Abstract
Aberrant methylation at the H19 paternal imprinted gene has been identified in different cohorts of infertile males. The causes of H19 methylation errors are poorly understood. In this study, we investigated the methylation status of the H19 gene in semen DNA samples from infertile males affected by MTHFR gene promoter hypermethylation. DNA from normal and abnormal semen samples harbouring MTHFR gene promoter hypermethylated, hmMTHFR-nor and hmMTHFR-abn, and without MTHFR methylation, MTHFR-nor and MTHFR-abn, were investigated for methylation status in the H19 locus using bisulfite-treated DNA PCR, followed by cloning and sequencing. The prevalence of H19 hypomethylated clones was 20% in hmMTHFR-nor and 0% in MTHFR-nor semen samples (p < 0.05), and 28% in hmMTHFR-abn compared with 16% in MTHFR-abn semen samples (p > 0.05). These results underscore the association between H19 methylation defects and hypermethylation of the MTHFR gene promoter in normal semen samples and suggest that aberrant methylation at H19 may occur in the normal sperm of infertile males affected by MTHFR gene dysfunction. These findings provide new insights into the mechanisms causing abnormal methylation in imprinted genes and, in turn, male infertility.
Introduction
Genomic imprinting is a mechanism that regulates gene expression in a parental origin-dependent way. DNA methylation is the chemical process that controls genomic imprinting. It consists in the attachment of methyl groups to cytosine residues at CpG dinucleotides, CpG islands, in specific DNA regions of imprinted genes, known as differentially methylated regions (DMRs), allowing for mono-allelic parent-specific gene expression.Citation1
Correctly imprinted maternal and paternal genes are needed to regulate major functions at the fetomaternal interface, such as nutrient transport, trophoblast proliferation, invasion and angiogenesis.Citation2 In rodents and humans, impaired methylation imprints generate small placentae with abnormalities in proliferation, apoptosis and trophoblast differentiation and miscarrige.Citation3-Citation8
In male gametes, DNA methylation at imprinted genes is established prior to entry into meiosis and is maintained throughout development.Citation9,Citation10 Three genetic loci have been found to have paternal imprinting in sperm cells, IGF2/H19, RASGRF1 and GTL2.Citation11 The best-characterized of these genes is IGF2/H19, which displays reciprocal paternal IGF2 and maternal H19 gene expression.Citation9 IGF2/H19 genes share common enhancers, located downstream of H19, whose activity is regulated by a DMR upstream of the H19 gene. In the maternal allele, H19 is unmethylated, allowing the CTCF insulator protein (CCCTC-binding factor) to bind to the DMR. This process prevents IGF2 from accessing common enhancers, thus inhibiting IGF2 and promoting H19 expression. In the paternal allele, H19 is methylated and CTCF binding is blocked, thus inactivating H19 and promoting IGF2 expression.Citation12
Recent studies have indicated that appropriate establishment of H19 imprinting plays a critical role in maintaining fertility, since H19 methylation defects have been identified in several groups of males experiencing infertility.Citation13-Citation16 The causes inducing H19 imprinting disorders in spermatogenetic cells are currently poorly understood.
In a recent study, we found that methylenetetrahydrofolate reductase (MTHFR) gene promoter hypermethylation is strongly associated with semen samples from infertile couples affected by the recurrence of spontaneous abortion (RSA) compared with semen samples from controls.Citation17 Since MTHFR is one of the key regulatory enzymes maintaining the bioavailability of endogenous methyl groups,Citation18 it was suggested that spermatogonial cells harboring MTHFR gene dysfunctions, due to promoter hypermethylation, resulted in a low methyl donor pool giving rise to spermatozoa affected by methylation errors at the paternal imprinted genes which, in turn, accounted for spontaneous abortion.Citation17
Taking advantage of our previous results, in this study we have investigated whether methylation defects at the H19 imprinted gene occurred in DNA semen samples from infertile males affected by MTHFR gene promoter hypermethylation.
