 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Analysis of epigenetic mechanisms, particularly DNA methylation, is of increasing interest for epidemiologic studies examining disease etiology and impacts of environmental exposures. The Infinium HumanMethylation450 BeadChip® (450K), which interrogates over 480 000 CpG sites and is relatively cost effective, has become a popular tool to characterize the DNA methylome. For large-scale studies, minimizing technical variability and potential bias is paramount. The goal of this paper was to evaluate the performance of several existing and novel color channel normalizations designed to reduce technical variability and batch effects in 450K analysis from a large population study. Comparative assessment of 10 normalization procedures included the GenomeStudio® Illumina procedure, the lumi smooth quantile approach, and the newly proposed All Sample Mean Normalization (ASMN). We also examined the performance of normalizations in combination with correction for the two types of Infinium chemistry utilized on the 450K array. We observed that the performance of the GenomeStudio® normalization procedure was highly variable and dependent on the quality of the first sample analyzed in an experiment, which is used as a reference in this procedure. While the lumi normalization was able to decrease batch variability, it increased variation among technical replicates, potentially reducing biologically meaningful findings. The proposed ASMN procedure performed consistently well, both at reducing batch effects and improving replicate comparability. In summary, the ASMN procedure can improve existing color channel normalization, especially for large epidemiologic studies, and can be successfully implemented to enhance a 450K DNA methylation data pipeline.
Introduction
Epigenetic mechanisms regulate gene expression without changes in DNA sequence and include DNA methylation, histone modifications, and non-coding RNAs.Citation1–Citation3 Growing evidence shows that epigenetics may be an interface through which environmental exposures affect gene expression and adverse health.Citation4 DNA methylation, an addition of a methyl group at the carbon-5 position of cytosine (5mC) in CpG dinucleotides, is the best-studied epigenetic mechanism. Several technologies, including new generation sequencing and genome-wide arrays, are currently available to study the DNA methylome.Citation5 However, sequencing technologies can be prohibitively expensive for use with population-based studies, which may require analysis of hundreds of samples in large data sets over multiple time points. Illumina’s 450K Methylation array has emerged as one of the preferred methodologies to study DNA methylation because of its optimal combination of genome-wide coverage (99% of RefSeq genes), comprehensive representation of functional gene sub-regions, good reproducibility across other platforms (r = 0.88 with Pyrosequencing),Citation6,Citation7 and relative affordability.
Before sources of biological variability in DNA methylation can be accurately assessed, it is critical to minimize technical variance and bias. Experiments involving hundreds of samples need to be run in several batches across a long time span, potentially exacerbating variation in instrumentation and assay chemistry. Differences between the measurement of the two colored probes (red and green), including labeling hybridization efficiency and chip scanning properties, can also introduce noise to methylation results. The Illumina proprietary software package (GenomeStudio) adjusts for this variability of color signals across an experiment, which we refer to as the Illumina First Sample Normalization (IFSN). In addition, other normalization methodologies have recently been proposed, including smooth quantile normalization from the lumi R packageCitation8 and other pipelines drawing on its infrastructure.Citation9,Citation10
Another class of adjustment that has received attention in the literature addresses the two different 450K Infinium array chemistries: Infinium I, which was previously implemented on an older Illumina methylation 27K array, and Infinium II, which was added as coverage expanded for the 450K array. Recent studies have demonstrated that the signals from the Infinium I and II assays are likely not completely comparable: Infinium I has a broader dynamic range of methylation values, called βs, that tend to be more stable and reproducible in comparison to Infinium II,Citation9,Citation11 potentially introducing a source of bias based on the type of probe used. Several correction and normalization methodologies have been proposed to adjust for differences between the two 450K Infinium chemistries including peak-based correction (PBC),Citation11 subset quantile normalization (SQN and SWAN),Citation9,Citation12 and β-mixture quantile normalization (BMIQ).Citation13 The PBC approach has been criticized in two recent publications for poor performance when its strong assumptions of bi-modality in β distributions are not met.Citation9,Citation12 However, a recent evaluation of the other available normalizations between Infinium I and II chemistries (SQN, SWAN, BMIQ) showed them to be comparable.Citation13 While initial assessment of each of these normalizations has been conducted,Citation13,Citation14 including evaluation of reductions in batch effects, the sample sizes used in these publications (ranging from 6–85 analyzed on 1–8 chips) have not been sufficient to detect the type of batch variability likely to occur in large population studies.
A complete 450K data preparation pipeline for epidemiologic analysis ideally includes several distinct components, including: subtraction of background signal, color channel normalization, checks for bisulfite conversion and extension efficiency, removal of poor performing CpG and SNP associated probes, and adjustment for Infinium chemistry. In the current study we conduct a focused assessment of the performance of color channel normalization procedures, one key aspect of 450K data preparation. Our study evaluates 10 procedures: two existing normalizations (IFSN and lumi), several variations on the method used by IFSN normalization, called reference factor (RN) based normalizations, and proposed here a new optimized All Sample Mean Normalization (ASMN) procedure. Our analysis uses a large data set of 432 samples (36 chips/5 plates) to identify which procedures most effectively minimize technical variation in population-based studies.
