Abstract
Methods to experimentally alter and functionally evaluate cytosine methylation in a site-specific manner have proven elusive. We describe a site-specific DNA methylation method, using synthetically methylated primers and high fidelity PCR coupled with ligation of reporter constructs. We applied this method to introduce methylated cytosines into fragments of the respective DAPK and RASSF1A promoters that had been cloned into luciferase reporters. We found that methylation of 3–7 residue CpG clusters that were 5′ adjacent to the transcription start site (TSS) of the DAPK gene produced up to a 54% decrease in promoter activity (p < 0.01). Similarly, for RASSF1A promoter reporter constructs, the methylation of either of two clusters of four CpGs each, but not an intervening cluster, produced a 63% decrease in promoter activity (p < 0.01), suggesting that precise mCpG position is crucial, and factors other than simple proximity to the TSS are at play. Chromatin immunoprecipitation analysis of these reporter constructs demonstrated that transcription factor Oct-1 and Sp1 preferentially bound the unmethylated vs. methylated DAPK or RASSF1A promoter reporter constructs at the functional CpG sites. Histone H1, hnRNP1, and MeCP2 showed preferential binding to methylated sequence at functional sites in these reporter constructs, as well as highly preferential (>8–80-fold) binding to native methylated vs. unmethylated chromatin. These results suggest that: (1) site-specific, precision DNA methylation of a reporter construct can be used for functional analysis of commonly observed gene promoter methylation patterns; (2) the reporter system contains key elements of the endogenous chromatin machinery.
Introduction
DNA methylation is an important process during normal development and homeostasis, and frequently becomes deregulated during disease pathogenesis, including carcinogenesis.Citation1,Citation2 DNA methylation profiles at the level of individual CpGs exhibit substantial variation across molecules, cells, tissues, and phenotypes within the normal human population.Citation3-Citation9 So far, little is known about the functional implications of the precise spatial distribution patterns of DNA methylation among tissues/individuals.
Functional analysis of individual CpG dinucleotides is desirable for better understanding of normal cellular physiology, inter-individual variation of DNA methylation, disease pathogenesis and for the optimization of cancer biomarker searches for robustness and precision, where it is critical to select CpG dinucleotides that exhibit a central role in gene silencing.
The high-resolution analysis of CpG methylation, including evaluating its functional implications, is an obligatory step in rational approaches to both epigenetic biomarker development, and targeted epigenetic interventions. Development of a site-specific DNA methylation technique is critical for functional analysis of individual CpG dinucleotides. In 2002, Curradi et al.Citation10 reported a patch methylation technique, but the method has not worked in our lab, and there are no further publications about this technique. Other techniques for experimental manipulation of DNA methylation have included siRNA directed DNA methylation, found in plant and yeast; there are a few papers supporting siRNA directed DNA methylation in human cells.Citation11-Citation15 Zinc finger-DNA methyltransferase has also been used for site-directed DNA methylation in vitro at low precision.Citation16-Citation19 Teng et al. reported that transfection of methylated DNA complementary to target DNA could induce the DNA methylation of the target region also at low precision.Citation20-Citation22 Recently, a CpG-free luciferase reporter vector was reported for functional analysis of DNA methylation using methyltransferase SssI.Citation23 Among the limitations of these techniques is that the methylation cannot be precisely targeted to individual single CpGs as a site-specific intervention. Recently, Arakawa et al. developed PCR base method for in vitro site-specific DNA methylation, but this method is only used for linear vectors, and is quite cumbersome.Citation24
Here we report a new protocol which is based on high fidelity PCR coupled with ligation using synthetically methylated primers for site specific methylation of reporter constructs; subsequent testing of the impact of specific methylation clusters was performed. We also explored the DNA-binding proteins associated with these methylation-specific effects. This PCR-ligation reaction technique could be used to make single or multiple CpG methylation changes in a reporter construct of interest. Therefore, it becomes possible to couple high-resolution observations of CpG methylation patternsCitation25-Citation27 to experimental recapitulation of the precise methylation sequence, and proceed to functional analysis. This technique could be useful in functional epigenetic studies, epigenetic biomarker development, and targeting for precise epigenetic interventions.
Results
Principle of site-specific DNA methylation
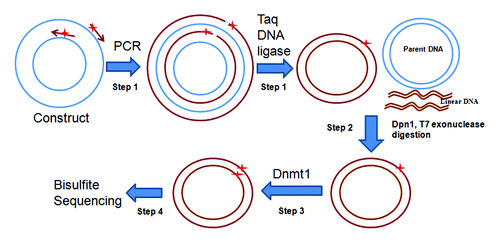
The principle of the method is shown in in four steps. Step 1, PCR-ligation coupled reaction: A reporter construct is used for site specific methylation modification and two primers with desired CpG methylation and 5′-end phosphorylated are synthesized for the amplification of the entire constructs. The primers are extended during temperature cycling by high fidelity DNA polymerase without strand-displacement activity, and generate new complementary strand DNA containing a nick, which can then be ligated by Taq DNA ligase. After 25 to 30 cycles, ~2 μg DNA product could be produced. Step 2, Dpn1 and T7 exonuclease digestion: Following temperature cycling, the product is purified and digested with Dpn1 and T7 exonuclease, in which the parental DNA template is digested by Dpn1 that recognizes 5′-Gm6ATC-3′ sequences in the parental plasmid derived from E. Coli; the linear DNA is then digested by T7 exonuclease. Step 3, Dnmt1 modification: The DNA is purified and treated with human DNA Methyltransferase (Dnmt1, New England Biolabs Inc.). Dnmt1 is specific for hemi-methylated DNA and is used to methylate the DNA strand complementary to the methylated primer. Step 4, Bisulfite sequencing: The methylated CpG site is verified by bisulfite genomic DNA sequencing.
Figure 1. Strategy for site-specific DNA methylation. Step1, Conventional reporter constructs are adapted as template. A pair of primers (arrows) containing synthetically methylated CpGs (stars) are phosphorylated at 5′ end. Both primers are extended by high-fidelity DNA polymerase to generate new complementary strand DNA containing a nick. The new complementary strand DNA is ligated to form circular DNA by Taq DNA ligase. Step 2, The parental DNA template is digested by Dpn1 which recognizes 5′-Gm6ATC-3′, which are initially derived from E. coli., and the resulting linear DNA is digested by T7 exonuclease. Step 3, Dnmt1 is used to methylate the remaining DNA strand complementary to the methylated primer. Step 4, The integrity of the intended methylated CpG sites is confirmed by bisulfite sequencing before transfection.
PCR-ligase coupled amplification of DAPK reporter constructs
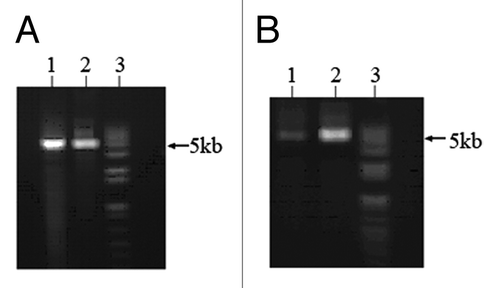
A 268 bp DAPK promoter fragment inserted into Luciferase reporter constructs (5011 bp) was effective as template. Two primers designed for effecting the site-specific DNA methylation by the PCR-ligation reaction included the synthetically methylated forward primer: 5′-GCCGCCTGCC GGCCGCCCTC CG-3′, in which the four bold C’s are methylated cytosine. Two high fidelity DNA polymerases (pfuUltra II Fusion HS DNA polymerase and Herculase II Fusion DNA polymerase were tested for PCR-ligase coupled amplification with Taq DNA ligase. PCR-ligation products were digested with Dpn1 and T7 exonuclease. Results showed that Herculase II Fusion DNA polymerase coupled with Taq DNA ligase produced high yield of circled DNA products ().