Results
COBRA analysis of H19 PCR products
In order to confirm that the sequencing results from PCR templates accurately reflected the overall methylation pattern of the amplified H19 from the sperm cells and leukocytes, a restriction analysis (COBRA) of the PCR products was performed. About half of the methylated and half of the unmethylated templates were obtained after DNA digestion with Taq I and Mlu I restriction enzymes in leukocytes in order to represent paternal and maternal alleles, indicating a lack of bias in the PCR reaction (). H19 PCR products, from DNA sperm belonging to the MTHFR group, yielded digested bands with both Taq I and Mlu I restriction enzymes, suggesting that most of the DNA in each semen sample was methylated at this locus (). In the hmMTHFR group, 7/10 (70%) semen DNA samples produced methylated and unmethylated bands: 3/5 (60%) from hmMTHFR-nor (1 with Taq I, 1 with Mlu I, and 1 with both Taq I and Mlu I enzymes) and 4/5 (80%) from hmMTHFR-abn (2 with Taq I, and 2 with both Taq I and Mlu I enzymes) ( and ).
Figure 1. Methylation status of the H19 imprinted gene in hmMTHFR and MTHFR semen DNA samples from infertile males. (A)Genomic structure of H19 locus and the GenBank accession number. Upper line: filled-in boxes and horizontal arrow indicate gene exons and orientation, respectively; filled-in horizontal arrows represent H19 DMR region and the white box represents the DMR sequence which was analyzed in this study. Lower line: DMR sequence includes 18 CpG islands (the CTCF-binding site 6 region from 4 to 8 CpG island is also shown). The horizontal arrows represent the primers. Vertical arrowheads indicate the unique bisulfite-PCR restriction enzyme sites, which were analyzed with Mlu I and Taq I. (B) Overall methylation status of H19 detected by COBRA assay in sperm from hmMTHFR and MTHFR semen DNA samples and in the control leukocyte DNA. The same bisulfite-treated DNA amplified by PCR and used for cloning and sequencing, was digested with Mlu I and Taq I restriction enzymes, which were cut only if the restriction site was methylated. Cases 2 and 3 from hmMTHFR semen samples show the unmethylated band with both Mlu I and Taq I enzyme digestion. MW, Molecular weight; C+, human leukocyte DNA.
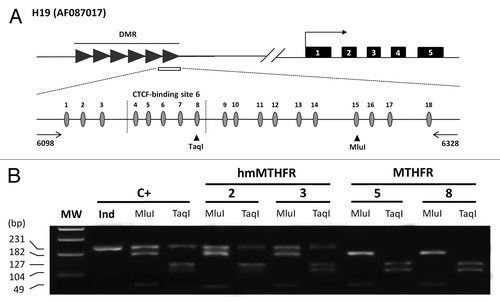
Table 1. COBRA analysis of H19 PCR products
Methylation status of H19 locus in hmMTHFR and MTHFR semen DNA samples
The methylation status of the H19 locus was investigated by sequencing analysis of the cloned PCR products. We reasoned that spermatozoa carrying MTHFR gene dysfunctions contained extensive methylation defects at the H19 locus, so that only H19 hypomethylated clones, i.e., clones showing 50% or more than 50% unmethylated CpG islands, were taken into account in this analysis.
A total of 200 clones were investigated for the DNA methylation status of H19 locus in sperm DNA samples from infertile males: 100 clones from the hmMTHFR group and 100 from the MTHFR group. We examined a total of 18 CpG sites in a 231 bp fragment of the H19 gene ().
In each semen sample, clones with H19 hypomethylation and complete methylation were analyzed ().
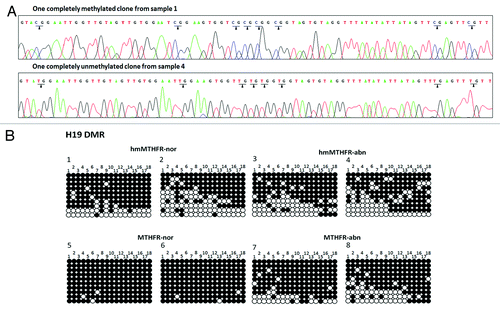
Figure 2. Sequencing analyses of clones from hmMTHFR and MTHFR semen samples. (A) DNA sequencing of H19 in two different clones hmMTHFR-nor (sample 1) and hmMTHFR-abn (sample 4). Arrowheads indicate the methylated CpG islands in sample 1 and unmethylated CpG islands in sample 4. (B) Bisulfite-PCR sequencing for H19 in two representative semen samples endowed with the lowest and the highest prevalence of H19 hypomethylated clones, respectively, from hmMTHFR-nor and hmMTHFR-abn, as well as MTHFR-nor and hmMTHFR-abn semen samples. Filled-in and clear circles represent methylated and unmethylated CpG islands, respectively. The 18 CpG islands within the H19 locus are numbered on the upper side of the circles.
