Abstract
Parkinson disease (PD) is a multifactorial neurodegenerative disorder with high incidence in the elderly, where environmental and genetic factors are involved in etiology. In addition, epigenetic mechanisms, including deregulation of DNA methylation have been recently associated to PD. As accurate diagnosis cannot be achieved pre-mortem, identification of early pathological changes is crucial to enable therapeutic interventions before major neuropathological damage occurs. Here we investigated genome-wide DNA methylation in brain and blood samples from PD patients and observed a distinctive pattern of methylation involving many genes previously associated to PD, therefore supporting the role of epigenetic alterations as a molecular mechanism in neurodegeneration. Importantly, we identified concordant methylation alterations in brain and blood, suggesting that blood might hold promise as a surrogate for brain tissue to detect DNA methylation in PD and as a source for biomarker discovery.
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disorder in the elderly affecting 2% of the population over 60 y old.Citation1 The classic form of PD is manifested clinically as a slowly progressive movement disorder with resting tremor and postural instability. Neuropathologically, PD is characterized by the formation of intracytoplasmic inclusions known as Lewy bodies, containing α-synuclein (α-syn)Citation2,Citation3 and by the loss of dopaminergic neurons mainly in the substantia nigra pars compacta, although neurodegeneration has been also reported to occur in the cortex and other brain areas, and these alterations correlate with non-motor symptoms. At the molecular level, accumulation of misfolded α-syn affects mitochondrial function, autophagyCitation4 and the expression of several functional groups of genes,Citation5,Citation6 although the molecular basis of transcriptional deregulation remain elusive.
Epigenetic processes control several neurobiological and cognitive functions, from early brain development and neurogenesisCitation7 to memory formation, learning and synaptic plasticity.Citation8 Altered epigenetic mechanisms have also been associated with neurological disorders, including Rett syndrome, autism, schizophrenia and Alzheimer, Huntington and Parkinson diseases.Citation9 We recently reported a decay on DNA methylation in the brain of PD subjects, associated with the interaction of α-syn with DNA methyltransferase 1 (DNMT1) that results in sequestration of DNMT1 in the cytoplasm.Citation10 Moreover, other studies showed that decreased methylation at SNCA intron 1 might contribute to deregulation of α-syn expression in sporadic PD cases,Citation11,Citation12 highlighting the involvement of aberrant epigenetic mechanisms in PD pathology.
PD is a multifactorial disease where environmental and genetic factors are intricately associated. Since accurate diagnosis cannot be achieved until clear motor features have developed, which occurs when 50% or more of pigmented neurons in the substantia nigra have been lost, increasing efforts are dedicated to identify early prodromal non-motor symptoms, including autonomic disturbances, olfactory dysfunctions, depression and sleep disorders that may contribute to the precocious diagnosis of PD and to enable therapeutic interventions before major neuropathological damage has occurred. Biomarkers, molecules that represent the particular signature of a pathological process and that can be easily accessed and quantified, represent a new diagnostic tool to monitor disease progression and therapy efficacy.Citation13 DNA methylation is currently used as a successful biomarker in several cancer types,Citation14–Citation16 and represents a highly promising biomarker for neurodegenerative disorders.Citation17,Citation18 As emerging techniques allow monitoring DNA methylation from easily accessible peripheral tissues like blood, an important question rises whether DNA methylation changes from peripheral blood leukocytes (PBLs) would correlate with the brain methylome. We investigated genome-wide DNA methylation changes in brain and blood samples from PD patients in comparison to control subjects. We report here differential methylation for several genes previously associated with PD pathology, supporting the role of epigenetic deregulation as a molecular pathological mechanism in neurodegeneration. Importantly, we identified concordant methylation alterations in a subset of genes in PD brain and blood, suggesting that PBLs might represent a good proxy for brain methylation alterations associated with PD and might constitute a new source for biomarker discovery and development.
Results and Discussion
We previously reported a significant decrease in DNA methylation in the frontal cortex of patients with Parkinson disease and the related disorder Dementia with Lewy bodies, associated with the retention of DNMT1 in the cytoplasm.Citation10 In the present study we further investigated the extent of this epigenetic deregulation by analyzing genome-wide DNA methylation profiles in postmortem frontal cortex samples and peripheral blood leukocytes (PBLs) from the same individuals on a cohort of PD patients (n = 5) in comparison to age-matched healthy control subjects (n = 6). The cases studied did not differ significantly regarding age, brain weight, postmortem interval and years of education. Most PD cases presented low scores on the Mini-Mental State Examination (MMSE) suggestive of cognitive impairment and probable cortical involvement in pathology (Table S1A). Although the most striking pathological feature of PD is the loss of dopaminergic neurons in the substantia nigra, neurodegeneration also occurs in the cortex,Citation19 and variation of the regional volume of frontal cortex is a characteristic of PD manifestations with cognitive impairment,Citation20,Citation21 underscoring the involvement of this brain structure in PD pathology.