Figure 2. PCR-ligase coupled amplification of DAPK reporter constructs. Two primers containing four methylated CpGs were used for PCR-ligase coupled amplification of DAPK reporter constructs (5011bp). (A) pfuUltra II Fusion HS DNA polymerase (lane 1) and Herculase II Fusion DNA polymerase (lane 2) could amplify the constructs along with Taq DNA ligase. (B) PCR-ligation products were digested with Dpn1 and T7 exonuclease, lane 1: pfuUltra II Fusion HS DNA polymerase product; lane 2: Herculase II Fusion DNA polymerase product; lane 3: 1kb plus DNA ladder.
Verification of site-specific methylation in DAPK promoter reporter constructs
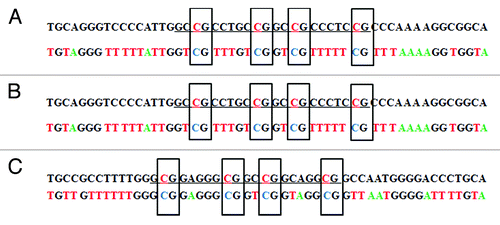
The methylation status of the newly-synthesized construct was confirmed with tag modified bisulfite sequencing (tBGS)Citation25 and then the constructs were transfected into A549 cells. Thirty-six hours after transfection, total DNA was extracted for methylation status reassessment of the reporter construct. After bisulfite treatment, the DAPK promoter was amplified with vector-specific primers and the methylation status was again determined by tag-modified bisulfite sequencing transfection. The results showed that only the intended four target CpG sites were specifically methylated in the constructs prior to transfection, and they remained methylated after transfection for at least 36 h (; Fig. S1).
Figure 3. Bisulfite sequencing of the DAPK promoter in site-specific methylated reporter constructs. Site-specific methylated constructs (MP2) were used for bisulfite treatment. The promoter region was amplified with vector specific primers flanking the promoter. PCR product was subject to direct Sanger sequencing.Citation25 (A) The sequencing result before transfection into A549 cells. (B) The sequencing result after transfection into A549 cells for 36 h. (C) The anti-sense strand sequencing result after transfection into A549 cells for 36 h. The upper sequence in each pair is genome sequence in which the underlined target sequence containing four methylated cytosines are in red characters. In the bisulfite sequencing, all unmethylated cytosines were converted to thymine. Only the four intended target cytosines were methylated in the bisulfite sequencing result. Corresponding chromatograms are depicted in the supplement (Fig. S1).
Functional analysis of DAPK promoter methylation
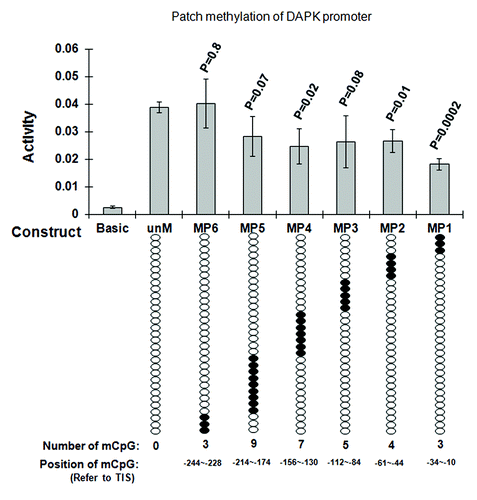
We created six site-specific methylated DAPK promoter reporter constructs (MP1-MP6) using six methylated PCR primers (Table S1) containing the same methylated CpGs clusters. As controls, unmethylated PCR primers were used for creating unmethylated DAPK promoter reporter constructs by the same protocol. Results showed that the methylation of the three CpGs (MP1) (out of 31 CpGs in the promoter) adjacent to the transcription start site (TSS) produced a 54% decrease in promoter activity (p = 0.002) as compared with the unmethylated DAPK promoter (). The methylation of four CpGs (MP2) at -44~-61 and seven CpGs at -130~-156 (MP4) decreased the promoter activity by 31% (p = 0.01) and 32% (p = 0.02). The methylation of the 12 CpG sites most upstream from the TSS had no significant effect on promoter activity. We then methylated the 12 CpGs combining the CpGs from each of three methylated oligos (MP1, MP2, MP3) together, to determine if there are any synergetic effects on the promoter activity to larger regions of methylation. Results showed that there were no synergetic effects of these combined CpGs on DAPK promoter activity repression (Fig. S2).
Figure 4. Functional analysis of CpG methylation impact on DAPK promoter activity. Regions ~60 bp in length (30 bp forward and 30 bp reverse primers fit end-to-end) were tested. The numbers of methylated CpG sites were determined by the endogenous sequence in that 60 bp region (e.g., four CpGs at -10, -23, and -34 for MP1, three CpGs for MP2, five CpGs for MP3, seven CpGs for MP4, nine CpGs for MP5 and three CpGs for MP6). Unmethylated DAPK reporter constructs (unM) and six site-specific methylated DAPK reporter constructs (MP1 to MP6) were transfected into A549 cells, PRL-TK which expresses Renilla luciferase was co-transfected as an internal control. The empty vector (Basic) was also transfected as negative control. All constructs were transfected in triplicates. After 36 h, luciferase activities were determined by dual luciferase assay. Firefly luciferase activity was normalized to Renilla luciferase activity (internal control). Here, the most 3′ cluster (the three CpGs methylated together by the MP1 patch, which covers the region -34 to -10, reference to TSS) appears to halve the promoter activity. P values represent t test comparisons with the unmethylated DAPK promoter insert (unM) control. KEY: Basic: the reporter construct without promoter; unM: The reporter construct with unmethylated DAPK promoter; MP1 to MP6: reporter constructs with DAPK promoter methylated at different CpG sites; mCpG: Methylated CpG.
Functional analysis of DNA methylation in RASSF1A promoter reporter constructs
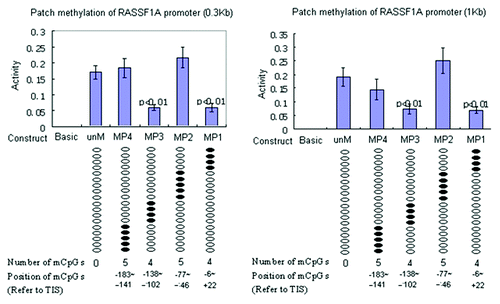
To further demonstrate our site specific methylation technique is reproducible and applicable to other genes, we tested it on RASSF1A promoter reporter constructs with two different fragment sizes of the same promoter sequence (~300 bp and 1 kb). We created four site-specific methylated RASSF1A promoter reporter constructs using four pairs of methylated PCR primers () containing 4 to 5 methylated CpGs each. Results showed that the methylation of two clusters of four CpGs each (out of 18 analyzed CpGs in the promoter) adjacent to the TSS produce a 63% decrease in promoter activity (p < 0.01), as compared with the unmethylated RASSF1A promoter (). The methylation of the other 10 CpG sites had no significant effect on promoter activity. The results were robust and identical, whether the smaller or larger encompassing region was cloned into the reporter vector.
Table 1. Proteins differentially binding to methylated or unmethylated DNA oligos repeating DAPK and RASSF1A proximal promoters by LC-MS/MS
Figure 5. Functional analysis of DNA methylation impact on RASSF1A promoter reporter constructs. Unmethylated RASSF1A reporter constructs (unM) and four site-specific methylated reporter constructs (MP1 to MP4) were transfected into A549 cells. PRL-TK, which expresses Renilla luciferase was co-transfected as an internal control. The empty vector (Basic) was also transfected as negative control. All constructs were transfected in triplicates. After 36 h, luciferase activities were determined by dual luciferase assay. Firefly luciferase activity was normalized to Renilla luciferase activity (internal control). Here, each of two clusters, each containing four methylated CpGs at +21,+15,+2,-6 and -138, -129,-125,-102, reference to TSS, appear to halve promoter activity. P values represent t test comparisons with the unmethylated RASSF1A promoter insert (unM) control. Basic, the reporter construct without promoter; unM, the reporter construct with unmethylated RASSF1A promoter; MP1 to MP4, reporter constructs with RASSF1A promoter methylated at different CpG sites; mCpG, methylated CpG.