During the first step, the overall distribution of the different clones within the hmMTHFR group and the MTHFR group was evaluated. H19 hypomethylation was detected in 24% of the clones from the hmMTHFR group and in 8% of the clones from the MTHFR group (p = 0.0038) (). Complete H19 methylation was found in 14% of the clones from the hmMTHFR group and in 45% of the clones in the MTHFR group and (p = 0.0001) ().
Figure 3. Frequency of H19 hypomethylated and completely methylated clones. (A) Frequency of hypomethylated (gray bars) and completely methylated (white bars) clones of H19 in hmMTHFR and MTHFR groups. * H19 frequency significantly different from MTHFR group; ∆H19 frequency significantly different from MTHFR group. (B) Frequency of H19 hypomethylated clones in hmMTHFR-nor and MTHFR-nor groups and in hmMTHFR-abn and MTHFR-abn groups. * H19 frequency significantly different from MTHFR-nor group; ∆H19 frequency significantly different from MTHFR-nor group; (C) Frequency of H19 completely methylated clones in hmMTHFR-nor and MTHFR-nor groups and in hmMTHFR-abn and MTHFR-abn groups. * H19 frequency significantly different from MTHFR-nor and hmMTHFR-abn groups; ∆H19 frequency significantly different from MTHFR-abn group.
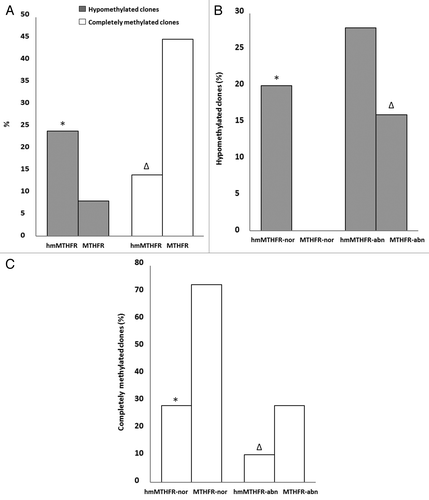
In the second step of our analysis, the methylation status of H19 gene was investigated in hmMTHFR-nor and hmMTHFR-abn semen samples as well as MTHFR-nor and MTHFR-abn semen samples. The prevalence of H19 hypomethylated clones was 20% in hmMTHFR-nor compared with 0% in MTHFR-nor semen samples (p = 0.0027) and was 28% in hmMTHFR-abn compared with 16% in MTHFR-abn semen samples (p = 0.2274) (). The frequency of completely methylated H19 clones was 28% in hmMTHFR-nor compared with 72% in MTHFR-nor semen samples (p < 0.0001) and was 10% in hmMTHFR-abn compared with 28% in MTHFR-abn semen samples (p < 0.0001).
Comparative statistical analyses between hmMTHFR-nor and hmMTHFR-abn, as well as MTHFR-nor and MTHFR-abn are reported in .