DNA methylation profiles in brain and blood samples from Parkinson disease patients
Methylome analysis was performed using the Infinium Human 450K beadchip and GenomeStudio (both from Illumina). Detection levels were similar among groups, with an average of 485,386 CpG detected at p < 0.01. We first compared the methylation profiles between brain and blood to investigate if PBLs could represent a good source to profile methylation changes associated with PD. We detected 2,908 CpG with differential methylation (DM) in the brain and 3,897 CpG in the blood of PD cases, representing about 0.6% and 0.8% of the total array probes interrogated, respectively (). A few recent reports raised some concern about the use of whole blood DNA on methylation studies because the heterogeneity of the white cell types, each having their own particular epigenome, might confound DNA methylation measurements and disease associations.Citation22,Citation23 Even though, we observed that the overall methylation patterns of brain and blood were similar, with more than 80% of the sites reported as DM being hypomethylated, in agreement with our previous findings ().Citation10 In addition, distribution of average β values across samples and tissues showed similar profiles with most CpG clustering in low-methylation (LMF, β values < 20%) and high-methylation fractions (HMF, β values > 80%) in both, brain and blood (). However, while there were no significant differences between brain and blood in the LMF, there were more CpG in the HMF in PD blood in comparison to control subjects’ blood and also to PD brains (One-way ANOVA corrected for multiple observations and Bonferroni post-hoc test, p < 0.05 and p < 0.001 respectively). Consistent with β-value distribution and previous reports,Citation24 CpG neighborhood context analysis and genomic location distribution was comparable between brain and blood samples, and showed that loci with decreased methylation were more likely to locate at CG islands (CGi) and associated with promoter regions including TSS1500, TSS200 and 1st exon sites; while CpG sites located farther away from islands (open sea) and at the gene bodies were more likely to present increased methylation (). Finally, analysis of annotated genes presenting DM in PD samples regarding Gene Ontology using Panther Classification System (www.pantherdb.org)Citation25 and clustering by Biological Process, also showed that the same functional groups were affected in brain and blood, with cell communication and cellular and metabolic processes being the more populated clusters (), and including genes related to apoptosis, a molecular pathway largely implicated in PD.Citation26
Table 1. Distribution of differentially methylated regions in brain and blood DNA from Parkinson disease patients
Figure 1. DNA methylation profiles of brain and blood in Parkinson disease cases. (A) Comparison of the fractions of differentially methylated loci that showed gain or loss of methylation in PD brain and blood. (B) Distribution of average β values across samples and tissues showing enrichment in low-methylation (LMF, β values < 20%) and high-methylation fractions (HMF, β values > 80%). IMF, intermediate methylated fraction (β values < 20% and > 80%). *Significant difference in PD blood in comparison to control subjects’ blood (p < 0.05) and to PD brains (p < 0.001, One-way ANOVA with correction for multiple observations and Bonferroni post-hoc test). (C) CpG neighborhood context analysis in brain and blood samples that showed increased methylation (IM) or decreased methylation (DM) in PD. CG islands Shores are defined as regions up to 2 Kb from the CGi Start or End; Shelves are defined as the next 2 Kb boundaries from CGi shores. (D) Genomic location distribution of differentially methylated loci in PD brain and blood samples. TSS1500, CpG sites within 200–1500 bp from the transcription-starting site (TSS). TSS200, CpG sites within 200 bp from the TSS. Body, gene body regions. (E) Gene ontology analysis of annotated genes showing differential methylation clustering by biological process.

We performed differential methylation analysis applying the Illumina Custom model after normalization to control probes present in the array and using the control subjects group as reference. We selected probes showing absolute Delta β-values > |0.2| at p < 0.01 (with FDR q < 0.001 to control for multiple comparisons) as DM, a threshold previously suggested to improve detection of DM sites in this array platform with 99% confidence.Citation27 We identified 317 probes in brain and 476 in blood with increased methylation while 2591 probes in brain and 3421 in blood showed decreased methylation in PD (). As gender is a known source of variation for DNA methylation in different tissues,Citation28,Citation29 we focused our analysis on autosomal loci, which represented about half of DMRs with increased methylation and about 5% of DM sites with decreased methylation in both, brain (Table S2) and PBLs (Table S3).