Protein binding analysis on methylated and unmethylated promoter
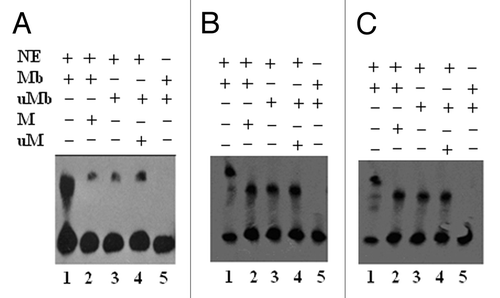
To begin to understand how the critical CpG clusters in DAPK and RASSF1A promoter impact on gene expression, we compared nuclear proteins binding to the methylated vs. unmethylated oligos, by performing gel-shift assays. The results () showed that the methylated DAPK probe (DAPK-MP1) did bind nuclear extract proteins, which could then be competitively inhibited by unlabeled methylated oligo DNA (, lanes 1, 2). The unmethylated probe (DAPK-P1) displayed some non-specific binding (, lanes 3, 4). Similarly, methylated RASSF1A probes (RASSF1A-MP1 and RASSF1A-MP3) also bind nuclear extract proteins, which could be partially inhibited by unlabeled methylated oligo DNA (, lanes 1, 2). The unmethylated RASSF1A probes (RASSF1A-MP1 and RASSF1A-MP3) also displayed some non-specific binding (, lanes 3, 4).
Figure 6. LightShift EMSA for biotin labeled methylated and unmethylated DAPK-MP1, RASSF1A –MP1, and MP3 oligos from the DAPK and RASSF1A promoter. (A): Lane 1: Nuclear extract proteins specifically bind to the methylated DAPK-MP1 probe. Lane 2: The binding of nuclear extract proteins to the methylated DAPK-MP1 was competed with unlabeled methylated DAPK-MP1. Lane 3: Nuclear extract proteins nonspecifically bind to unmethylated DAPK-P1 probe. Lane 4: The nonspecific binding of nuclear extract proteins to the unmethylated DAPK-P1 was not competed with unlabeled unmethylated DAPK-P1. Lane 5: Methylated DAPK-MP1 only as negative control. (B): Lane 1: Nuclear extract proteins specifically bind to the methylated RASSF1A-MP1. Lane 2: The binding of nuclear extract proteins to the methylated RASSF1A-MP1 was partially inhibited by unlabeled methylated RASSF1A-MP1. Lane 3: Nuclear extract proteins bind to unmethylated RASSF1A-P1. Lane 4: The binding of nuclear extract proteins to the unmethylated RASSF1A-P1 was competed with unlabeled unmethylated RASSF1A-P1. Lane 5: RASSF1A-MP1 only as negative control. (C): Lane 1: Nuclear extract proteins specifically bind to the methylated RASSF1A-MP3. Lane 2: The binding of nuclear extract proteins to the methylated RASSF1A-MP3 was partially inhibited by unlabeled methylated RASSF1A-MP3. Lane 3: Nuclear extract proteins bind to unmethylated RASSF1A-P3. Lane 4: The binding of nuclear extract proteins to the unmethylated RASSF1A-P3 was competed with unlabeled unmethylated RASSF1A-P3. Lane 5: RASSF1A-MP3 only as negative control. NE: nuclear extracts; Mb: methylated DNA labeled with biotin; M: methylated DNA unlabeled; uMb: unmethylated DNA labeled with biotin; uM: unmethylated DNA unlabeled.
Identification of proteins binding to the methylated oligo
In order to identify the responsible proteins binding to the methylated oligos (DAPK -MP1, RASSF1A-MP1, and RASSF1A-MP3), we immobilized the methylated and unmethylated DNA oligo to the Dynabeads (Invitrogen), and pulled down the binding proteins by affinity purification. The results of LC-MS/MS analysis (Fig. S3) showed that: for DAPK promoter there are two proteins (histone H1 and hnRNPH1) specifically binding to the methylated DAPK-MP1 (), and five proteins non-differentially binding to both methylated DAPK-MP1 and unmethylated DAPK-P1 (Table S2). For the RASSF1A promoter, there are five proteins (including histone H1 and hnRNPH1) differentially binding to the methylated RASSF1A-MP1, and two proteins specifically binding to the methylated RASSF1A-MP3 (). There are additional four proteins specifically binding to the unmethylated RASSF1A-P1 and nine proteins specifically binding to the unmethylated RASSF1A-P3 (). There are 10 proteins non-differentially binding to both methylated RASSF1A-MP1 and unmethylated RASSF1A -P1, 14 proteins non-differentially binding to both methylated RASSF1A-MP3 and unmethylated RASSF1A –P3 (Table S2).
Chromatin immunoprecipitation analysis of reporter constructs containing functional CpG sites
In silico sequence analyses revealed that the three CpG sites methylated in the DAPK-MP1 encompass a putative binding site of transcription factor Oct-1, and that there are 16 putative Sp1 binding sites in the DAPK promoter and 17 putative Sp1 binding sites in the RASSF1A promoter. We did not find any transcription factors (Oct-1 or Sp-1) nor known methylated CpG binding proteins in the LC-MS/MS results; only histone H1 and hnRNP uniquely bound the methylated DNA oligos (DAPK-MP1 and RASSF1A-MP1) containing the functional CpG sites. We considered the limitation of detectability of LC-MS/MS for lower abundant proteins, which is about 200 pmol.Citation52 We therefore performed ChIP assay in A549 cells transfected with DAPK-MP1, RASSF1A-MP1, and RASSF1A-MP3 reporter constructs using anti-Histone H1 and anti-hnRNP, anti-Oct1, anti-Sp1, and anti-MeCP2 antibodies (; Fig. S4). The results of real time PCR using reporter construct specific primers indicated that Oct-1 was uniquely bound to unmethylated DAPK construct and not the methylated DAPK-MP1. Sp1 was also preferentially bound to promoters in unmethylated reporter constructs of both DAPK and RASSF1A; the binding ability to methylated DAPK-MP1, RASSF1A-MP1, and RASSF1A-MP3 reporter constructs was reduced to 82%, 10%, and 16% of their unmethylated counterparts (; Fig. S4). MeCP2 was bound to each of methylated promoter constructs (Fig. S4). The histone H1 and hnRNPH1 were highly preferentially bound to methylated constructs (; Fig. S4), which is consistent with the results of ChIP assay from the unmethylated promoter in native chromatin from NHBE cells and methylated promoter in native chromatin from MCF7 cells (Table S3; Fig. S5). The ChIP results therefore indicate that the reporter system contains many elements of the endogenous chromatin machinery.
Table 2. Real time PCR results for ChIP assay
Discussion
DNA methylation sequencing in a variety of systems and contexts reveals complex patterns of promoter methylation. We have here shown that methylation of certain specific CpG sites drive the repression of a reporter gene more than other CpGs and confirmed some of the proteins mediating that effect. The methylation reporter method used is novel in that synthetically methylated oligos are precisely incorporated into the reporter constructs by a PCR + ligation reaction to create precisely methylated clusters, mimicking same of the detail of in vivo chromatin patterns we have previously observed.Citation25,Citation53
The site-specific DNA methylation technique reported here is based on PCR coupled with DNA ligation, and is suitable for a circular vector. In our method, because the PCR is coupled to a Taq DNA ligase reaction, the reaction conditions of the DNA polymerase must be compatible with Taq DNA ligase. Therefore, we chose a compatible DNA polymerase for the DNA ligase combination base on the yield of the circular PCR product. Our method is quite different from a recently reported method for linear reporter vector creation,Citation24 in which PCR and ligation are in different reactions, the PCR products are prepared in single-strand form before ligation, and that strategy, as a consequence, is somewhat more complex to execute.Citation24
For our primer design, the target methylation CpGs are integrated into primers, and the 5′ end must be phosphorylated for DNA ligation. The primer size could be up to 60 bp, as tested in our lab to date. The forward and reverse primers do not have to be 5′ end adjacent since the DNA extension is circular, but the extension time should be adjusted to the reporter construct size. In PCR-ligation DNA products, some products may have one strand ligated and circular, the other strand is still nicked because of ligation inefficiency. Addition of a ligation step using ligase specific buffer may increase circular DNA up to 20% (not shown).