Methylation status of CTCF-binding site 6
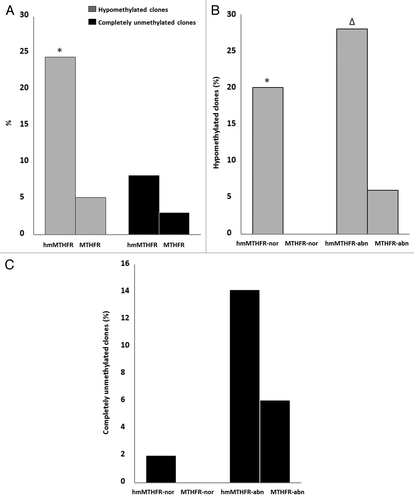
The CTCF-binding site 6 includes 5 CpG islands, from 4 to 8 CpG, within the H19 locus. Hypomethylation (i.e., almost 3 unmethylated CpG islands) and complete non-methylation of the CTCF-binding site 6 region of H19 locus was evaluated in each single clone from the hmMTHFR and MTHFR group. In the hmMTHFR group and MTHFR group, the CTCF-binding site 6 region was hypomethylated in 24% and 5% of the clones, respectively (p = 0.0002), whereas the CTCF-binding site 6 was completely unmethylated in 8% of the clones from the hmMTHFR group and in 3% of the clones from the MTHFR group (p = 0.2134).
In hmMTHFR-nor and in MTHFR-nor semen samples, the prevalence of clones with hypomethylated CTCF-binding site 6 was 20% and 0% (p = 0.0027), respectively, whereas the frequency of completely unmethylated clones was 2% in hmMTHFR-nor and 0% in MTHFR-nor semen samples (p = 0.3149). In hmMTHFR-abn and MTHFR-abn semen samples, hypomethylation occurred in 28% and in 6% of the clones, respectively (p = 0.0078), whereas completely unmethylated clones were found in 14% of hmMTHFR-abn and in 6% of MTHFR-abn semen samples (p = 0.3173) ().
Figure 4. Frequency of clones with hypomethylation and complete unmethylation of CTCF-binding site 6. (A) Frequency of clones with hypomethylation (gray bars) and complete unmethylation (black bars) of CTCF binding site 6 in hmMTHFR and MTHFR groups. *The frequency of clones with hypomethylation of CTCF binding site 6 is significantly different from MTHFR group; (B) Frequency of clones with hypomethylation of CTCF binding site 6 in hmMTHFR-nor and MTHFR-nor groups and in hmMTHFR-abn and MTHFR-abn groups. * The frequency of clones with hypomethylation of CTCF binding site 6 is significantly different from MTHFR-nor group; ∆ The frequency of clones with hypomethylation of CTCF binding site 6 is significantly different from MTHFR-abn group; (C) Frequency of clones with complete unmethylation of CTCF binding site 6 in hmMTHFR-nor and MTHFR-nor groups and in hmMTHFR-abn and MTHFR-abn groups.
Discussion
Methylation loss at the H19 imprinted gene has been detected in semen samples from infertile males suggesting that H19 imprinting errors represent a risk factor for male infertility.Citation13-Citation16,Citation19 The causes inducing aberrant methylation at H19 are poorly understood.
This study shows for the first time that extensive methylation defects at the H19 imprinted gene occur in sperm DNA of infertile males harboring hypermethylation of MTHFR gene promoter. The methylation status of H19 locus was investigated in two different groups of sperm DNA samples, one with hypermethylated MTHFR gene promoter, the hmMTHFR group, and one without promoter methylation of the MTHFR gene, the MTHFR group.
The prevalence of H19 hypomethylated clones was higher in the hmMTHFR group than in the MTHFR group (p < 0.05). On the contrary, the prevalence of completely methylated H19 clones was higher in the MTHFR group than in the hmMTHFR group (p < 0.05). These results indicate that methylation defects at the H19 locus frequently occur in semen samples harboring hypermethylation at the MTHFR gene promoter.