Results
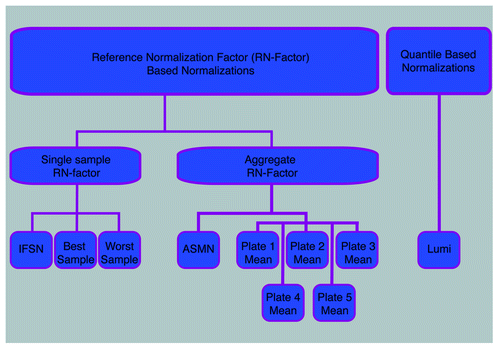
To assess color channel normalization performance, 10 procedures were implemented on 450K data from a large epidemiologic cohort. These procedures fell into two distinct methodological categories: (1) reference normalization factor (RN-factor) based and (2) quantile based methods (). The first category included nine variations of RN-factor based procedures and the second category was represented by the lumi smooth quantile normalization.
Figure 1. Flowchart of normalizations implemented. Ten color channel normalization procedures were implemented. Nine of those procedures were reference normalization factor (RN-factor) based methods that use the n = 93 normalization control probes assayed in every sample on the 450K chip for adjustment. Of the RN-factor based methods, three methods used the RN-factors from a single sample: the Illumina first sample normalization (IFSN), the best performing sample normalization, and the worst performing sample normalization. The remaining six RN-factor based procedures use aggregated RN-factors across different groups of samples, including the mean RN-factors for each plate of the experiment (Plates1–5 Means) and the all sample mean normalization (ASMN) that uses the mean RN-factors for all experimental samples. The remaining normalization, the lumi procedure, uses a quantile-based methodology instead of RN-factors.
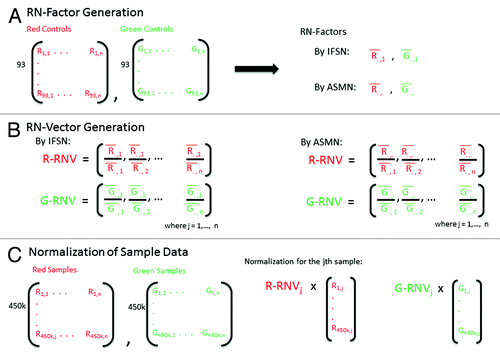
RN-factor based normalizations utilize the mean values from the red and green normalization control probes included on the 450K chip as RN-factors in their adjustment (). These RN-factors are used to compute two vectors of length n (RN-vectors), containing the ratio of each sample’s mean red and green control probe values to that of the RN-factor of the same color (). Sample normalization occurs by multiplying the jth sample’s red and green signals by the corresponding elements from the red and green RN-vectors ( and Methods).
Figure 2. Reference normalization factor (RN-factor) based color channel normalization for the 450K methylation array. (A) The 450K chip includes n = 93 normalization control probes in both assay colors (red and green). The mean values of these sites are used to create RN-factors for normalizing both color channels over all samples (i.e., an experiment). The Illumina first sample normalization (IFSN) method uses the first sample’s mean red and green control probes as RN-factors ( and
). The all sample mean normalization (ASMN) method instead uses the mean read and green control probes taken across all control sites and all samples in a given experiment (
and
) as RN-factors. (B) A set of sample-wise normalization values, taken as the ratio of the RN-factor to each sample’s mean control probe values, is then computed. This results in a vector of length n normalization values for each color channel (R-RNV and G-RNV). (C) Color channel normalization of sample data occurs by multiplying the each of the jth sample’s red and green signals by the jth normalization value from the corresponding RN-vector (where j = 1,2,…, n).
Among the RN-factor based normalizations, procedures differed by which control probe observations were used to calculate the RN-factors (). There were two groups of RN-factor procedures: a) those using only the control probe values from a single sample (IFSN, best sample, worst sample) and b) those using RN-factors aggregated across groups of samples (mean by each of 5 assay plates, ASMN). shows each step of RN-factor based normalization for both a single sample (IFSN) and an aggregate procedure (ASMN).
Performance of the normalization procedures was evaluated by three criteria. First, we assessed the stability of RN-factor based normalizations when using RN-factors from samples of varying quality, or when using RN-factors aggregated across batches (i.e., assay plates) or an entire experiment (i.e., ASMN). The other two criteria included evaluation of repeatability of technical replicates and reduction in batch variation.
RN-factor based normalization stability
The majority of samples from our cohort proved to be of good quality with less than one percent of CpG sites with detection p-values equal or greater than 0.05. However, nine of the 432 samples were considered of lower quality (> 1% of CpG sites with detection p values ≥ 0.05). When we plotted the signal intensity of normalization control probe against quality of methylation calls (measured by number of detectable CpG sites), we found that samples with low red and green control signals also had lower quality methylation calls (). The correlation between control probe signal intensity and number of detectable CpG sites was 0.76 (P < 0.0005) for both red and green signals. If according to the Illumina IFSN algorithm () the first sample on which the entire experiment is normalized happens to be one of low quality, the overall results and interpretation of the data may be negatively affected. Thus, ASMN was developed to increase normalization stability and robustness by non-arbitrarily drawing on observations from all Illumina internal controls and study samples (described in detail in Methods).
Figure 3. Plot of mean red (A) and green (B) signal intensity of normalization control probes (n = 93) by number of detected CpG sites in the 450K array sample data (n = 432). For both color channels, samples with lower intensity readings in their normalization control probes tended to have more poor performing CpG sites in their samples.