We made the arbitrary choice to test promoter sequences from two tumor suppressor genes. DAPK (Death-associated protein kinase) is a serine/threonine, microfilament-bound kinase involved in interferon-, TNF- or FAS-induced apoptosis.Citation54,Citation55 Loss of function of this kinase could significantly affect cellular apoptosis. Methylation of the DAPK promoter is seen in both lung adenocarcinoma and squamous-cell carcinoma at prevalences ranging from 30–48%,Citation56-Citation58 and appears to occur early in carcinogenesis.Citation59,Citation60
When we inserted a 300bp DNA fragment from the DAPK promoter directly upstream of the transcription start site into the luciferase reporter construct and progressively methylated the 31 CpGs in individual clusters of convenience, encompassing every CpG site in the fragment, we found that three CpGs (out of 31 CpGs in the promoter fragment) adjacent to the transcription start site (TSS) produce the most significant decrease in promoter activity as compared with the unmethylated DAPK promoter. Other clusters of four methylated CpGs at one site (-44~-61) and five methylated CpGs at another (-54~-112) also decreased the promoter activity. When methylating all 11 proximate CpGs from DAPK-MP1, DAPK-MP2, and DAPK-MP3 together in one reporter, there were no apparent synergetic effects on the promoter activity. We speculate that this lack of synergy may be due to the fact that these CpGs are very close together (within 100 bp), and it is plausible that one large complex of DNA methylation proteins could potentially block the entire 100 bp region from transcription factors access. This is a hypothesis requiring further testing.
We also verified the methylation reporter using part of the RASSF1A promoter. This member of the RAS superfamily of GTP-binding proteins has an important role in signal-transduction pathways that controls cell proliferation, differentiation, and death,Citation61,Citation62 and it has been reported that RASSF1A could be inactivated in 40–60% and 100% of non-small-cell and small-cell lung cancers, respectively.Citation63,Citation64
Two promoter DNA fragments (~1000 bp, and a ~300 bp subfragment of the first) from the same RASSF1A gene promoter sequence were each inserted into identical reporter constructs, and the same cluster of methylation was created in each by site-specific methylation. The results indicate that the methylation effects on the promoter activities were reproducible, and not affected by the size of inserted promoter, suggesting some robustness to broader sequence context. There were two specific regions (MP1 and MP3) that showed reduction of reporter expression, in both the larger and smaller RASSF1A promoter fragment constructs, spanning another (MP2) where no such effect was apparent. This non-response to the MP2 cluster methylation suggests sequence and/or position specificity of the methylation effect on reporter expression, and that the effect is not attributable to mere proximity to the TSS alone. Recent reportsCitation65,Citation66 showed that RASSF1A methylation could be a candidate biomarker for lung cancer. We compared the methylated CpG sites, Deng et al.Citation65 examined and some were included in our MP1 and MP2 region, Fraipont et al.Citation66 checked the CpGs downstream of the TIS.
As to local mechanism of methylation impact on reporter expression, the proteins associating with the methylation effects in the DAPK and RASSF1A reporters tested were initially explored by EMSA for the relevant methylated/unmethylated oligo pair. The subsequent LC-MS/MS analysis detected differential binding of histone H1 and hnRNPH1, but did not find methylated CpG binding proteins (MeCP) or any transcription factors, we presume due to sensitivity limits of LC-MS/MS. We therefore proceeded to test expected transcription factors (Oct-1 for DAPK, Sp1 for both) and MeCP2 with ChIP of the reporter constructs themself. Sp1 is a sequence-specific transcription factor that recognizes GC boxes.Citation67 In silico sequence analyses revealed that the TATAless, GC-rich promoter of the DAPK and RASSF1A contained many Sp1 binding sites, but only one Oct-1 binding site in the DAPK-MP1 region of interest. Oct1 is widely expressed in adult tissues, and is implicated in crucial and versatile biological events, such as embryogenesis, neurogenesis, immunity, and body glucose and amino acid metabolism.Citation68 ChIP analysis demonstrated that MeCP2 uniquely bound to the methylated reporter constructs and transcription factor Oct-1 and Sp1 could bind to unmethylated DNA but the binding was inhibited by DNA methylation at these binding sites. Histone H1 is an abundant and essential component of chromatin and has strong competition with MeCP for nucleosome binding sites.Citation69,Citation70 .There are papers in the literature suggesting that certain histone H1 subclasses bind preferentially to GC-rich DNACitation71-Citation75 and other H1 subclasses bind primarily to AT rich sequences.Citation76,Citation77 hnRNPH1 has not previously been associated with DNA methylation regulation. One can speculate this RNA binding protein is a logical regulatory point for pre-mRNA processing, mRNA metabolism, and transport, but this clearly requires further dissection. Overall, the results indicate that the reporter system contains many elements of the endogenous chromatin regulatory machinery.
In conclusion, we have developed a simple PCR-ligation based method for site-specific methylation in reporter constructs. This method has three advantages: complete precision in site specificity of methylation, simplicity, and high methylation efficiency. The limitation is that the methylation alterations are limited to the two PCR primer regions, and therefore strategies to circumvent this size constraint need to be developed. This method can be used to aid the validation of specific CpG methylation sites as physiologically relevant, patho-physiologically relevant, and/or as potential disease biomarkers. This technical development could also help guide development of sequence-specific alteration of DNA methylation for preventive and therapeutic purposes.
Materials and Methods
Construction of promoter-luciferase constructs
The respective 5′ flanking regions of the DAPK and RASSF1A genes were amplified from genomic DNA of normal human bronchial epithelial cells (NHBE; Lonza, Inc.) by high fidelity PCR. PCR reactions were prepared in 50-μl volumes containing 1× Pfx Amplification buffer (invitrogen) 0.3 μM of each primer, 0.3 mM of each of the four deoxynucleoside triphosphates (dNTPs), 200 ng of genomic DNA. One unit of AccuPrime™ Pfx DNA polymerase (Invitrogen). Thermocycling was conducted using either the GeneAmp® PCR System 9600 (Applied Biosystems) for 95 °C 5 min, 35 cycles of 95 °C 15 s, 60 °C 30 s and 68 °C 1 min. Three pairs of primers were used (the underlined sequences are Sac1 and Nco1 restriction sites for cloning): DAPK-F: CATTATAGAG CTCGCGTGGG TGTGGGGCGA GTG and DAPK-R: TAATTCATCC ATGGCGTTGG CTCGGTCCGG CTGT for amplification of 268 bp (-243~+25) DAPK promoter. In order to test sequence context, two differently sized fragments (288 bp and 1023 bp) of the proximal RASSF1A promoter were obtained using two pairs of primers: RASSF1A-F1: CATTATAGAG CTCAAGTGTG TTGCTTCAGC AAACC and RASSF1A-R1: TAATTCACCA TGGCGGCGGG AAGGAGCTGA GG for amplification of 288 bp (-266~+22) RASSF1A promoter. RASSF1A-F2: CATTATAGAG CTCCCTAGAT CCCAGAAATC TGGGAG, and RASSF1A-R1 for amplification of 1023 bp (-1000~+22) RASSF1A promoter. PCR products were purified from an agarose gel and digested with Sac1 and Nco1. The three digested fragments were cloned into the Sac1/Nco1 site of the promoterless pGL3-basic firefly luciferase reporter vector (Promega) to produce three constructs. All constructs were sequenced prior to use in order to ensure a correct insertion sequence and direction.