Previous data has indicated that H19 methylation loss results in abnormal spermatogenesis.Citation13-Citation16 Complete demethylation or a low level of methylation at the H19 locus have been detected in semen samples with abnormal semen parameters such as oligozoospermia, astenozoospermia and teratozoospermia, suggesting that aberrant methylation of this gene may represent a risk factor for male infertility.Citation13-Citation16 In this study, the hmMTHFR and MTHFR group included semen samples with normal and abnormal semen parameters, thus allowing the prevalence of H19 hypomethylated clones to be determined in normal and abnormal sperm. In accordance with previous studies,Citation13-Citation16 the prevalence of H19 hypomethylated clones was higher in MTHFR-abn than in MTHFR-nor semen samples (p < 0.05). On the contrary, the frequency of H19 hypomethylated clones was approximately equal in hmMTHFR-abn (28%) and in hmMTHFR-nor (20%) semen samples (p > 0.05). To our knowledge, this is the first study showing extensive methylation defects, i.e., hypomethylated clones, at H19 locus in normal sperm samples from infertile males. While some previous studies have reported methylation errors at the H19 locus in normal sperm, these errors usually involved only a few CpG sites within the H19 gene. Furthermore, comparative analyses of the prevalence of H19 hypomethylated clones between semen samples from the hmMTHFR group and MTHFR group showed that H19 hypomethylated clones occurred only in hmMTHFR-nor when compared with MTHFR-nor semen samples (p < 0.05) and equally in hmMTHFR-abn compared with MTHFR-abn semen samples (p > 0.05). These results clearly underscore the association between H19 methylation defects and the hypermethylation of the MTHFR gene promoter in normal semen samples, suggesting that the methylation status of H19 gene in normal sperm could be affected by MTHFR gene dysregulation. Nevertheless, despite statistical analysis between hmMTHFR-abn and MTHFR-abn semen samples being non-significant, probably due to the low sample size used in this study, we cannot exclude that imprinting defects of H19, even in abnormal sperm, may be due to MTHFR gene dysfunction since prevalence of H19 hypomethylated clones in hmMTHFR-abn was almost 2-fold greater than in MTHFR-abn semen samples. Finally, the detection of H19 hypomethylated clones in MTHFR-abn semen samples indicates that other different mechanisms are involved in H19 methylation defects in abnormal sperm as previously suggested.Citation20-Citation25
Infertility is a reproductive health problem that affects approximately 15% of couples. Half of these cases are due to male factors,Citation26 and about 30% of male infertility cases are idiopathic, since the molecular mechanisms underlying the defects remain unknown.Citation27 In these patients, sperm with normal semen parameters is frequently observed. Our results indicate that normal sperm harbouring dysfunctions of the MTHFR gene, hmMTHFR-nor, is affected by wide-ranging methylation defects at the H19 locus. Moreover, we verified that most of these methylation defects occurred within the CTCF-binding site 6 region which was hypomethylated or completely unmethylated in 7% and 23% of the clones, respectively. As is already known, CTCF-binding site methylation allows the IGF2 maternal allele to be expressed which is required for normal pre-implantation of the embryo as well as fetal growth and survival.Citation28-Citation30 Therefore, spermatozoa with abnormal methylation of CTCF binding site 6 have a high risk of causing biallelic inactivation of the IGF2 gene in the human embryo, which could lead to negative effects on embryo development and pregnancy outcome.
Consistently, hypomethylation or complete unmethylation at the CTCF-binding site 6 region has been previously reported in semen samples from normozoospermic males in infertile couples affected by RSA.Citation31 Interestingly, in our previous study we detected that hypermethylation of the MTHFR gene frequently occurred in normal semen samples from RSA males compared with controls. These data strongly support the hypothesis that one possible mechanism involved in some cases of idiopathic male infertility is the lack of MTHFR gene activity leading to unmethylation of the CTCF-binding site which in turn affects the fertilizing potential of normal sperm. In addition, low levels of methyl donor pools in spermatogenetic cells may also compromise the methylation status of some other paternal imprinted genes. In this context, we speculate that spermatozoa with imprinting errors could be selectively discarded and may not be able to fertilize oocytes or give rise to embryos that would fail to develop normally, as previously suggested by studies performed in mice using azacytidine to induce hypomethylation in sperm.Citation32
In conclusion, this study has shown that extensive methylation defects at the H19 imprinted gene occur in semen samples from infertile males harboring hypermethylation of the MTHFR gene. This association was strongly evident in semen samples with normal semen parameters suggesting that the methylation status of the H19 gene in normal sperm may be affected by dysfunctions of the MTHFR gene. These findings provide new insights into the mechanisms causing abnormal methylation at imprinted genes and, in turn, in some cases of idiopathic male infertility.