Concordant variation of differentially methylated loci in PD brain and blood
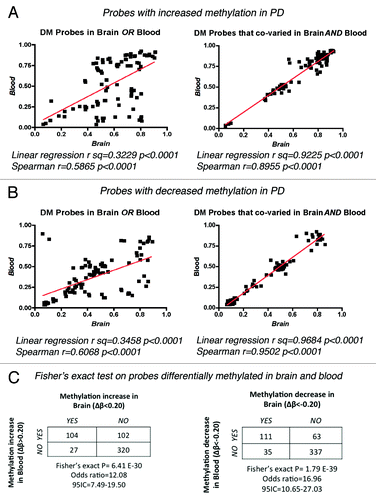
We further refined our analysis to the fraction of autosomal probes showing differential methylation and associated with annotated genes, which corresponded to a total of 174 genes in the brain (84 with increased and 90 with decreased methylation in PD) and 233 annotated genes in blood (127 with increased and 106 with decreased methylation in PD, ). Strikingly, 30% (124/407) of the total autosomal annotated genes with DM presented concordant changes in methylation between brain and blood (63 loci with increased methylation and 61 with decreased methylation respectively, and Table S4). We performed bootstrap simulations to test the probability of selecting the same 124 probes from both pools of differentially methylated loci in PD brain and blood only by chance (Fig. S1). Random sample generation on 10,000 independent iterations, showed only n = 6 common probes that appear in both sets at high frequency, precluding the possibility that we could select 124 genes with concordant methylation alterations by chance. This statistic analysis with a bootstrap approachCitation30 supported the observation for both, genes with increased and decreased methylation. In addition, we analyzed the possible correlation between changes in methylation recorded in brain and blood, and represented by the individual β-values per probe for each PD case studied (). Linear regression analysis and Spearman correlation coefficients calculated for the 20 top probes with DM in only one tissue or co-varying in both, showed a tight correlation of methylation levels for the probes with concordant methylation changes in both, brain and blood. Once more, this observation holds true for the probes with increased () and decreased methylation (). Finally, we tested the probability that the changes in methylation observed in blood were independent from the changes in methylation observed for the same probes in the brain by performing a Fisher’s exact probability test on DM probes that showed increased or decreased methylation in PD brain and/or blood (). In both scenarios, the null hypothesis was rejected, supporting a biologically meaningful relationship between the variation of methylation in brain and PBLs. Taken in all, this analyses suggest that a number of methylation changes in PD is shared between brain and blood, positioning these 124 genes that co-varied among tissues as candidates for biomarker discovery.
Figure 2. Correlation of β-values between brain and blood in PD samples. Linear regression and Spearman correlation coefficients were calculated for the methylation values (β) of individual PD samples for the top 20 DM probes that showed increased (A) or decreased (B) methylation in only one tissue or that co-varied in both brain and blood. (C) Fisher’s exact probability test on DM probes testing the null hypothesis that the observed changes in methylation in brain were independent of the changes on the same probes in blood.
In order to determine the potential association of these co-varying DM genes with PD pathology, we mined the 30 top changing loci against curated genomic data repositories using NextBio® Disease Atlas (www.nextbio.com).Citation31 Strikingly, the expression of 73% of DM loci (22/30) was reported deregulated in PD by at least one study, including 4 genes highly associated with PD risk in GWAS (HLA-DQA1, GFPT2, MAPT and MIR886; ), suggesting that alterations in DNA methylation might represent a common molecular mechanism underlying transcriptional deregulation and might be highly relevant to PD pathology. Noteworthy, genetic polymorphisms in GSST1, encoding glutathione S-transferase and GSTTP1, involved in conjugation of electrophiles and protection against reactive oxygen species have been associated with increased risk to PD after exposure to Paraquat, a widely used herbicide that we reported recently to alter adult neurogenesis in PD models.Citation32 In addition, the expression of major histocompatibility complex class II-related transcript HLA-DQA1 is altered by overexpression of HDAC1, a histone deacetylase increased in prefrontal cortex of patients with schizophrenia and related psychiatric disorders,Citation33 suggesting that this gene is under a complex epigenetic regulation and its imbalance might trigger cognitive and behavioral pathologies. Furthermore, MIR886 is a highlighted candidate among co-varying genes with decrease methylation, for which more than 8 probes mapping to adjacent CpGs showed significant decay in PD. MIR886 has recently been renamed as Vault RNA 2–1 (vtRNA2–1), precursor of svtRNA2–1a, a small non-coding RNA whose upregulation induces neuronal dysfunction and was reported as an early event in PD pathology.Citation34
Table 2. Differentially methylated autosomal genes that co-varied in brain and blood DNA from PD patients
Despite concerns regarding the use of whole blood as a source for DNA methylation profiling, significant co-variation between brain and blood methylomes was lately reported after weighted correlation network analysis of 2,442 Illumina DNA methylation arrays.Citation35 This meta-analysis identified an age-related co-methylation module that can be found in diverse independent data sets. Biological evaluation showed that module membership is associated with CGi status and autosomal chromosome location, two features that characterized our co-varying genes. In addition, functional enrichment analysis revealed that the aging-related consensus module includes genes involved in nervous system development, neuron differentiation and neurogenesis and also genes known to be downregulated in Alzheimer disease. We identified 10 genes from our co-varying group among the top 1000 members of the aging-related methylation module. Importantly, all of them were associated to PD at NextBio Disease Atlas: SLC12A5,Citation36–Citation38 ABCA3,Citation39 FHIT,Citation36 FAT1,Citation38 CPLX2,Citation36–Citation38,Citation40,Citation41 APBA1,Citation37,Citation38,Citation41 MAGI2,Citation36,Citation38,Citation42,Citation43 CNTNAP2,Citation38,Citation39 ATP8A2,Citation36–Citation40,Citation44 and SMOC2,Citation38 reinforcing the idea that detection of differential methylation events pertinent to PD pathology is feasible from blood samples, and also rising the possibility of common risk factors for PD and age-associated brain changes mediated by methylation of select genes.