Overall strategy of site-specific DNA methylation
To methylate the specific CpG site in the promoter of reporter constructs, we used a site-specific DNA methylation technique, where a pair of primers containing synthetically methylated CpGs is employed to amplify the recombinant reporter construct with high-fidelity PCR ().
The two primers are 5′-end adjacent for the amplification of the entire constructs, and 5′-end phosphorylated for ligation. The primers are extended during temperature cycling by high fidelity DNA polymerase without strand-displacement activity, and generate new complementary strand DNA containing a nick, which can then be ligated by Taq DNA ligase. Following temperature cycling, the parental DNA template is digested by Dpn1 which recognizes methylated 5′-Gm6ATC-3′ sequences in the parental constructs, which are initially derived from E. coli. The linear DNA is digested by T7 exonuclease. After purification, the product is treated with human DNA Methyltransferase (Dnmt1, New England Biolabs Inc.). Dnmt1 is specific for hemi-methylated DNA) and is used to methylate the DNA strand complementary to the methylated primer. The methylated CpG site is verified by bisulfite genomic DNA sequencing.
Site-specific methylation of promoter reporter construct
We created six site-specific methylated DAPK promoter reporter constructs and eight site-specific methylated RASSF1A promoter reporter constructs using PCR primers containing 3 to 9 methylated CpGs each (). As controls, unmethylated PCR primers were used for creating unmethylated promoter reporter constructs by the same protocol. The PCR-ligation reaction was performed in 50 µl 1× Herculase II reaction buffer (Stratagene), 1mM NAD+ (NEB), 0.5 µl Herculase II fusion DNA polymerase, 160 U Taq DNA ligase and 200 µM each dNTP, using 10 ng reporter constructs as template, 250nM each primer. Reaction condition: denature at 98 °C for 2 min, then 98 °C × 20 s, 60 °C × 30 s, 72 °C × 3 min, and 65 °C ×5 min for 26 cycles. The PCR-ligation reaction is the critical step in this site-specific DNA methylation protocol. The denature temperature 98 °C is necessary for supercoiled plasmid templates. The DNA polymerase should have no strand displacement activity. The reaction condition should be compatible with tag DNA ligase (e.g., pH value, salt concentration). The PCR-ligation product was purified with QIAquick PCR purification kit (Qiagen) and subject to digestion with Dpn1 and T7 exonuclease in NEB buffer 2 overnight for removing the parental DNA and non-circular DNA. The circular DNA constructs containing the methylated primer were purified with QIAquick PCR purification kit (Qiagen) and treated with Dnmt1 to methylate the complementary strand of the methylated primer. Dnmt1 treatment was performed in 50 μl reaction containing 50 mM Tris-HCl, 1 mM Dithiothreitol, 1 mM EDTA, 5% Glycerol, 100 μg/ml Bovine Serum Albumin and 160 μM S-adenosylmethionine, 2 μg DNA, 4 units of Dnmt1, pH 7.8 and incubated at 37 °C for 4 h. The removing of linear DNA and parent DNA by Dpn1 and T7 exonuclease before Dnmt1 treatment may increase the efficiency of Dnmt1 due to the removing of unnecessary substrate. The site-specific methylated constructs were subject to bisulfite treatment and the promoter region was amplified for DNA sequencing. Site-specific methylated and unmethylated promoter reporter constructs were transfected into A549 cells. After 36 h incubation, promoter activity was determined by dual luciferase assay.
Cell culture and transfection
A549 cells were maintained in F-12K nutrient mixture medium (Invitrogen) supplemented with 10% (v/v) FBS 10 µg/ml gentamicin at 37 °C in a humidified 5% CO2 atmosphere. Transfections were performed in triplicate in 24-well plates. Each well was plated with 2 × 105 cells 24h before transfection. Transfection was performed using 0.5 μg of reporter gene construct, together with 20ng of reference PRL-TK vector (Promega) expressing Renilla luciferase constitutively, which serves as an internal control of transfection efficiency. DNA was combined with 3 μl of Lipofectamine LTX and 1 μl of plus reagent in Opti-MEM reduced serum medium (Invitrogen). After a 36 h incubation, the cells were lysed by Passive Lysis Buffer (Promega). The activities of firefly and Renilla luciferases were determined by Dual-Luciferase reporter assay system (Promega). Firefly luciferase activity was then numerically normalized to Renilla luciferase activity.
GC tag-modified bisulfite genomic DNA sequencing (tBGS) Citation 25
Bisulfite treatment was performed with DNA methylation kit (Zymo Research), with the reaction condition optimized to 50 °C for 20 h. DNA was eluted in 10 µl of elution buffer. Bisulfite-treated genomic DNA was amplified by two step PCR as described previously.Citation25 Step 1: PCR amplification with the vector specific primers. Step 2: Nest PCR using tag-modified primers: an 18–45 base tag containing at least 10 cytosines is added to the 5′end of each of the forward primer, similarly, an 18–45 base tag containing at least 10 guanines is added to the 5′end of each of the reverse primer. The product of the first step PCR is used as template. The nested PCR product is purified by DNA gel extraction kit (Qiagen), and subject to directly sequencing, using the tag as sequencing primer. The GC tag-modified primer was used because for bisulfite treated DNA, due to the bisulfite conversion of unmethylated C ≥ U in the template, there is low GC content in the PCR product. This is especially apparent when the target sequence is unmethylated.Citation19 This low GC content PCR product often produces C-noisy signal background in sense strand sequencing result, or G noisy signal background in antisense strand sequencing result, making the result impossible to interpret. Therefore, we used GC enriched tag-modified PCR primers, which yields a 5′ and 3′ GC-tagged PCR product that sufficiently enhances the GC content to allow successful direct PCR product sequencing.
Electrophoretic mobility shift assay (EMSA)
Synthesized methylated and unmethylated double strand oligo DNA containing critical functional CpG sites of DAPK and RASSF1A promoter were end labeled with biotin (Integrated DNA Technologies). Nuclear extracts were prepared from A549 cells, using NE-PER nuclear extraction kit (Pierce Biotechnology). EMSA was performed with LightShift Chemiluminescent EMSA Kit (Pierce Biotechnology).
LC-MS/MS analysis
Approximately 9.0 mg of magnetic Dynabeads (Invitrogen) was incubated with 5.0 μmol biotin labeled DNA for 10 min at room temperature in the binding and washing buffer containing 5mM Tris-HCl (pH7.5), 0.5 mM EDTA and 1 M NaCl. The DNA-coupled magnetic Dynabeads (Invitrogen) were washed with 500 µl of binding and washing buffer twice and incubated with 4 μg of poly(deoxyinosinic-deoxycytidylic; Pierce Biotechnology) and 4 mg of nuclear extracts from A549 cells for 30 min at 37 °C in Binding buffer (Pierce Biotechnology). The Dynabead-DNA–protein complex was precipitated, and washed with 500 µl of Binding buffer (Pierce Biotechnology) three times and then suspended in 50 µl of 25 mM buffer ABC (ammonium bicarbonate, pH 8.0). The bound proteins were released by incubating at 95 °C for 5min. and reduced using TCEP (tris[2-carboxyethyl]phosphine, final concentration 10 mM) for 30 min at room temperature, then alkylated using iodoacetamide (final concentration of 50 mM) for 30 min at room temperature in the dark. The final volume was adjusted to 100 μl using 25 mM buffer ABC for all approaches, then 1 μl of 1% ProteaseMaxTM trypsin enhancer and 200 ng of trypsin (Promega) were added and the digestion was performed at 37 °C overnight with gentle shaking. The reaction was stopped by adding 1 μl of 50% trichloroacetic acid (TCA) and centrifuged at 20 000 × g for 30 min. The supernatant was transferred to a new tube and directly analyzed by LC-MS/MS.
LC-MS/MS results were created from the raw mass spectra using Proteome Discoverer 1.2, merged and searched against the mammalian NCBI database (May 27, 2011) using the in-house Mascot Protein Search engine. The following parameters for searches were used: trypsin 2 missed cleavages; fixed modification of carbamidomethylation (Cys); variable modifications of deamidation (Asn and Gln) and oxidation (Met); monoisotopic masses; peptide mass tolerance of 3.0 Da; product ion mass tolerance of 0.6 Da. Proteins are considered identified having at least two bold red significant (p < 0.05) peptides.
