson S, et al. Identification and functional validation of HPV-mediated hypermethylation in head and neck squamous cell carcinoma. Genome Med 2013; 5:15; http://dx.doi.org/10.1186/gm419; PMID: 23419152
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, et al. Ensembl 2012. Nucleic Acids Res 2012; 40:Database issue D84 - 90; http://dx.doi.org/10.1093/nar/gkr991; PMID: 22086963
- Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 2012; 9:215 - 6; http://dx.doi.org/10.1038/nmeth.1906; PMID: 22373907
- Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods 2012; 9:473 - 6; http://dx.doi.org/10.1038/nmeth.1937; PMID: 22426492
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108 - 12; http://dx.doi.org/10.1038/nature07829; PMID: 19295514
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013; 153:320 - 34; http://dx.doi.org/10.1016/j.cell.2013.03.036; PMID: 23582323
- Thomas-Chollier M, Hufton A, Heinig M, O’Keeffe S, Masri NE, Roider HG, et al. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nat Protoc 2011; 6:1860 - 9; http://dx.doi.org/10.1038/nprot.2011.409; PMID: 22051799
- Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet 2001; 10:699 - 703; http://dx.doi.org/10.1093/hmg/10.7.699; PMID: 11257102
- Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 2013; 41:Database issue D377 - 86; http://dx.doi.org/10.1093/nar/gks1118; PMID: 23193289
- Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 2002; 3:11 - 20; http://dx.doi.org/10.1038/nrm714; PMID: 11823794
- Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer 2004; 4:118 - 32; http://dx.doi.org/10.1038/nrc1276; PMID: 14964308
- Kreimer AR, Clifford GM, Boyle P, Franceschi S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev 2005; 14:467 - 75; http://dx.doi.org/10.1158/1055-9965.EPI-04-0551; PMID: 15734974
- Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond) 2006; 110:525 - 41; http://dx.doi.org/10.1042/CS20050369; PMID: 16597322
- Collins SI, Constandinou-Williams C, Wen K, Young LS, Roberts S, Murray PG, et al. Disruption of the E2 gene is a common and early event in the natural history of cervical human papillomavirus infection: a longitudinal cohort study. Cancer Res 2009; 69:3828 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-08-3099; PMID: 19401452
- Schache AG, Liloglou T, Risk JM, Filia A, Jones TM, Sheard J, et al. Evaluation of human papilloma virus diagnostic testing in oropharyngeal squamous cell carcinoma: sensitivity, specificity, and prognostic discrimination. Clin Cancer Res 2011; 17:6262 - 71; http://dx.doi.org/10.1158/1078-0432.CCR-11-0388; PMID: 21969383
- Butcher LM, Beck S. AutoMeDIP-seq: a high-throughput, whole genome, DNA methylation assay. Methods 2010; 52:223 - 31; http://dx.doi.org/10.1016/j.ymeth.2010.04.003; PMID: 20385236
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754 - 60; http://dx.doi.org/10.1093/bioinformatics/btp324; PMID: 19451168
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al, 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078 - 9; http://dx.doi.org/10.1093/bioinformatics/btp352; PMID: 19505943
- Chavez L, Jozefczuk J, Grimm C, Dietrich J, Timmermann B, Lehrach H, et al. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res 2010; 20:1441 - 50; http://dx.doi.org/10.1101/gr.110114.110; PMID: 20802089
- Nix DA, Courdy SJ, Boucher KM. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics 2008; 9:523; http://dx.doi.org/10.1186/1471-2105-9-523; PMID: 19061503
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106; http://dx.doi.org/10.1186/gb-2010-11-10-r106; PMID: 20979621
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26:841 - 2; http://dx.doi.org/10.1093/bioinformatics/btq033; PMID: 20110278
- Thirlwell C, Eymard M, Feber A, Teschendorff A, Pearce K, Lechner M, et al. Genome-wide DNA methylation analysis of archival formalin-fixed paraffin-embedded tissue using the Illumina Infinium HumanMethylation27 BeadChip. Methods 2010; 52:248 - 54; http://dx.doi.org/10.1016/j.ymeth.2010.04.012; PMID: 20434562
- Carén H, Djos A, Nethander M, Sjöberg RM, Kogner P, Enström C, et al. Identification of epigenetically regulated genes that predict patient outcome in neuroblastoma. BMC Cancer 2011; 11:66; http://dx.doi.org/10.1186/1471-2407-11-66; PMID: 21314941
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009; 19:1639 - 45; http://dx.doi.org/10.1101/gr.092759.109; PMID: 19541911