Material and Methods
Semen samples
The 20 semen samples used in this study had been collected from males (men aged 36.1 ± 1.2) who were in couples reporting fertility problems. DNA from these 20 semen samples had been previously analyzed for the methylation status of MTHFR gene promoter:Citation17 10 semen DNA samples had MTHFR promoter hypermethylation (i.e., nearly 100% of CpG islands were methylated within the promoter region), hereafter indicated as hmMTHFR group, and 10 semen DNA samples had MTHFR promoter region unmethylated hereafter indicated as MTHFR group. In both the hmMTHFR and MTHFR group, 5 semen samples were with normal semen parameters (≥ 15 × 106 sperm/ml; ≥ 32% sperm rapid progressive motility and ≥ 4% normal sperm morphology), hmMTHFR-nor and MTHFR-nor, and 5 semen samples were with abnormal semen parameters, hmMTHFR-abn (1 oligoastenozoospermic, 1 teratozoospermic, 2 astenozoospermic and 1 oligozoospermic), and MTHFR-abn (1 oligoteratozoospermic, 1 teratozoospermic, 1 astenozoospermic and 2 oligozoospermic).
Bisulfite treated DNA PCR of H19 locus
The methylation assay was performed at the DMR of the H19 imprinted gene. 150 ng of sodium bisulfite-treated sperm DNA was amplified at the H19 locus by hemi-nested Methylation-Specific PCR using the primers F6005, 5′-AGGTGTTTTAGTTTTATGGATGATGG-3′, R6326, 5′-TCCTATAAATATCCTATTCCCAAATAACC-3′ and F6115, 5′-TGTATAGTATATGGGTATTTTTGGAGGTTT-3′, which amplify a sequence of 231 bp (GenBank accession number AF087017, position 6098–6328) containing 18 CpG islands (). The PCR program was: 10 min of denaturation at 94°C followed by 35 cycles of 30s at 94°C, 30s at 60°C and 30s at 72°C and a final extension for 5 min at 72°C.Citation10,Citation15
The H19 amplified sequence contains the CTCF-binding site 6 region, which includes 5 CpG islands, from 4 to 8 CpG island ().
Combine Bisulfite Restriction Analysis (COBRA)
To confirm that sequencing results did not reflect a cloning bias, bisulfite-treated DNA PCR samples from sperm cells and human leukocytes (positive control of the reaction) were subjected to restriction analysis (COBRA) with the Taq I and Mlu I enzymes.Citation13,Citation19 These enzymes can only cleave to methylated DNA sequences so that the undigested and digested products indicated unmethylated and methylated templates, respectively. Digestion products were visualized by DNA electrophoresis on a 2.5% agarose gel.
DNA cloning and sequencing
Amplified products were purified with the QIAquick PCR Purification Kit (Qiagen) and then cloned with the TOPO TA cloning kit (Invitrogen), using the Turbo Competent E. coli bacteria strain (EuroClone) and the pCR 2.1-TOPO vector (Invitrogen), according to manufacturer’s instructions. Selection of bacterial clones containing the fragment of interest was performed using selective LB growth medium with ampicillin (100 μg/ml, Sigma-Aldrich). For each DNA sample, 10 positive clones were selected for sequencing analysis. Single clones were sequenced using automated ABI Prism Genetic (Analyzer Applied Biosystems).
Statistical analysis
Chi-square trend test was performed with Yates' correction to analyze the association between methylation patterns of MTHFR and H19. P-values < 0.05 were considered statistically significant.