We further validated the findings from the Infinium methylation array by an independent technique. We performed methylation-sensitive restriction/qPCR on brain and blood samples on 5 genes from the co-varying group for which Epitect Methyl II assays (Qiagen) were available to match the CGi covered by array probes. We included MRI1 and TMEM9 as candidates with increased methylation () and GSST1, TUBA3E and KCNH1 as genes with decreased methylation in the qPCR analysis (Fig. S2C–E). Array findings for 80% of tested genes (4/5) were validated by qPCR methods (Fig. S2).
The array platform selected for this study contains multiple probes mapping adjacent to each other and covering different CpGs per annotated gene. We detected probes associated with the same gene and showing opposite changes in methylation for only 6 genes out of 407 genes for which we observed DM in brain and/or blood, including HLA-DRB1, LRKK1, MMEL1, HLA-DQB1, OR12D3 and VAV2 (denoted by an * in Tables S2–4). Although all the probes reached significance in our stringent analysis, still the biological interpretation of the effects of these changes in the context of transcriptional regulation need to be further explored.
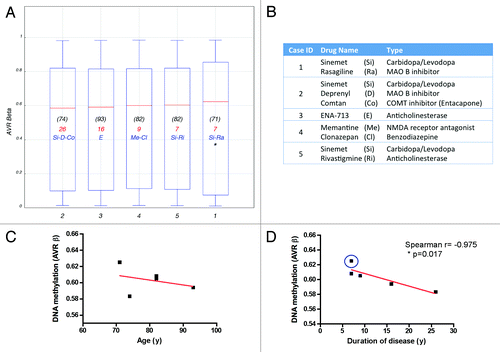
As mentioned before, clear diagnosis of PD is achieved in the clinic when movement symptoms have clearly being established, and patients are usually started in anti-parkinsonian treatments soon after. All of the PD cases included in our cohort received different drug regimens and the duration of disease also varied among cases (Table S1B; ), therefore the question might arise whether the observed epigenetic alterations are due to ongoing PD pathology or rather an effect of medication or age, as the later was associated with DNA methylation decline.Citation45 Almost no systematic investigation was conducted so far on the impact of pharmacological treatments on DNA methylation, in particular regarding levodopa/carbidopa, the most widely used dopamine replacement drugs. We plotted the individual methylation values for each PD patient studied (expressed as the average β-value from all interrogated probes/individual). Methylation profiles did not significantly differed among patients () and no clear association with therapy was evident. Interestingly, higher methylation levels were observed for one patient with a record of cancer, which is known to greatly alter methylationCitation46 suggesting that disease might override epigenetic changes due to age and potentially to drugs. Noteworthy, while average β-values did not correlated with age (), a high Spearman correlation coefficient was observed for duration of disease (), suggesting that methylation changes might be driven by PD pathological processes.
Figure 3. Effects of drug regimen, age and disease duration on DNA methylation. (A) Box plot showing the average β-value of the total probes interrogated per individual PD case. Age at time of death is indicated in (black). Duration of disease, calculated as years elapsed since first PD diagnosis until time of death, is indicated in red. Drug treatment most relevant to PD pathology is indicated in blue. (B) Medication details (additional information is presented in Table S1B). *Denotes a patient with record of cancer. Correlation of DNA methylation (average β-value) with age (C) or disease duration (D) plotted per individual PD subject studied. Blue circle identifies the patient with cancer record. Case ID corresponds with .
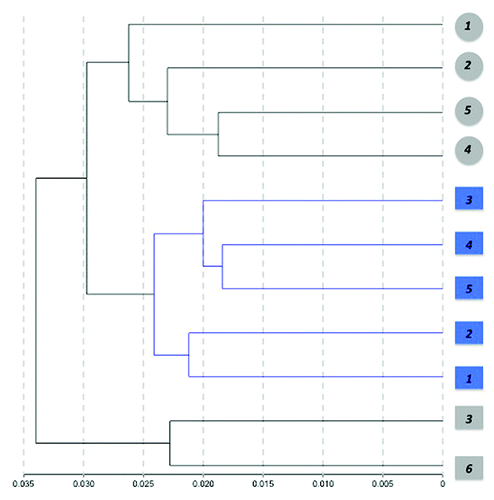
Finally, we performed unsupervised hierarchical clustering of the individual methylation profiles detected from blood using absolute correlation () as a test of the potential capacity of methylation profiles to differentiate between control and diseased subjects. Interestingly, the resulting dendogram showed a clear separation between control and PD cases. Although samples were also clustered by gender, male control subjects were closer to female controls than to PD males. The existence of a distinctive DNA methylation pattern than can separate control from disease subjects was previously reported for the related disorder Dementia with Lewy bodies, where an unsurpevised clustering showed a distinct methylation profile in brain.Citation47
Figure 4. Hierarchical clustering of samples based on individual methylation profiles detected from blood. Dendogram showing the unsupervised hierarchical clustering of the 11 cohort subjects using absolute correlation. Control cases (gray) and PD patients (blue) were grouped based on Pearson correlations of whole genome methylation profiles using a 1 - |r| distance measure and nesting with average linkage method. An average of 485,386 CpG sites/group with detection p < 0.001 were featured in the analysis. Females are represented by circles and males by squares. Numbers represent the individual sample ID.