Chromatin Immunoprecipitation (ChIP) analysis
Approximately 106 cells were transfected with promoter reporter constructs using Lipofectamine as described above. After 36 h, cells were treated with 1% formaldehyde (final concentration, v/v) for 10 min at room temperature to cross-link proteins to DNA. Chromatin immunoprecipitation was performed using Pierce Agarose ChIP kit (Pierce Biotechnology). Briefly, the cross-linked DNA was fragmented with Micrococcal nuclease and precipitated with antibodies: anti-Histone H1 (Thermo Scientific), anti-hnRNP (Thermo Scientific), Anti-Oct1 (Santa Cruz), anti-Sp1 (Santa Cruz), and anti-MeCP2 (LifeSpan Biosciences). The target DNA was detected by real time PCR, using reporter construct specific primers: Forward: 5′-TATCGATAGG TACCGAGCTC-3′; Reverse: 5′-GTTTTTGGCG TCTTCCATGG-3′; After 45 cycles of real time PCR, the precipitated DNA was normalized to input DNA by 1/2∆CT, (∆CT equals CT of the immunoprecipitated DNA minus CT of input DNA), The ratio of the immunoprecipitated DNA from methylated constructs vs. that from the unmethylated constructs was calculated by 1/2∆∆CT (∆∆CT equals ∆CT of the immunoprecipitated DNA from methylated constructs minus ∆CT of the immunoprecipitated DNA from unmethylated constructs). The PCR product size for DAPK promoter reporter construct was 308 bp. The PCR product size for DAPK reporter construct was 308 bp and for RASSF1A construct was 328 bp. Final PCR product was analyzed on 1.5% agarose gel.
Data analysis
The promoter activities of the various methylated reporter constructs were determined by Dual-Luciferase reporter assay system (Promega). Firefly luciferase activity was then normalized to Renilla luciferase activity (internal transfection efficiency control). T test was used to compare triplicate luciferase levels of the variously methylated promoters with that of unmethylated promoters, using SigmaSTAT software.
Additional material
Download Zip (795.5 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by NIH grants: RC1 CA145422; R21 CA 121068; R03 CA132145; R01 CA106186; 1 R21 CA104812.
Acknowledgments
David Reynolds and the Genomics Core at Albert Einstein College of Medicine for help with DNA sequencing; Edward Nieves, Myrasol Callaway, and Berta Burd Proteomics Core of Albert Einstein College of Medicine for the LC-MS/MS analysis. Also, we offer gratitude to Drs Matthew Scharff, Eric Bouhassira, Vern Schramm, Quan Du, Joseph Locker, and Arthur Skoltchi for helping to clarify previous drafts of the manuscript.
Patent
Patent application for this method was submitted in 2011.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/26195
References
- Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007; 447:425 - 32; http://dx.doi.org/10.1038/nature05918; PMID: 17522676
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature 2007; 447:433 - 40; http://dx.doi.org/10.1038/nature05919; PMID: 17522677
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 2012; 44:40 - 6; http://dx.doi.org/10.1038/ng.969; PMID: 22120008
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Res 2010; 20:320 - 31; http://dx.doi.org/10.1101/gr.101907.109; PMID: 20133333
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315 - 22; http://dx.doi.org/10.1038/nature08514; PMID: 19829295
- Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, Smith AD. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 2011; 146:1029 - 41; http://dx.doi.org/10.1016/j.cell.2011.08.016; PMID: 21925323
- Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR, et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell 2011; 44:17 - 28; http://dx.doi.org/10.1016/j.molcel.2011.08.026; PMID: 21924933
- Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, Zheng H, Yu J, Wu H, Sun J, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol 2010; 8:e1000533; http://dx.doi.org/10.1371/journal.pbio.1000533; PMID: 21085693
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O’Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011; 471:68 - 73; http://dx.doi.org/10.1038/nature09798; PMID: 21289626
- Curradi M, Izzo A, Badaracco G, Landsberger N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol Cell Biol 2002; 22:3157 - 73; http://dx.doi.org/10.1128/MCB.22.9.3157-3173.2002; PMID: 11940673
- Kawasaki H, Taira K. Induction of DNA methylation and gene silencing by short interfering RNAs in human cells. Nature 2004; 431:211 - 7; http://dx.doi.org/10.1038/nature02889; PMID: 15311210
- Schoft VK, Chumak N, Mosiolek M, Slusarz L, Komnenovic V, Brownfield L, Twell D, Kakutani T, Tamaru H. Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep 2009; 10:1015 - 21; http://dx.doi.org/10.1038/embor.2009.152; PMID: 19680290
- Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004; 305:1289 - 92; http://dx.doi.org/10.1126/science.1101372; PMID: 15297624
- Castanotto D, Tommasi S, Li M, Li H, Yanow S, Pfeifer GP, Rossi JJ. Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells. Mol Ther 2005; 12:179 - 83; http://dx.doi.org/10.1016/j.ymthe.2005.03.003; PMID: 15963934
- Suzuki K, Shijuuku T, Fukamachi T, Zaunders J, Guillemin G, Cooper D, Kelleher A. Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J RNAi Gene Silencing 2005; 1:66 - 78; PMID: 19771207
- Meister GE, Chandrasegaran S, Ostermeier M. Heterodimeric DNA methyltransferases as a platform for creating designer zinc finger methyltransferases for targeted DNA methylation in cells. Nucleic Acids Res 2010; 38:1749 - 59; http://dx.doi.org/10.1093/nar/gkp1126; PMID: 20007601
- Li F, Papworth M, Minczuk M, Rohde C, Zhang Y, Ragozin S, Jeltsch A. Chimeric DNA methyltransferases target DNA methylation to specific DNA sequences and repress expression of target genes. Nucleic Acids Res 2007; 35:100 - 12; http://dx.doi.org/10.1093/nar/gkl1035; PMID: 17151075
- Smith AE, Ford KG. Specific targeting of cytosine methylation to DNA sequences in vivo. Nucleic Acids Res 2007; 35:740 - 54; http://dx.doi.org/10.1093/nar/gkl1053; PMID: 17182629
- Smith AE, Hurd PJ, Bannister AJ, Kouzarides T, Ford KG. Heritable gene repression through the action of a directed DNA methyltransferase at a chromosomal locus. J Biol Chem 2008; 283:9878 - 85; http://dx.doi.org/10.1074/jbc.M710393200; PMID: 18272522
- Hsiao SH, Lee KD, Hsu CC, Tseng MJ, Jin VX, Sun WS, Hung YC, Yeh KT, Yan PS, Lai YY, et al. DNA methylation of the Trip10 promoter accelerates mesenchymal stem cell lineage determination. Biochem Biophys Res Commun 2010; 400:305 - 12; http://dx.doi.org/10.1016/j.bbrc.2010.08.048; PMID: 20727853