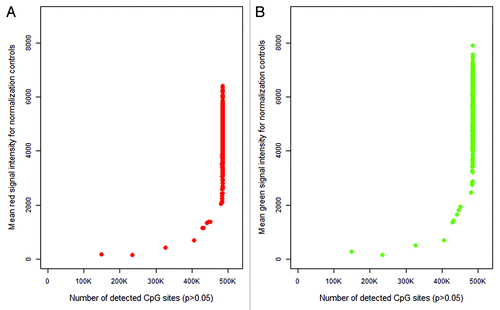
Although we observed a positive association between control probe signal intensity and number of detectable CpG sites, this relationship appeared to exhibit a threshold effect (). Samples with fewer numbers of detectable CpG sites also had lower mean red and green control probe values, but increases in probe signal intensities above 2000 for red and 3000 for green did not appear to contribute to additional gains in CpG detection. While both the number of detectable CpG sites and the mean control probe signal intensity provide information regarding assay quality, only the former was a measure designed for this purpose. This makes it difficult to distinguish what constitutes a “better” sample or a “better” mean control probe value from those above the control signal threshold. We implemented the ASMN procedure to draw from the central tendency of this distribution rather than the tail, since we did not have convincing evidence to prefer higher mean control probe values above the threshold. Further, drawing from the center of this distribution made the ASMN more stable and less susceptible to variation in sample quality. To confirm that the mean was an appropriate measure of central tendency, we also performed the normalization using median RN-factors, but obtained similar results (data not shown).
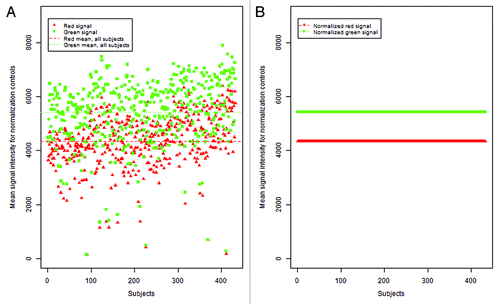
We found little available rationale, including information in the Illumina reference manual, to support the preferential use of control probe data from the first sample over other samples beyond convenience.Citation15 The IFSN approach carries an unstated assumption: the control probe values from any sample should perform equally well at reducing color bias and batch variation. This is not always the case, for instance shows a broad range of variability in the normalization control values among 432 samples analyzed in this study. Furthermore, this variability also suggests that the particular sample used for normalization may affect the normalization quality of all samples. After performing normalization, RN-factor based methods bring all data observations to the same scale. illustrates this effect by showing that, after normalization, the previously dispersed normalization control values () become standardized to the values of the red and green RN-factors.
Figure 4. Mean control probe color signal intensity before and after normalization. (A) Distribution of mean green and red normalization controls (93 controls per signal color per sample) as included in the 450K chip over 432 DNA samples. Each point, red triangle or green square, represents the average of the normalization controls for that signal color per sample prior to implementation of color channel normalization. (B) Following adjustment using a reference normalization factor (RN-factor) based normalization, the average normalization controls for all samples are ‘forced’ to be the same level, making observations across samples comparable. Here, ASMN normalization was performed which uses the mean red and green signal for all samples for adjustment.
In , we compared the normalized βs given an unadjusted β of 0.10 for all samples normalizing either on the lowest or highest quality sample. In general, normalization using the poorest quality sample (sample 411 in this data set) yielded much lower methylation βs. Further, normalization using the poor quality samples led to much larger variability in βs ( and ), particularly at extreme methylation values. shows that when we normalize using a high quality sample (e.g., sample 355), or if we normalize over a summary measure (mean over one plate or all samples), the βs do not change drastically after normalization and remain in the high, medium, and low range. However, when we normalize over the low quality sample, all three βs (low, medium, and high) decreased and the normalized value for high βs became much lower (~0.6 vs. ~0.9). These results could bias downstream analyses as the power to detect differences in methylation would be lessened, highlighting the importance of choosing a reliable normalization procedure based on high quality samples.
Figure 5. Plot of normalized DNA methylation (βs) given an unadjusted β of 0.1 (Signal A = 5000 and Signal B = 570) for all 432 samples. Open circles represent data normalized using the sample with the least detectable sites (sample 411, the lowest quality sample). Filled circles were normalized using the sample with the most detectable sites (sample 355, the highest quality sample).
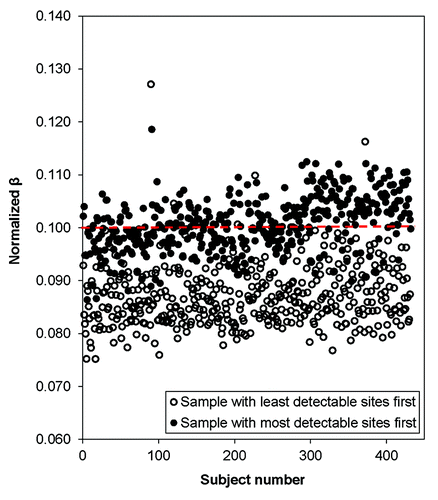
Figure 6. Average percent change of methylation values, βs, after normalization by best and worth performing samples. Mean percent change in βs, values ranging from 0.1 to 0.9, based on normalization by the lowest quality sample (largest amount of CpG sites with p < 0.05) and the highest quality sample (least amount of CpG sites with p < 0.05) over all samples (n = 432). While normalization by the highest quality sample changed the βs only slightly (< 10% on average), normalization by the lowest quality sample tended to change the low and high methylation βs substantially (> 10% on average).