| Abbreviations: | ||
| MTHFR | = | Methylenetetrahydrofolate reductase |
| DMR | = | differentially methylated regions |
| Combine Bisulfite Restriction Analysis | = | COBRA |
Acknowledgments
Supported in part by grants from the Fondazione Cassa di Risparmio di Cento, Cento; FAR projects, University of Ferrara, Ferrara.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Koerner MV, Barlow DP. Genomic imprinting-an epigenetic gene-regulatory model. Curr Opin Genet Dev 2010; 20:164 - 70; http://dx.doi.org/10.1016/j.gde.2010.01.009; PMID: 20153958
- Georgiades P, Watkins M, Burton GJ, Ferguson-Smith AC. Roles for genomic imprinting and the zygotic genome in placental development. Proc Natl Acad Sci U S A 2001; 98:4522 - 7; http://dx.doi.org/10.1073/pnas.081540898; PMID: 11274372
- Piedrahita JA. The role of imprinted genes in fetal growth abnormalities. Birth Defects Res A Clin Mol Teratol 2011; 91:682 - 92; http://dx.doi.org/10.1002/bdra.20795; PMID: 21648055
- Serman L, Vlahović M, Sijan M, Bulić-Jakus F, Serman A, Sincić N, et al. The impact of 5-azacytidine on placental weight, glycoprotein pattern and proliferating cell nuclear antigen expression in rat placenta. Placenta 2007; 28:803 - 11; http://dx.doi.org/10.1016/j.placenta.2007.04.001; PMID: 17509679
- Diplas AI, Lambertini L, Lee MJ, Sperling R, Lee YL, Wetmur J, et al. Differential expression of imprinted genes in normal and IUGR human placentas. Epigenetics 2009; 4:235 - 40; PMID: 19483473
- Lim AL, Ferguson-Smith AC. Genomic imprinting effects in a compromised in utero environment: implications for a healthy pregnancy. Semin Cell Dev Biol 2010; 21:201 - 8; http://dx.doi.org/10.1016/j.semcdb.2009.10.008; PMID: 19879952
- Dória S, Sousa M, Fernandes S, Ramalho C, Brandão O, Matias A, et al. Gene expression pattern of IGF2, PHLDA2, PEG10 and CDKN1C imprinted genes in spontaneous miscarriages or fetal deaths. Epigenetics 2010; 5:444 - 50; http://dx.doi.org/10.4161/epi.5.5.12118; PMID: 20484977
- Biniszkiewicz D, Gribnau J, Ramsahoye B, Gaudet F, Eggan K, Humpherys D, et al. Dnmt1 overexpression causes genomic hypermethylation, loss of imprinting, and embryonic lethality. Mol Cell Biol 2002; 22:2124 - 35; http://dx.doi.org/10.1128/MCB.22.7.2124-2135.2002; PMID: 11884600
- Biermann K, Steger K. Epigenetics in male germ cells. J Androl 2007; 28:466 - 80; http://dx.doi.org/10.2164/jandrol.106.002048; PMID: 17287457
- Kerjean A, Dupont JM, Vasseur C, Le Tessier D, Cuisset L, Pàldi A, et al. Establishment of the paternal methylation imprint of the human H19 and MEST/PEG1 genes during spermatogenesis. Hum Mol Genet 2000; 9:2183 - 7; http://dx.doi.org/10.1093/hmg/9.14.2183; PMID: 10958657
- Li JY, Lees-Murdock DJ, Xu GL, Walsh CP. Timing of establishment of paternal methylation imprints in the mouse. Genomics 2004; 84:952 - 60; http://dx.doi.org/10.1016/j.ygeno.2004.08.012; PMID: 15533712
- Arney KL. H19 and Igf2--enhancing the confusion?. Trends Genet 2003; 19:17 - 23; http://dx.doi.org/10.1016/S0168-9525(02)00004-5; PMID: 12493244
- Kobayashi H, Sato A, Otsu E, Hiura H, Tomatsu C, Utsunomiya T, et al. Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum Mol Genet 2007; 16:2542 - 51; http://dx.doi.org/10.1093/hmg/ddm187; PMID: 17636251
- Marques CJ, Carvalho F, Sousa M, Barros A. Genomic imprinting in disruptive spermatogenesis. Lancet 2004; 363:1700 - 2; http://dx.doi.org/10.1016/S0140-6736(04)16256-9; PMID: 15158633
- Marques CJ, Costa P, Vaz B, Carvalho F, Fernandes S, Barros A, et al. Abnormal methylation of imprinted genes in human sperm is associated with oligozoospermia. Mol Hum Reprod 2008; 14:67 - 74; http://dx.doi.org/10.1093/molehr/gam093; PMID: 18178607