- Hsu CC, Li HP, Hung YH, Leu YW, Wu WH, Wang FS, Lee KD, Chang PJ, Wu CS, Lu YJ, et al. Targeted methylation of CMV and E1A viral promoters. Biochem Biophys Res Commun 2010; 402:228 - 34; http://dx.doi.org/10.1016/j.bbrc.2010.09.131; PMID: 20933502
- Teng IW, Hou PC, Lee KD, Chu PY, Yeh KT, Jin VX, Tseng MJ, Tsai SJ, Chang YS, Wu CS, et al. Targeted methylation of two tumor suppressor genes is sufficient to transform mesenchymal stem cells into cancer stem/initiating cells. Cancer Res 2011; 71:4653 - 63; http://dx.doi.org/10.1158/0008-5472.CAN-10-3418; PMID: 21518779
- Klug M, Rehli M. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 2006; 1:127 - 30; http://dx.doi.org/10.4161/epi.1.3.3327; PMID: 17965610
- Arakawa T, Ohta T, Abiko Y, Okayama M, Mizoguchi I, Takuma T. A polymerase chain reaction-based method for constructing a linear vector with site-specific DNA methylation. Anal Biochem 2011; 416:211 - 7; http://dx.doi.org/10.1016/j.ab.2011.05.017; PMID: 21669180
- Han W, Cauchi S, Herman JG, Spivack SD. DNA methylation mapping by tag-modified bisulfite genomic sequencing. Anal Biochem 2006; 355:50 - 61; http://dx.doi.org/10.1016/j.ab.2006.05.010; PMID: 16797472
- Yamada N, Nishida Y, Tsutsumida H, Hamada T, Goto M, Higashi M, Nomoto M, Yonezawa S. MUC1 expression is regulated by DNA methylation and histone H3 lysine 9 modification in cancer cells. Cancer Res 2008; 68:2708 - 16; http://dx.doi.org/10.1158/0008-5472.CAN-07-6844; PMID: 18413738
- Taylor KH, Kramer RS, Davis JW, Guo J, Duff DJ, Xu D, Caldwell CW, Shi H. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res 2007; 67:8511 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-07-1016; PMID: 17875690
- Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 2008; 319:819 - 21; http://dx.doi.org/10.1126/science.1149250; PMID: 18258916
- Izzo A, Kamieniarz K, Schneider R. The histone H1 family: specific members, specific functions?. Biol Chem 2008; 389:333 - 43; http://dx.doi.org/10.1515/BC.2008.037; PMID: 18208346
- Happel N, Doenecke D. Histone H1 and its isoforms: contribution to chromatin structure and function. Gene 2009; 431:1 - 12; http://dx.doi.org/10.1016/j.gene.2008.11.003; PMID: 19059319
- Honoré B, Rasmussen HH, Vorum H, Dejgaard K, Liu X, Gromov P, Madsen P, Gesser B, Tommerup N, Celis JE. Heterogeneous nuclear ribonucleoproteins H, H’, and F are members of a ubiquitously expressed subfamily of related but distinct proteins encoded by genes mapping to different chromosomes. J Biol Chem 1995; 270:28780 - 9; http://dx.doi.org/10.1074/jbc.270.48.28780; PMID: 7499401
- Masuda A, Shen XM, Ito M, Matsuura T, Engel AG, Ohno K. hnRNP H enhances skipping of a nonfunctional exon P3A in CHRNA1 and a mutation disrupting its binding causes congenital myasthenic syndrome. Hum Mol Genet 2008; 17:4022 - 35; http://dx.doi.org/10.1093/hmg/ddn305; PMID: 18806275
- Wu Q, Gou Y, Wang Q, Jin H, Cui L, Zhang Y, He L, Wang J, Nie Y, Shi Y, et al. Downregulation of RPL6 by siRNA inhibits proliferation and cell cycle progression of human gastric cancer cell lines. PLoS One 2011; 6:e26401; http://dx.doi.org/10.1371/journal.pone.0026401; PMID: 22043320
- Manojlovic Z, Stefanovic B. A novel role of RNA helicase A in regulation of translation of type I collagen mRNAs. RNA 2012; 18:321 - 34; http://dx.doi.org/10.1261/rna.030288.111; PMID: 22190748
- Lin L, Li Y, Pyo HM, Lu X, Raman SN, Liu Q, Brown EG, Zhou Y. Identification of RNA helicase A as a cellular factor that interacts with influenza A virus NS1 protein and its role in the virus life cycle. J Virol 2012; 86:1942 - 54; http://dx.doi.org/10.1128/JVI.06362-11; PMID: 22171255
- Kuroda T, Murayama A, Katagiri N, Ohta YM, Fujita E, Masumoto H, Ema M, Takahashi S, Kimura K, Yanagisawa J. RNA content in the nucleolus alters p53 acetylation via MYBBP1A. EMBO J 2011; 30:1054 - 66; http://dx.doi.org/10.1038/emboj.2011.23; PMID: 21297583
- Owen HR, Elser M, Cheung E, Gersbach M, Kraus WL, Hottiger MO. MYBBP1a is a novel repressor of NF-kappaB. J Mol Biol 2007; 366:725 - 36; http://dx.doi.org/10.1016/j.jmb.2006.11.099; PMID: 17196614
- Sun J, Lee KJ, Davis AJ, Chen DJ. Human Ku70/80 protein blocks exonuclease 1-mediated DNA resection in the presence of human Mre11 or Mre11/Rad50 protein complex. J Biol Chem 2012; 287:4936 - 45; http://dx.doi.org/10.1074/jbc.M111.306167; PMID: 22179609
- Fell VL, Schild-Poulter C. Ku regulates signaling to DNA damage response pathways through the Ku70 von Willebrand A domain. Mol Cell Biol 2012; 32:76 - 87; http://dx.doi.org/10.1128/MCB.05661-11; PMID: 22037767
- Screaton GR, Cáceres JF, Mayeda A, Bell MV, Plebanski M, Jackson DG, Bell JI, Krainer AR. Identification and characterization of three members of the human SR family of pre-mRNA splicing factors. EMBO J 1995; 14:4336 - 49; PMID: 7556075
- Phadke M, Krynetskaia N, Mishra A, Krynetskiy E. Accelerated cellular senescence phenotype of GAPDH-depleted human lung carcinoma cells. Biochem Biophys Res Commun 2011; 411:409 - 15; http://dx.doi.org/10.1016/j.bbrc.2011.06.165; PMID: 21749859
- Orloff M, Peterson C, He X, Ganapathi S, Heald B, Yang YR, Bebek G, Romigh T, Song JH, Wu W, et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA 2011; 306:410 - 9; http://dx.doi.org/10.1001/jama.2011.1029; PMID: 21791690
- Almeida-Vega S, Catlow K, Kenny S, Dimaline R, Varro A. Gastrin activates paracrine networks leading to induction of PAI-2 via MAZ and ASC-1. Am J Physiol Gastrointest Liver Physiol 2009; 296:G414 - 23; http://dx.doi.org/10.1152/ajpgi.90340.2008; PMID: 19074642
- Privette Vinnedge LM, McClaine R, Wagh PK, Wikenheiser-Brokamp KA, Waltz SE, Wells SI. The human DEK oncogene stimulates β-catenin signaling, invasion and mammosphere formation in breast cancer. Oncogene 2011; 30:2741 - 52; http://dx.doi.org/10.1038/onc.2011.2; PMID: 21317931
- Kappes F, Waldmann T, Mathew V, Yu J, Zhang L, Khodadoust MS, Chinnaiyan AM, Luger K, Erhardt S, Schneider R, et al. The DEK oncoprotein is a Su(var) that is essential to heterochromatin integrity. Genes Dev 2011; 25:673 - 8; http://dx.doi.org/10.1101/gad.2036411; PMID: 21460035
- Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, Pommier Y. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J Biol Chem 2012; 287:12848 - 57; http://dx.doi.org/10.1074/jbc.M111.333963; PMID: 22375014
- Hubert L Jr., Lin Y, Dion V, Wilson JH. Topoisomerase 1 and single-strand break repair modulate transcription-induced CAG repeat contraction in human cells. Mol Cell Biol 2011; 31:3105 - 12; http://dx.doi.org/10.1128/MCB.05158-11; PMID: 21628532
- Bee A, Brewer D, Beesley C, Dodson A, Forootan S, Dickinson T, Gerard P, Lane B, Yao S, Cooper CS, et al. siRNA knockdown of ribosomal protein gene RPL19 abrogates the aggressive phenotype of human prostate cancer. PLoS One 2011; 6:e22672; http://dx.doi.org/10.1371/journal.pone.0022672; PMID: 21799931