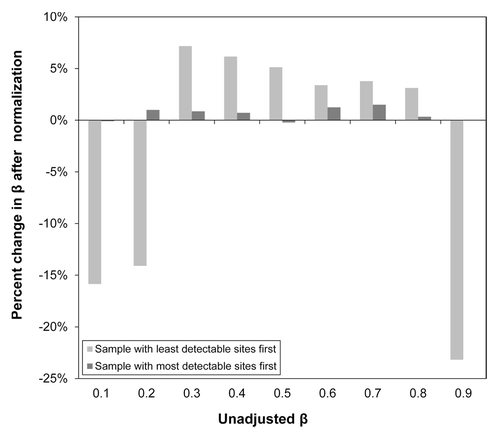
Table 1. Reference normalization factors and methylation (βs) for a single sample by normalization procedure
Repeatability and batch variability
When examining repeatability of replicates, we assessed the reduction in root mean squared error (root-MSE) between replicates by each normalization procedure (). All RN-factor based color channel normalization procedures resulted in lower mean root-MSE between replicates. The greatest reduction was a decrease of 10.83%, occurring with normalization using the RN-factors of the best performing sample in the experiment (sample 355 here). Those normalizations that used an aggregate RN-factor, such as the ASMN and the single plate-mean normalizations, each elicited similar reductions in root-MSE between replicates, all producing approximately a 10% reduction compared with un-normalized data. Of the color channel normalizations evaluated, the lumi normalization performed worst, actually slightly increasing mean replicate root-MSE (0.58%). However, both data sets that utilized an additional normalization technique for Infinium chemistry adjustment (BMIQ) saw increases in root-MSE compared with un-normalized. While this effect was relatively small for the ASMN combined with BMIQ normalization (a 1.21% increase), the lumi normalization followed by BMIQ produced a sizeable increase in mean replicate root-MSE (11.96%), indicating a decrease in repeatability. The changes in correlation observed for each of the normalization procedures relative to un-normalized results largely followed similar trends as those observed for root-MSE (). However, due to the bounded nature of the correlation coefficient, the magnitude of the effect was not as large.
Table 2. Repeatability of technical replicates by improvement of root mean squared error (root-MSE) and mean Spearman correlation (R2) compared for un-normalized results
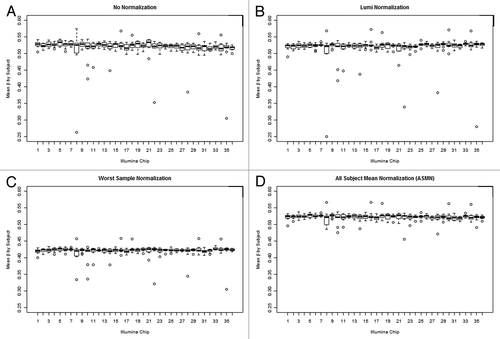
Visual assessment of batch variability also identified important differences between normalization procedures (). Color channel normalization is expected to increase comparability of mean chip values and decrease batch variability over un-normalized βs, as seen in . Lumi smooth quantile normalization () appears to retain many of the extreme points and batch trends observed in the raw βs, as does using the worst performing sample’s RN-factor values, which also decreases the real scale of the β distribution (). The box plots of mean sample β using normalization by ASMN, shown in , demonstrate a reduction in the number of outlier samples and batch-related variability.
Figure 7. Box plots of sample mean methylation by normalization methods. Box plots of mean per-sample methylation (β) for all sites interrogated on the 450K array (n = 485 512) by color channel normalization methods. Plots are shown for (A) un-normalized results and three different normalization methods, (B) lumi smooth quantile normalization, (C) normalization using the worst performing sample’s reference normalization factor values (sample 411), and (D) using the all sample mean normalization (ASMN) method. Each chip assays 12 samples, so every box plot contains 12 observations in total.
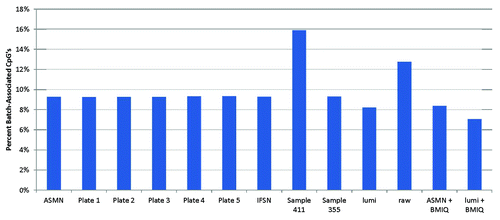
In the site-level analysis of batch-associated variability, the “raw” un-normalized βs showed a relatively high percentage of CpG sites that were associated with the chip batch (12.8%) () compared with other normalizations. Other mean RN-factor based color channel normalization procedures, including ASMN and each of the plate mean RN-factor normalization procedures showed fewer batch associated sites than raw βs and the percentage of sites were largely consistent across these procedures. When using only one sample’s control probe values, sample quality appeared to influence the amount of batch variability across the experiment. For instance, the best performing sample (by fewest number of non-detectable CpG sites) and a well-performing first-experiment sample used in the IFSN, both had percentages of batch-association comparable with aggregate RN-factor based procedures. However, the worst performing sample had the highest level of batch association. The lumi procedure also showed a reduction in the percentage of batch-associated sites compared with un-normalized results, having even a slightly lower percentage than the aggregate RN-factor based procedures. Additionally, the number of batch- associated sites was further reduced for both the lumi and ASMN when they had been followed by the BMIQ adjustment for Infinium assay chemistry.