In conclusion, we report here genome-wide alterations in DNA methylation associated with PD pathology in brain and blood tissues. Although DNA extracted from whole blood represents a complex mixture from different cell types and is highly variable among individuals, we detected similar methylation patterns between both tissues. Importantly, we identified a set of genes whose methylation coordinately varies in brain and blood, with most of these loci previously being implicated in PD pathology. Notably, the individual methylation profiles obtained from blood were able to separate control subjects from PD cases, highly suggesting that peripheral blood leukocytes might represent a good surrogate for brain tissue and opening a new source for biomarker discovery in PD.
Materials and Methods
Samples
All investigations involving human samples have been conducted according to the principles expressed in the Declaration of Helsinki. Approval was obtained from the Institutional Review Board of the University of California San Diego (UCSD), project 111085X. Written informed consent was obtained from all participating subjects, where appropriate.
Human brain samples
Postmortem human brain samples (frontal cortex) were provided by the Alzheimer Disease Research Center at UCSD and the Layton Aging and Alzheimer Research Center from Oregon Health and Science University from patients diagnosed with Parkinson disease (PD, n = 5) and non-demented age-matched control subjects (CT, n = 6). All cases were clinically characterized during life and histopathologically post mortem. Cases with a history of trauma, hemorrhage or infarction were excluded from the study. Post mortem intervals were no longer than 8 h. Genomic DNA was extracted from 25 mg of brain tissue with DNeasy Blood and Tissue kit (Qiagen, # 69504). Genomic DNA isolated from peripheral blood leukocytes (PBLs) was directly obtained from the brain repositories.
DNA methylation microarray
Briefly, 500 ng genomic DNA were converted by bisulfite treatment using the EZ DNA Methylation kit (Zymogen, # D5001) according to the manufacturer’s instructions. Genome-wide methylation profiling was performed on 200 ng of bisulfite-treated DNA using the Illumina Infinium HumanMethylation 450k BeadChip (Illumina, # WG-314-1003) at the Case Western Reserve University Department of Genetics and Genome Sciences Genomics Core, following standardized protocols at the facility and according to the manufacturer's instructions. Beadchips were scanned on an Illumina HiScan SQ. The methylation status of a specific CpG site was expressed as β values, calculated as the ratio of the fluorescence intensity signals of the methylated (M) and unmethylated (U) alleles, β = Max(M,0)/[Max(M,0) + Max(U,0) + 100]. β values range between 0 (non-methylated) and 1 (completely methylated). The Illumina GenomeStudio Software (version 2011.1) was used to assess quality and extract the DNA methylation signals from scanned arrays. Methylation data was extracted as raw signals with no background subtraction and data was normalized to control probes present on the array. An average of 485 386 probes were detected per sample at p < 0.001 and were incorporated into downstream analysis. Differential methylation analysis was performed using Illumina custom model and threshold for CpG selection was Delta β scores > ∣0.2∣ at p < 0.01 and FDR q < 0.001.
Methylation-sensitive restriction qPCR
To independently validate array findings, we performed methylation-sensitive restriction qPCR analysis using EpiTect Methyl II PCR assays (Sa Biosciences/Qiagen). The method is based on the detection of remaining input DNA after cleavage with methylation-sensitive and methylation-dependent restriction enzymes. Following digestion, the remaining DNA is quantified by real-time PCR in each individual enzyme reaction using primers that flank a CpG region of interest that comprise the restriction sites. We performed digestions on 400 ng of brain and PBL genomic DNA using EpiTect Methyl II DNA restriction kit (Qiagen, # 335452) as indicated in the manufacturer’s protocol. Digested DNA was used as template for qPCR Assay using RT2 SYBR® Green qPCR Mastermixes (Qiagen, # 330509) under standard amplification conditions. Cataloged Epitect II Methyl PCR primers used were GSST1 (EPHS109721-1A); MRI1 (EPHS107138-1A); KCNH1 (EPHS101243-1A), TMEM9 (EPHS101170-1A) and TUBA3E (EPHS108566-1A), all from Qiagen.
Statistical analysis
Statistical analysis was performed using One-way ANOVA corrected for multiple comparisons followed by the Bonferroni’s post hoc test or Student’s t-test (unpaired; two-tailed) with a significance of p < 0.05 (Prism Graph Pad Software) as indicated. Linear regression, Spearman’s correlation and Fisher’s exact probability test were calculated with Prism Graph Pad Software.