- Kobayashi T, Sasaki Y, Oshima Y, Yamamoto H, Mita H, Suzuki H, Toyota M, Tokino T, Itoh F, Imai K, et al. Activation of the ribosomal protein L13 gene in human gastrointestinal cancer. Int J Mol Med 2006; 18:161 - 70; PMID: 16786168
- Lines MA, Huang L, Schwartzentruber J, Douglas SL, Lynch DC, Beaulieu C, Guion-Almeida ML, Zechi-Ceide RM, Gener B, Gillessen-Kaesbach G, et al, FORGE Canada Consortium. Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am J Hum Genet 2012; 90:369 - 77; http://dx.doi.org/10.1016/j.ajhg.2011.12.023; PMID: 22305528
- McCloskey A, Taniguchi I, Shinmyozu K, Ohno M. hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science 2012; 335:1643 - 6; http://dx.doi.org/10.1126/science.1218469; PMID: 22461616
- Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem 2007; 389:1017 - 31; http://dx.doi.org/10.1007/s00216-007-1486-6; PMID: 17668192
- Han W, Wang T, Reilly AA, Keller SM, Spivack SD. Gene promoter methylation assayed in exhaled breath, with differences in smokers and lung cancer patients. Respir Res 2009; 10:86; http://dx.doi.org/10.1186/1465-9921-10-86; PMID: 19781081
- Cohen O, Feinstein E, Kimchi A. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J 1997; 16:998 - 1008; http://dx.doi.org/10.1093/emboj/16.5.998; PMID: 9118961
- Cohen O, Inbal B, Kissil JL, Raveh T, Berissi H, Spivak-Kroizaman T, Feinstein E, Kimchi A. DAP-kinase participates in TNF-alpha- and Fas-induced apoptosis and its function requires the death domain. J Cell Biol 1999; 146:141 - 8; PMID: 10402466
- Tang X, Khuri FR, Lee JJ, Kemp BL, Liu D, Hong WK, Mao L. Hypermethylation of the death-associated protein (DAP) kinase promoter and aggressiveness in stage I non-small-cell lung cancer. J Natl Cancer Inst 2000; 92:1511 - 6; http://dx.doi.org/10.1093/jnci/92.18.1511; PMID: 10995806
- Kim DH, Nelson HH, Wiencke JK, Christiani DC, Wain JC, Mark EJ, Kelsey KT. Promoter methylation of DAP-kinase: association with advanced stage in non-small cell lung cancer. Oncogene 2001; 20:1765 - 70; http://dx.doi.org/10.1038/sj.onc.1204302; PMID: 11313923
- Toyooka S, Toyooka KO, Miyajima K, Reddy JL, Toyota M, Sathyanarayana UG, Padar A, Tockman MS, Lam S, Shivapurkar N, et al. Epigenetic down-regulation of death-associated protein kinase in lung cancers. Clin Cancer Res 2003; 9:3034 - 41; PMID: 12912953
- Schwartz AG, Prysak GM, Bock CH, Cote ML. The molecular epidemiology of lung cancer. Carcinogenesis 2007; 28:507 - 18; http://dx.doi.org/10.1093/carcin/bgl253; PMID: 17183062
- Belinsky SA, Liechty KC, Gentry FD, Wolf HJ, Rogers J, Vu K, Haney J, Kennedy TC, Hirsch FR, Miller Y, et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res 2006; 66:3338 - 44; http://dx.doi.org/10.1158/0008-5472.CAN-05-3408; PMID: 16540689
- Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene 1998; 17:11 Reviews 1395 - 413; http://dx.doi.org/10.1038/sj.onc.1202174; PMID: 9779987
- Downward J. The ins and outs of signalling. Nature 2001; 411:759 - 62; http://dx.doi.org/10.1038/35081138; PMID: 11459043
- Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 2000; 25:315 - 9; http://dx.doi.org/10.1038/77083; PMID: 10888881
- Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst 2001; 93:691 - 9; http://dx.doi.org/10.1093/jnci/93.9.691; PMID: 11333291
- Li W, Deng J, Jiang P, Zeng X, Hu S, Tang J. Methylation of the RASSF1A and RARβ genes as a candidate biomarker for lung cancer. Exp Ther Med 2012; 3:1067 - 71; PMID: 22970018
- de Fraipont F, Levallet G, Creveuil C, Bergot E, Beau-Faller M, Mounawar M, Richard N, Antoine M, Rouquette I, Favrot MC, et al, Intergroupe Francophone de Cancérologie Thoracique. An apoptosis methylation prognostic signature for early lung cancer in the IFCT-0002 trial. Clin Cancer Res 2012; 18:2976 - 86; http://dx.doi.org/10.1158/1078-0432.CCR-11-2797; PMID: 22434665
- Zhang Q, Sun L, Yin L, Ming J, Zhang S, Luo W, Qiu X. CCL19/CCR7 upregulates heparanase via specificity protein-1 (Sp1) to promote invasion of cell in lung cancer. Tumour Biol 2013; Forthcoming PMID: 23649655
- Zhao FQ. Octamer-binding transcription factors: genomics and functions. [Landmark Ed] Front Biosci (Landmark Ed) 2013; 18:1051 - 71; http://dx.doi.org/10.2741/4162; PMID: 23747866
- Ghosh RP, Horowitz-Scherer RA, Nikitina T, Shlyakhtenko LS, Woodcock CL. MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol Cell Biol 2010; 30:4656 - 70; http://dx.doi.org/10.1128/MCB.00379-10; PMID: 20679481
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997; 88:471 - 81; http://dx.doi.org/10.1016/S0092-8674(00)81887-5; PMID: 9038338
- Maclean JA, Bettegowda A, Kim BJ, Lou CH, Yang SM, Bhardwaj A, Shanker S, Hu Z, Fan Y, Eckardt S, et al. The rhox homeobox gene cluster is imprinted and selectively targeted for regulation by histone h1 and DNA methylation. Mol Cell Biol 2011; 31:1275 - 87; http://dx.doi.org/10.1128/MCB.00734-10; PMID: 21245380
- Giambra V, Volpi S, Emelyanov AV, Pflugh D, Bothwell AL, Norio P, Fan Y, Ju Z, Skoultchi AI, Hardy RR, et al. Pax5 and linker histone H1 coordinate DNA methylation and histone modifications in the 3′ regulatory region of the immunoglobulin heavy chain locus. Mol Cell Biol 2008; 28:6123 - 33; http://dx.doi.org/10.1128/MCB.00233-08; PMID: 18644860
- Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE, Stein A, Woodcock CL, Skoultchi AI. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 2005; 123:1199 - 212; http://dx.doi.org/10.1016/j.cell.2005.10.028; PMID: 16377562
- Wellman SE, Sittman DB, Chaires JB. Preferential binding of H1e histone to GC-rich DNA. Biochemistry 1994; 33:384 - 8; http://dx.doi.org/10.1021/bi00167a049; PMID: 8286360
- Wellman SE, Song Y, Mamoon NM. Sequence preference of mouse H1(0) and H1t. Biochemistry 1999; 38:13112 - 8; http://dx.doi.org/10.1021/bi9914917; PMID: 10529182
- Bonnefoy E, Bandu MT, Doly J. Specific binding of high-mobility-group I (HMGI) protein and histone H1 to the upstream AT-rich region of the murine beta interferon promoter: HMGI protein acts as a potential antirepressor of the promoter. Mol Cell Biol 1999; 19:2803 - 16; PMID: 10082546
- Käs E, Izaurralde E, Laemmli UK. Specific inhibition of DNA binding to nuclear scaffolds and histone H1 by distamycin. The role of oligo(dA).oligo(dT) tracts. J Mol Biol 1989; 210:587 - 99; http://dx.doi.org/10.1016/0022-2836(89)90134-4; PMID: 2614835
- Hisaoka M, Ueshima S, Murano K, Nagata K, Okuwaki M. Regulation of nucleolar chromatin by B23/nucleophosmin jointly depends upon its RNA binding activity and transcription factor UBF. Mol Cell Biol 2010; 30:4952 - 64; http://dx.doi.org/10.1128/MCB.00299-10; PMID: 20713446