Figure 8. Percent of 450K array CpG sites associated with chip batch (p < 0.01) shown by normalization method. Normalization methods include: All sample mean normalization (ASMN), normalization by reference normalization factors (RN-factors) taken as the mean control probe values for each of the plates (1–5) run, Illumina first sample normalization (IFSN), normalization by the worst performing sample’s RN-factors (sample 411) and the best performing sample’s RN-factors (sample 355), lumi smooth quantile normalization, raw un-normalized results, and both the ASMN and lumi normalization followed by β-mixture quantile normalization (BMIQ). Batch association was evaluated by ANOVA for each of the n = 485 512 CpG sites interrogated.
Finally, the ASMN normalization procedure has been compiled into an R package that will be freely available in an open-source distribution in the bioconductor repository for bioinformatics software (www.bioconductor.org).
Discussion
In this study, we implemented and evaluated the performance of 10 variations of color channel normalization for Illumina 450K methylation data from a large epidemiologic study. In addition to using two common color channel normalization procedures (IFSN and lumi), we also implemented our preferred new ASMN normalization procedure, and several additional strategies to evaluate the range of performance that could be achieved with RN-factor based procedures. We specifically examined the ability of these normalization procedures to reduce major sources of technical variability by assessment of (1) batch effects and (2) performance of technical replicates included in the experiment. We found that the ASMN procedure outperformed the Illumina recommended IFSN algorithm, and further, that ASMN consistently performed well while the performance of IFSN varied depending on sample quality. We observed comparable performance between normalizations using the RN-factors from the best performing sample and ASMN, while the latter had the added benefit of not relying on data mining. We also found that the ASMN procedure was better at increasing repeatability between technical replicates than the commonly used lumi approach and had similar benefits for reducing batch effects. Lastly, we confirmed that the advantages of ASMN normalization compared with lumi were retained even after adjustment for differences in Infinium chemistry using the popular BMIQ algorithm. These findings suggest that the ASMN procedure is an improvement over existing strategies for color channel normalization, especially for large epidemiologic studies. Thus, its implementation in conjunction with other data cleaning steps in any 450K methylation data pipeline is warranted.
Improved performance in repeatability and reduction of batch effects were observed for ASMN when compared with the IFSN procedure recommended by Illumina. While some of these gains in performance were relatively small in scale, as when comparing the number of batch associated CpG’s found for each procedure, they were consistent across all performance measures. Further, our parallel assessment of normalization by using the RN-factor values for both the best and worst performing samples showed the range of possible performance that could have been garnered with the IFSN strategy. While the best sample’s performance was largely comparable with the ASMN, the worst sample’s performance was drastically worse, even seeming to introduce batch variability compared with un-normalized results (). This range of performance demonstrated that while some samples may perform satisfactorily when used in normalization, others may introduce bias to results. The likelihood of a poor performing normalization by the IFSN strategy is essentially a random draw from the range of sample qualities included in a given experiment. The ASMN procedure provides a convenient and more reliable alternative, since its performance is stable over a given experiment. In addition, the use of ASMN instead of normalization by RN-factors from the best performing sample provides a robust methodology that does not rely on prior access to data or data mining.
Comparison of the ASMN procedure to the lumi normalization showed that ASMN had increased repeatability across all metrics evaluated. In fact, lumi often performed only marginally better than using raw un-normalized results (). While lumi did not effectively improve repeatability, it did provide substantial reductions in batch effects, outperforming both ASMN and the best sample RN-factor normalizations in this regard. One possible explanation for this inconsistent performance may be over-fitting of the lumi algorithm, which aggressively coerces the distribution of normalization targets to have identical quantiles as the reference distribution. In turn, this may reduce the number of possible methylation values and minimize batch effects, even while not addressing the repeatability issues. Further, since the loss in batch variability does not co-occur with gains in repeatability, the apparent benefits of this approach may actually come at the cost of artificially reduced biological variability.
When we examined the performance of the lumi and ASMN procedures followed by adjustment for differences in Infinium chemistry using the BMIQ algorithm, we continued to observe benefits of using ASMN rather than lumi. While in general both lumi + BMIQ and ASMN + BMIQ performed well at reducing batch variability, neither of these combined strategies saw improved performance of technical replicates compared with data sets receiving only color channel normalization. Again, the lumi + BMIQ data set exhibited the same trend seen in the lumi color normalization alone: much lower batch variability with increased variability between technical replicates. As such, it seems likely that the issue of the lumi algorithm over-fitting is retained even when followed by BMIQ normalization. The ASMN + BMIQ data set, like the data set receiving the ASMN normalization alone, had consistent performance in reducing technical variability. While some of the gain in repeatability between replicates afforded by the ASMN was lessened with addition of BMIQ, it was previously demonstrated that adjustment for Infinium chemistry is neededCitation9,Citation11,Citation13 and, thus, BMIQ has to remain in the 450K data processing pipeline.
It is important to clarify that the assessment we present here is focused on performance of color channel normalization in particular, and isn’t a comprehensive evaluation of all the processing steps needed prior to analysis of biological effects from 450K array data. Several additional data processing steps have been suggested in the literature and are freely available as R packages, including filtering out SNP-associated probes included in the 450K assay and adjusting for the Infinium I and II chemistries.Citation9,Citation10,Citation13 To confirm that improved performance would be retained in the context of a full pipeline, we also performed SNP-filtration prior to ASMN normalization and observed similar gains (data not shown). Our results indicate that color channel normalization should indeed be performed in addition to SNP-filtering and Infinium chemistry adjustment (BMIQ), and should be included in any robust Infinium data processing pipeline.