Differential methylation analysis
The Illumina custom model was applied, under the assumption that β-values are normally distributed among replicates. False discovery rate was computed according with the method of Benjamini-HochbergCitation48 to correct for multiple comparisons.
Correlation of methylation values between brain and blood
We report linear regression and the Spearman correlation coefficients of β-methylation values in brain and blood for each individual of the PD group. We compared the individual β values of the top 20 probes showing concordant methylation changes in brain and blood and the 20 top probes showing differential methylation in only one tissue (selected as the 10 top probes with DM in brain and the 10 top probes with DM in blood).
We also tested the probability that increased or decreased methylation in blood was independent than increased or decreased methylation of the same probes in the brain by performing a Fisher’s exact probability test on differentially methylated probes showing changes in methylation in PD brain and/or blood.
Bootstrap analysis
We performed bootstrap simulations to test the probability of selecting the same 124 probes from both pools of differentially methylated probes in PD brain and blood only by chance. We independently analyzed probes sets with increased and decreased methylation in PD. We generated a random sample from the 131 probes with increased methylation in brain and from the 206 that gained methylation in PD blood, and computed how many probes were present on both sets. The procedure was repeated 10 000 times. For the 10 000 iterations, most of the bootstrapped samples had a low number of co-varying probes, with n = 6 being the number of probes that reached the highest frequency (p < 0.001). Similar results were obtained when the same procedure was performed for the group of 148 probes showing decreased methylation in brain and 174 probes that lost methylation in PD blood, and testing for the probability of finding the same 61 probes in both sets.
| Abbreviations: | ||
| PD | = | Parkinson disease |
| CpG | = | cytosine-phosphate-guanine dinucleotide |
| DMR | = | differentially methylated regions |
| GCi | = | CpG islands |
Additional material
Download Zip (709 KB)Acknowledgments
We thank Dr Robin Guariglia, Dr Randy Woltjer and Dr Joseph Quinn at the Layton Aging and Alzheimer Research Center from OHSU, supported by NIH grant NIA-AG08017 and Dr Edward Koo and Mr. Floyd Sarzosa at the Shiley-Marcos Alzheimer Disease Research Center from UCSD, supported by NIH grant AG051331, for contributing the tissues analyzed in this study. We want to thank specially to donors and their families for their invaluable contribution. We also thank Dr Nicholas Beckloff and Dr Simone Edelheit at the CWRU Department of Genetics and Genome Sciences Genomics Core Laboratory from Case Western Reserve University for DNA processing and microarray hybridization. We also thank statistician James Proudfoot from UCSD Clinical Translational Research Institute for assistance with bootstrap analysis. This work was supported by NIH grants AG5131 and AG18440 to EM.
Submitted
06/04/13
Revised
07/17/13
Accepted
07/23/13
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
EM contributed to study design, advised on neuropathological selection of cases included and contributed to write the paper. WD performed microarray validation experiments by qPCR. DG analyzed the clinical data and contributed to the discussion. PD designed and supervised the study; obtained DNA from postmortem samples, analyzed the data and wrote the paper.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/epigenetics/article/25865
References
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 2003; 39:889 - 909; http://dx.doi.org/10.1016/S0896-6273(03)00568-3; PMID: 12971891
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997; 388:839 - 40; http://dx.doi.org/10.1038/42166; PMID: 9278044
- Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol 1998; 152:367 - 72; PMID: 9466562
- Levy OA, Malagelada C, Greene LA. Cell death pathways in Parkinson’s disease: proximal triggers, distal effectors, and final steps. Apoptosis 2009; 14:478 - 500; http://dx.doi.org/10.1007/s10495-008-0309-3; PMID: 19165601
- Simunovic F, Yi M, Wang Y, Macey L, Brown LT, Krichevsky AM, et al. Gene expression profiling of substantia nigra dopamine neurons: further insights into Parkinson’s disease pathology. Brain 2009; 132:1795 - 809; http://dx.doi.org/10.1093/brain/awn323; PMID: 19052140
- Yacoubian TA, Cantuti-Castelvetri I, Bouzou B, Asteris G, McLean PJ, Hyman BT, et al. Transcriptional dysregulation in a transgenic model of Parkinson disease. Neurobiol Dis 2008; 29:515 - 28; http://dx.doi.org/10.1016/j.nbd.2007.11.008; PMID: 18191405
- Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 2011; 14:1345 - 51; http://dx.doi.org/10.1038/nn.2900; PMID: 21874013
- Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci 2010; 13:1319 - 23; http://dx.doi.org/10.1038/nn.2666; PMID: 20975755
- Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol 2009; 8:1056 - 72; http://dx.doi.org/10.1016/S1474-4422(09)70262-5; PMID: 19833297
- Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, et al. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem 2011; 286:9031 - 7; http://dx.doi.org/10.1074/jbc.C110.212589; PMID: 21296890
- Jowaed A, Schmitt I, Kaut O, Wüllner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci 2010; 30:6355 - 9; http://dx.doi.org/10.1523/JNEUROSCI.6119-09.2010; PMID: 20445061
- Matsumoto L, Takuma H, Tamaoka A, Kurisaki H, Date H, Tsuji T, et al. CpG Demethylation Enhances Apha-Synuclein Expression and Affects the Pathogenesis of Parkinson's Disease. PLoS ONE 2010; 5:1 - 9; http://dx.doi.org/10.1371/journal.pone.0015522
- Graeber MB. Biomarkers for Parkinson’s disease. Exp Neurol 2009; 216:249 - 53; http://dx.doi.org/10.1016/j.expneurol.2008.12.017; PMID: 19166835
- Belinsky SA, Klinge DM, Dekker JD, Smith MW, Bocklage TJ, Gilliland FD, et al. Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clin Cancer Res 2005; 11:6505 - 11; http://dx.doi.org/10.1158/1078-0432.CCR-05-0625; PMID: 16166426
- He Q, Chen HY, Bai EQ, Luo YX, Fu RJ, He YS, et al. Development of a multiplex MethyLight assay for the detection of multigene methylation in human colorectal cancer. Cancer Genet Cytogenet 2010; 202:1 - 10; http://dx.doi.org/10.1016/j.cancergencyto.2010.05.018; PMID: 20804913
- Kim H, Kwon YM, Kim JS, Lee H, Park JH, Shim YM, et al. Tumor-specific methylation in bronchial lavage for the early detection of non-small-cell lung cancer. J Clin Oncol 2004; 22:2363 - 70; http://dx.doi.org/10.1200/JCO.2004.10.077; PMID: 15197197
- Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 2012; 13:679 - 92; http://dx.doi.org/10.1038/nrg3270; PMID: 22945394
- Jain K. The Handbook of Biomarkers. Humana Press 2010.
- Burn DJ. Cortical Lewy body disease and Parkinson’s disease dementia. Curr Opin Neurol 2006; 19:572 - 9; http://dx.doi.org/10.1097/01.wco.0000247607.34697.a2; PMID: 17102696
- Lee SH, Kim SS, Tae WS, Lee SY, Lee KU, Jhoo J. Brain volumetry in Parkinson’s disease with and without dementia: where are the differences?. Acta Radiol 2013; •••; http://dx.doi.org/10.1177/0284185113476029; PMID: 23474765
- Zarei M, Ibarretxe-Bilbao N, Compta Y, Hough M, Junque C, Bargallo N, et al. Cortical thinning is associated with disease stages and dementia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2013; 84:875 - 82; http://dx.doi.org/10.1136/jnnp-2012-304126; PMID: 23463873
- Adalsteinsson BT, Gudnason H, Aspelund T, Harris TB, Launer LJ, Eiriksdottir G, et al. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS One 2012; 7:e46705; http://dx.doi.org/10.1371/journal.pone.0046705; PMID: 23071618
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 2012; 7:e41361; http://dx.doi.org/10.1371/journal.pone.0041361; PMID: 22848472
- Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 2012; 13:R43; http://dx.doi.org/10.1186/gb-2012-13-6-r43; PMID: 22703893
- Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 2013; 41:Database issue D377 - 86; http://dx.doi.org/10.1093/nar/gks1118; PMID: 23193289
- Nakamura T, Cho DH, Lipton SA. Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp Neurol 2012; 238:12 - 21; http://dx.doi.org/10.1016/j.expneurol.2012.06.032; PMID: 22771760
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288 - 95; http://dx.doi.org/10.1016/j.ygeno.2011.07.007; PMID: 21839163
- Kibriya MG, Raza M, Jasmine F, Roy S, Paul-Brutus R, Rahaman R, et al. A genome-wide DNA methylation study in colorectal carcinoma. BMC Med Genomics 2011; 4:50; http://dx.doi.org/10.1186/1755-8794-4-50; PMID: 21699707
- Naumova OY, Lee M, Koposov R, Szyf M, Dozier M, Grigorenko EL. Differential patterns of whole-genome DNA methylation in institutionalized children and children raised by their biological parents. Dev Psychopathol 2012; 24:143 - 55; http://dx.doi.org/10.1017/S0954579411000605; PMID: 22123582
- Efron BaT. R. An Introduction to the Bootstrap. Monographs on Statistics and Applied Probability 1993; 57:1 - 399
- Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, et al. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One 2010; 5:5; http://dx.doi.org/10.1371/journal.pone.0013066; PMID: 20927376
- Desplats P, Patel P, Kosberg K, Mante M, Patrick C, Rockenstein E, et al. Combined exposure to Maneb and Paraquat alters transcriptional regulation of neurogenesis-related genes in mice models of Parkinson’s disease. Mol Neurodegener 2012; 7:49; http://dx.doi.org/10.1186/1750-1326-7-49; PMID: 23017109
- Jakovcevski M, Bharadwaj R, Straubhaar J, Gao G, Gavin DP, Jakovcevski I, et al. Prefrontal Cortical Dysfunction After Overexpression of Histone Deacetylase 1. Biol Psychiatry 2013; •••; http://dx.doi.org/10.1016/j.biopsych.2013.03.020; PMID: 23664640
- Miñones-Moyano E, Friedländer MR, Pallares J, Kagerbauer B, Porta S, Escaramís G, et al. Upregulation of a small vault RNA (svtRNA2-1a) is an early event in Parkinson disease and induces neuronal dysfunction. RNA Biol 2013; 10:10; http://dx.doi.org/10.4161/rna.24813; PMID: 23673382
- Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol 2012; 13:R97; http://dx.doi.org/10.1186/gb-2012-13-10-r97; PMID: 23034122
- Kumaran R, van der Brug M, Vandrovcova J, Ding J, Sharma S, Renton A, Dillman A, Lees A, Cookson MR, Bandopadhyay R. . Gene expression changes across multiple regions of the Parkinson’s disease brain. Gene Expression Omnibus (GEO) NCBI2011:Series GSE28894.