- Boissonnas CC, Abdalaoui HE, Haelewyn V, Fauque P, Dupont JM, Gut I, et al. Specific epigenetic alterations of IGF2-H19 locus in spermatozoa from infertile men. Eur J Hum Genet 2010; 18:73 - 80; http://dx.doi.org/10.1038/ejhg.2009.117; PMID: 19584898
- Rotondo JC, Bosi S, Bazzan E, Di Domenico M, De Mattei M, Selvatici R, et al. Methylenetetrahydrofolate reductase gene promoter hypermethylation in semen samples of infertile couples correlates with recurrent spontaneous abortion. Hum Reprod 2012; 27:3632 - 8; http://dx.doi.org/10.1093/humrep/des319; PMID: 23010533
- Trimmer EE. Methylenetetrahydrofolate reductase: biochemical characterization and medical significance. Curr Pharm Des 2013; 19:2574 - 93; http://dx.doi.org/10.2174/1381612811319140008; PMID: 23116396
- Kobayashi H, Hiura H, John RM, Sato A, Otsu E, Kobayashi N, et al. DNA methylation errors at imprinted loci after assisted conception originate in the parental sperm. Eur J Hum Genet 2009; 17:1582 - 91; http://dx.doi.org/10.1038/ejhg.2009.68; PMID: 19471309
- Bourc’his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 2004; 431:96 - 9; http://dx.doi.org/10.1038/nature02886; PMID: 15318244
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 2004; 429:900 - 3; http://dx.doi.org/10.1038/nature02633; PMID: 15215868
- Yaman R, Grandjean V. Timing of entry of meiosis depends on a mark generated by DNA methyltransferase 3a in testis. Mol Reprod Dev 2006; 73:390 - 7; http://dx.doi.org/10.1002/mrd.20430; PMID: 16362968
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69:915 - 26; http://dx.doi.org/10.1016/0092-8674(92)90611-F; PMID: 1606615
- Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005; 308:1466 - 9; http://dx.doi.org/10.1126/science.1108190; PMID: 15933200
- Stouder C, Paoloni-Giacobino A. Transgenerational effects of the endocrine disruptor vinclozolin on the methylation pattern of imprinted genes in the mouse sperm. Reproduction 2010; 139:373 - 9; http://dx.doi.org/10.1530/REP-09-0340; PMID: 19887539
- de Kretser DM. Male infertility. Lancet 1997; 349:787 - 90; http://dx.doi.org/10.1016/S0140-6736(96)08341-9; PMID: 9074589
- Filipponi D, Feil R. Perturbation of genomic imprinting in oligozoospermia. Epigenetics 2009; 4:27 - 30; http://dx.doi.org/10.4161/epi.4.1.7311; PMID: 19106644
- Fedoriw AM, Stein P, Svoboda P, Schultz RM, Bartolomei MS. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science 2004; 303:238 - 40; http://dx.doi.org/10.1126/science.1090934; PMID: 14716017
- Rand E, Ben-Porath I, Keshet I, Cedar H. CTCF elements direct allele-specific undermethylation at the imprinted H19 locus. Curr Biol 2004; 14:1007 - 12; http://dx.doi.org/10.1016/j.cub.2004.05.041; PMID: 15182675
- Olek A, Walter J. The pre-implantation ontogeny of the H19 methylation imprint. Nat Genet 1997; 17:275 - 6; http://dx.doi.org/10.1038/ng1197-275; PMID: 9354788
- Ankolkar M, Patil A, Warke H, Salvi V, Kedia Mokashi N, Pathak S, et al. Methylation analysis of idiopathic recurrent spontaneous miscarriage cases reveals aberrant imprinting at H19 ICR in normozoospermic individuals. Fertil Steril 2012; 98:1186 - 92; http://dx.doi.org/10.1016/j.fertnstert.2012.07.1143; PMID: 22959455
- Oakes CC, Kelly TL, Robaire B, Trasler JM. Adverse effects of 5-aza-2′-deoxycytidine on spermatogenesis include reduced sperm function and selective inhibition of de novo DNA methylation. J Pharmacol Exp Ther 2007; 322:1171 - 80; http://dx.doi.org/10.1124/jpet.107.121699; PMID: 17581917