While other studies have examined normalization strategies for 450K data, to date they have focused on reducing differences between the Infinium I and II chemistries, and have been under-powered to evaluate the batch effects that are likely to occur in large association studies.Citation9,Citation11,Citation12 A main advantage of our approach was a large sample size and inclusion of many technical replicates for rigorous evaluation of normalization performance. Only one other evaluation has been published to date examining normalization of data from the 450K assay for anything approaching a population study.Citation14 This study based on 85 samples, found that a pipeline that included lumi color adjustment, followed by BMIQ performed the best at reducing batch variability and increasing repeatability. However, Marabita et al. mostly directed their comparison to performance between Infinium I and II chemistry adjustment. Further, they did not consider an option we propose here as ASMN, which we tested alongside the lumi + BMIQ procedure (that they preferred). Our study’s capacity to detect true batch effects was much larger than the Marabita study, which examined only 85 samples analyzed on eight BeadChips. The numerous BeadChips and plates analyzed in our study (nsamples = 432, nchips = 36, nplates = 5) are more representative of the scale of batch effects that would be encountered in large population or case-control studies. Also, Marabita et al. only examined repeatability with n = 16 total replicates (n = 8 pairs) while our assessment included more than double that number of replicates (n = 38 from n = 15 samples).
In summary, we implemented the most comprehensive comparative evaluation of color channel normalization procedures for the 450K assay to date. The large sample size and the many technical replicates included in the analysis allowed for careful assessment of sources of technical variability, including those that are likely to be unique to large epidemiologic studies. Our results show that the ASMN normalization procedure that we introduced is an excellent alternative to the two leading color channel normalization strategies, Illumina’s IFSN and lumi. ASMN reduced technical variability compared with the IFSN procedure and did not encounter the performance trade-offs of the lumi approach. As ASMN relies on a predefined measure of central tendency among control values, it is a stable and robust approach to normalization. Further, the ASMN procedure yielded reductions in technical variability beyond normalization for Infinium chemistry type alone by BMIQ. These findings suggest that, especially for large epidemiologic studies, the ASMN color channel normalization is a valuable component to be included in a 450K methylation data pipeline.
Materials and Methods
Samples
DNA was isolated from a convenience set of blood clots from 408 healthy children participating in a longitudinal birth cohort study, using QIAamp DNA blood kits from Qiagen according to the manufacturer’s protocol. Following isolation, all samples were checked for DNA quantity and quality by Nanodrop 2000 spectrophotometer. Samples were retained if they produced high yield and good DNA quality (as assessed by 260/280 ratio exceeding 1.6) and concentrations were adjusted to 50ng/ul. DNA aliquots of 1ug were bisulfite converted using Zymo Bisulfite conversion Kits (Zymo Research). Study protocols were approved by the University of California, Berkeley Committee for Protection of Human Subjects
Illumina Infinium HumanMethylation450 DNA methylation assay
DNA samples were whole genome amplified, enzymatically fragmented, purified, and applied to the 450K BeadChips according to the Illumina methylation protocol.Citation6,Citation16 BeadChips were processed with robotics and analyzed using the Illumina Hi-Scan system. Each 450K BeadChip can fit n = 12 samples in total, and these chips are usually run combined onto plates in sets of 8, for a batch of n = 96 samples. To accommodate all of the samples analyzed in this experiment, 36 BeadChips were run across 5 plates. BeadChips included on the same plate (up to n = 8 BeadChips per plate) were analyzed simultaneously, and time between plate runs was approximately one week using the same batch of all reagents and chips.
Data extraction
Sample data were extracted using Illumina GenomeStudio software (version XXV2011.1, Methylation Module 1.9) methylation module. This provides raw intensities for both red and green color channels, detection p-values as a measure of assay performance, and βs calculated from raw signals for all samples at all 485 577 assayed probes. Data cleaning performed prior to evaluation of different normalization procedures included background correction of raw signal intensities according to Illumina recommendations using GenomeStudio software. The background is determined as the lowest 5th percentile of the 600 negative controls included in the assay and was subtracted from the probe intensities. Also, the n = 65 SNP probes noted in the Illumina manual were filtered out, leaving 485 512 CpG sites for analysis.
Simultaneous to sample data extraction, control probe data extraction was also performed using the GenomeStudio software. This additional matrix contains raw signal observations for all of the probes included as controls in the design of the 450K assay. Such control values include negative controls (for background subtraction), extension controls, staining controls, bisulfite controls, and n = 93 normalization control probes among others. The normalization control probe pairs are targeted to non-variable regions of stable housekeeping genes and are the observations used to calculate the RN-factors used RN-factor based normalization procedures ().
Quality assurance/quality control (QA/QC)
Of the samples selected for analysis, 14 samples were randomly chosen to be included as technical replicates. Replicates were designed to maximize the capacity to detect multiple forms of bias across the experiment. As such, 3 pairs of samples were included as intra-chip replicates, 6 pairs were included as intra-plate replicates, 4 pairs were DNA isolation replicates, and 1 sample was an inter-plate replicate run 7 times across all plates of the experiment. Furthermore, one internal control sample (DNA from a Jurkat cell-line) was run on each sample plate, replicated 5 times in total. Including the Jurkat DNA, 15 sets of replicates were included throughout the experiment, comprising n = 38 QA/QC samples. The location of samples on assay wells for each of the Illumina BeadChips was randomized.