- Moran LB, Duke DC, Deprez M, Dexter DT, Pearce RK, Graeber MB. Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson’s disease. Neurogenetics 2006; 7:1 - 11; http://dx.doi.org/10.1007/s10048-005-0020-2; PMID: 16344956
- Roth R.. Human body index - transcriptional profiling. Gene Expression Omnibus (GEO) NCBI2007:Series GSE7307.
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, et al, Global PD Gene Expression (GPEX) Consortium. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2010; 2:52ra73; http://dx.doi.org/10.1126/scitranslmed.3001059; PMID: 20926834
- Durrenberger PF, Grünblatt E, Fernando FS, Monoranu CM, Evans J, Riederer P, et al. Inflammatory Pathways in Parkinson’s Disease; A BNE Microarray Study. Parkinsons Dis 2012; 2012:214714; PMID: 22548201
- Lesnick TG, Papapetropoulos S, Mash DC, Ffrench-Mullen J, Shehadeh L, de Andrade M, et al. A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet 2007; 3:e98; http://dx.doi.org/10.1371/journal.pgen.0030098; PMID: 17571925
- Lewandowski NM, Ju S, Verbitsky M, Ross B, Geddie ML, Rockenstein E, et al. Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc Natl Acad Sci U S A 2010; 107:16970 - 5; http://dx.doi.org/10.1073/pnas.1011751107; PMID: 20837543
- Muller KE, LaVange L. M., Ramey, S.L. and Ramey, C.T. Power calculations for general multivariate models including repeated measures applications. J Am Stat Assoc 1992; 87:1209 - 26; http://dx.doi.org/10.1080/01621459.1992.10476281
- Zhang Y, James M, Middleton FA, Davis RL. Transcriptional analysis of multiple brain regions in Parkinson’s disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am J Med Genet B Neuropsychiatr Genet 2005; 137B:5 - 16; http://dx.doi.org/10.1002/ajmg.b.30195; PMID: 15965975
- Bollati V, Schwartz J, Wright R, Litonjua A, Tarantini L, Suh H, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev 2009; 130:234 - 9; http://dx.doi.org/10.1016/j.mad.2008.12.003; PMID: 19150625
- Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet 2001; 10:687 - 92; http://dx.doi.org/10.1093/hmg/10.7.687; PMID: 11257100
- Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, et al. A DNA methylation fingerprint of 1628 human samples. Genome Res 2012; 22:407 - 19; http://dx.doi.org/10.1101/gr.119867.110; PMID: 21613409
- Benjamini YaHY. Controlling the False Discovery Rate: A Practical and Powerful Approch to Multiple Testing. J R Stat Soc Ser A Stat Soc 1995; 57:289 - 300
- Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet 2011; 7:e1002141; http://dx.doi.org/10.1371/journal.pgen.1002141; PMID: 21738487
- Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 2010; 74:97 - 109; http://dx.doi.org/10.1111/j.1469-1809.2009.00560.x; PMID: 20070850
- Spencer CC, Plagnol V, Strange A, Gardner M, Paisan-Ruiz C, Band G, et al, UK Parkinson’s Disease Consortium, Wellcome Trust Case Control Consortium 2. Dissection of the genetics of Parkinson’s disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet 2011; 20:345 - 53; http://dx.doi.org/10.1093/hmg/ddq469; PMID: 21044948
- Stenvinkel P, Karimi M, Johansson S, Axelsson J, Suliman M, Lindholm B, et al. Impact of inflammation on epigenetic DNA methylation - a novel risk factor for cardiovascular disease?. J Intern Med 2007; 261:488 - 99; http://dx.doi.org/10.1111/j.1365-2796.2007.01777.x; PMID: 17444888
- Eisenhart C. The assumptions underlying the analysis of variance. Biometrics 1947; 3:1 - 21; http://dx.doi.org/10.2307/3001534; PMID: 20240414