In addition to replicates, the Illumina GenomeStudio software provides an internal measure of assay quality for each CpG site interrogated: a detection p-value. This value represents the chance that the signals produced from a given site were not distinguishable from background. Thus, a small detection p-value would indicate that the fluorescent signals at a particular CpG site were likely above background levels. Illumina suggests using a detection p-value cutoff of 0.05 above which a CpG site should be excluded from analysis.
Color channel batch normalization procedures
Including all QA/QC samples, a total of n = 432 samples were assayed. We refer to this as the total samples included in our “experiment.” Further, we define “batch” effects as occurring at two different levels: (1) the BeadChip level (which includes observations from n = 12 samples) and, (2) the plate level (which includes n = 8 BeadChips and n = 96 samples). Our experiment includes 36 BeadChip batches and 5 plate batches. Unless otherwise specified, all batch analysis was conducted at the BeadChip level.
To evaluate the performance of different procedures adjusting for color channel bias across batches, we implemented 10 different normalization procedures to background subtracted signals, creating a total of ten different data sets. These 10 procedures fell into two methodological categories: (1) reference normalization factor (RN-factor) based and (2) quantile based methods (). The nine RN-factor based procedures utilize the values of the n = 93 normalization control probe to construct RN-factors () and differ by which observations are used to calculate RN-factors. There were two groups of RN-factor based methods: (1) those using only the RN-factors from a single sample and (2) those using aggregated RN-factors ().
Each of the 10 color channel normalization procedures are described below:
(1) The Illumina first sample normalization (IFSN) is the standard color channel normalization recommended by Illumina. This procedure uses the mean of the first sample’s normalization control probe values (both red and green) to calculate the RN-factors.
(2) Another single-sample RN-factor normalization was performed: one using the RN-factors for the best performing sample in the experiment (sample number 355). As described in QA/QC above, the best performing samples was determined by having the highest number of CpG sites meeting a detection p-value threshold less than 0.05.
(3) A single-sample RN-factor normalization was also performed using the RN-factors for the worst performing sample in the experiment (sample number 411). The worst performing sample in the experiment was determined by having the least number of CpG sites meeting a detection p-value threshold less than 0.05.
(4) The all sample mean normalization (ASMN) strategy that we developed uses the means of the RN-factors of all samples in the experiment (in this case n = 432) as the RN-factors.
(5–9) Beyond calculating the RN-factors as the mean over all samples in the experiment, we also performed normalization by averaging over different sub-groups within the experiment, namely each of the 5 plate-batches in which the experiment was run. RN-factors calculated as the mean RN-factors by each plate created 5 different mean-plate RN-factors and 5 output data sets. These procedures essentially set 1 plate batch as the baseline to which all other batches are normalized.
(10) Lastly, one non-RN-factor based color channel normalization, the lumi smooth quantile normalization procedure, was also implemented. This approach involves local polynomial smoothing followed by an interpolation step. The procedure assumes that the distributions of data within each color channels are identical and coerces the distribution of each target color channel to have identical quantiles to the reference distribution.
To further confirm the stability of the ASMN procedure, an additional data set was generated which removed n = 16 667 CpG sites that potentially include common (minor allele frequency > 5%) SNPs prior to ASMN normalization. SNP list was obtained using the HapMap project population most comparable to our cohort.Citation17 All measures of normalization performance were retained following removal of possible SNP-associated CpGs (data not shown).
Adjustment for Infinium chemistry
In addition to the ten data sets created by implementing different color channel batch normalization strategies, we also implemented an adjustment procedure (BMIQ) to account for the systematically different performances of the Infinium I and II chemistries to two of our color channel normalized data sets. We applied BMIQ to the ASMN and lumi normalized data sets (numbers 2 and 6 above) to evaluate how reduction of batch variability would be impacted by adding this needed correction for assay chemistry. The BMIQ normalization procedure is a model-based strategy that applies a three-state β mixture model to assign methylation states, followed by quantile normalization using the parameters of these β distributions.Citation13
Statistical analysis
After extraction of raw values was conducted using the Illumina Genome Studio software, all subsequent statistical analysis was performed using the R statistical computing software. The lumi smooth quantile normalization was implemented using the lumi package.Citation8 The BMIQ algorithm was implemented using the freely available code cited in Teschendorff et al.Citation13
Repeatability was assessed by comparison of the performance of the 15 sets of technical replicates distributed broadly across all of the chips run for the experiment. We take our use of the term “repeatability” from the Wild, Vineis, and Garte (2008) text, meaning the “ability to yield the same results… each time the test is conducted in the same laboratory.”Citation18 Standard deviations and coefficients of variation were calculated for all CpG sites run on the Infinium assay (n = 485 512 CpG sites total). The means of these measures, taken for both Infinium I and II assays separately, were taken across all replicate sets for each of the color channel normalizations conducted as a measure of procedure stability ().
Table 3. Mean standard deviation (SD) and coefficient of variation (CV) between 15 sets of replicates by type of Illumina infinium chemistry (Inf I and InfII) and different normalization procedures
Further, the root mean squared error (root-MSE) was computed between all sets of technical replicates for each of the normalization procedures evaluated. This provided an estimate of technical error in the same scale as the measurement taken, in this case on the zero-to-one scale of methylation βs. For raw, un-normalized β values, the mean root-MSE among all 15 sets of technical replicates was 0.0499 β units. Using this value as a reference, we compared the mean replicate root-MSE across each of the different normalization procedures to this standard expressed as a percentage change from the mean root-MSE for the un-normalized data set. Spearman correlation coefficients were also calculated for all replicates sets and averaged by normalization procedure as an additional measure of replicate comparability.
Batch variability was also evaluated for each of the normalization procedures implemented. Box plots of mean per-sample β for all sites interrogated on the 450K array were constructed to visualize trends in means by batch across the entire experiment. Plots are shown for three different color channel normalization procedures (lumi, worst sample RN-factor, and ASMN) by the Illumina chip batch on which they were analyzed (). Beyond visual assessment of batch trends, a site-level analysis of batch-associated variability was conducted for each of the normalizations utilized. Batch variability across chips was evaluated by ANOVA for each of the CpG sites. A site was considered “batch-associated” if the p-value associated with effect of analysis chip was less than or equal to 0.01. Levels of batch association were compared between each normalization procedure by taking the number of CpG sites meting the p ≤ 0.01 criteria for batch association as a percentage of total sites on the 450K assay.
| Abbreviations: | ||
| 450K | = | Infinium HumanMethylation450 BeadChip® |
| ASMN | = | all sample mean normalization |
| PBC | = | peak-based correction |
| SQN | = | subset quantile normalization |
| SWAN | = | subset quantile within array normalization |
| BMIQ | = | beta-mixture quantile normalization |
| IFSN | = | Illumina first sample normalization |
| RN-Factor | = | reference normalization factor |
| RNV | = | reference normalization vector |
| Root-MSE | = | root mean squared error |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Drs Farren Briggs and Vitaly Volberg for their helpful contributions to the discussion. This publication was made possible by grants 2P01ES009605-14 and 1R01ES021369-01 A1 from the National Institute of Environmental Health Science (NIEHS) and RD 83451301 from the Environmental Protection Agency (EPA). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIEHS.
References
- Foley DL, Craig JM, Morley R, Olsson CA, Dwyer T, Smith K, Saffery R. Prospects for epigenetic epidemiology. Am J Epidemiol 2009; 169:389 - 400; http://dx.doi.org/10.1093/aje/kwn380; PMID: 19139055
- Pennisi E. Behind the scenes of gene expression. Science 2001; 293:1064 - 7; http://dx.doi.org/10.1126/science.293.5532.1064; PMID: 11498570
- Ho SM, Tang WY. Techniques used in studies of epigenome dysregulation due to aberrant DNA methylation: an emphasis on fetal-based adult diseases. Reprod Toxicol 2007; 23:267 - 82; http://dx.doi.org/10.1016/j.reprotox.2007.01.004; PMID: 17317097
- Tammen SA, Friso S, Choi SW. Epigenetics: the link between nature and nurture. Mol Aspects Med 2012; 34:753 - 64; PMID: 22906839
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 2010; 11:191 - 203; http://dx.doi.org/10.1038/nrg2732; PMID: 20125086
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288 - 95; http://dx.doi.org/10.1016/j.ygeno.2011.07.007; PMID: 21839163
- Roessler J, Ammerpohl O, Gutwein J, Hasemeier B, Anwar SL, Kreipe HH, Lehmann U. Quantitative cross-validation and content analysis of the 450k DNA methylation array from Illumina, Inc. BMC Res Notes 2012; 5:210; http://dx.doi.org/10.1186/1756-0500-5-210; PMID: 22546179
- Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24:1547 - 8; http://dx.doi.org/10.1093/bioinformatics/btn224; PMID: 18467348
- Touleimat N, Tost J. Complete pipeline for Infinium(®) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012; 4:325 - 41; http://dx.doi.org/10.2217/epi.12.21; PMID: 22690668
- Schalkwyk L, Pidsley R, Wong C, Touleimat N, Defrance M, Teschendorff A, et al. wateRmelon: Illumina 450 methylation array normalization and metrics. R package version 0.99.16. 2013.
- Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450K technology. Epigenomics 2011; 3:771 - 84; http://dx.doi.org/10.2217/epi.11.105; PMID: 22126295
- Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol 2012; 13:R44; http://dx.doi.org/10.1186/gb-2012-13-6-r44; PMID: 22703947
- Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013; 29:189 - 96; http://dx.doi.org/10.1093/bioinformatics/bts680; PMID: 23175756
- Marabita F, Almgren M, Lindholm ME, Ruhrmann S, Fagerström-Billai F, Jagodic M, Sundberg CJ, Ekström TJ, Teschendorff AE, Tegnér J, et al. An evaluation of analysis pipelines for DNA methylation profiling using the Illumina HumanMethylation450 BeadChip platform. Epigenetics 2013; 8:333 - 46; http://dx.doi.org/10.4161/epi.24008; PMID: 23422812
- Illumina. GenomeStudio Methylation Module v1.8 User Guide November, 2010, Accessed 5/1/2013.
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692 - 702; http://dx.doi.org/10.4161/epi.6.6.16196; PMID: 21593595
- International HapMap Project. . www.hapmap.ncbi.nlm.nih.gov.
- Vineis P, Garte S. Biomarker Validation. In: C. Wild, Vineis P, Garte S, eds. Molecular Epidemiology of Chronic Diseases. Chichester, UK: John Wiley & Sons, Ltd, 2008:pg72.