
Abstract
Preeclampsia (PE) is the major pregnancy-induced hypertensive disorder responsible for maternal and fetal morbidity and mortality that can be associated with intrauterine growth restriction (IUGR). PE and IUGR are thought to be due to a placental defect, occurring early during pregnancy. Several placental microRNAs (miRNAs) have been shown to be deregulated in the context of placental diseases and could thus play a role in the pathophysiology of PE. Here, we show that pri-miR-34a is overexpressed in preeclamptic placentas and that its placental expression is much higher during the first trimester of pregnancy than at term, suggesting a possible developmental role. We explored pri-miR-34a regulation and showed that P53, a known activator of miR-34a, is reduced in all pathological placentas and that hypoxia can induce pri-miR-34a expression in JEG-3 cells. We also studied the methylation status of the miR-34a promoter and revealed hypomethylation in all preeclamptic placentas (associated or not with IUGR), whereas hypoxia induced a hypermethylation in JEG-3 cells at 72 h. Despite the overexpression of pri-miR-34a in preeclampsia, there was a striking decrease of the mature miR-34a in this condition, suggesting preeclampsia-driven alteration of pri-miR-34a maturation. SERPINA3, a protease inhibitor involved in placental diseases, is elevated in IUGR and PE. We show here that miR-34a overexpression in JEG-3 downregulates SERPINA3. The low level of mature miR-34a could thus be an important mechanism contributing to SERPINA3 upregulation in placental diseases. Overall, our results support a role for miR-34a in the pathophysiology of preeclampsia, through deregulation of the pri-miRNA expression and its altered maturation.
Introduction
Preeclampsia (PE) is a common disease of human gestation, affecting 5 to 10% of pregnanciesCitation1 and is one of the first causes of pregnancy-associated maternal mortality and morbidity. This disease is defined by the occurrence of gestational hypertension and proteinuria. The only definitive treatment is to deliver the placenta (and the fetus), often leading to the birth of premature babies. PE can be associated with intrauterine growth retardation (IUGR), in around one third of the cases. Despite the late onset of the symptoms, the causes are thought to occur early during the first trimester of pregnancy, notably with an abnormal placentation leading to shallow invasion of the maternal uterine arteries and hypoperfusion (responsible of a pathological hypoxia).Citation2
Others and we have shown previously that several serine protease inhibitors (SERPIN) are transcriptionally modified in the context of placental diseases.Citation3,Citation4 Indeed, these proteins are involved in various pathways affected in these diseases, such as inflammation, coagulation, fibrinolysis, complement activation, and phagocytosis. In particular, we have recently shown that SERPINA3 is upregulated in PE and even more in PE+IUGR and IUGR placentas. Furthermore, its promoter is hypomethylated in PE and IUGR placentas in a region possibly binding several transcription factors (such as HIF1α, Hypoxia Inducible Factor).Citation4,Citation5 SERPINA3 is a specific inhibitor of elastase, a member of the matrix metalloprotease family, a family of factors known to play a crucial role during the implantation process, suggesting that this protein could play a role in the pathophysiology of placental diseases. In addition, we have shown an association between SNPs located in this gene and preeclampsia, these SNPs regulating the fixation of transcription factors according to the methylation status.Citation6 Outside DNA methylation, epigenetic regulation of transcription can be achieved by modulating the histone code and, notably, by shifting the cell content in small non-coding RNAs.
Among those, microRNAs (miRNAs) are well-known post-transcriptional regulators. They are encoded in different parts of the genome, such as intergenic regions, but also intronic regions; they can also be polycistronic. Their precursors are generally transcribed by RNAPol II. First, they are transcribed as primary-miRNA (pri-miRNA), constituted by a hairpin and flanking sequences. This pri-miRNA is processed by the complex Drosha (an RNase III enzyme), into a precursor-miRNA (pre-miRNA), which is only composed of the hairpin. The pre-miRNA is then exported to the cytoplasm by the protein Exportin 5. In the cytoplasm, the pre-miRNA is processed by the Dicer complex, a second RNase III enzyme, into a mature miRNA-miRNA* duplex. This double stranded molecule is composed of a mature guide strand miRNA and a passenger strand miRNA. The mature miRNA is then loaded into RISC (RNA-induced silencing complex), while the passenger strand miRNA is degraded. miRNA in RISC can interact with the UTR of its mRNA targets and could function via two silencing mechanisms: mRNA-targeted degradation or mRNA-targeted translational inhibition.Citation7,Citation8
Numerous miRNAs have already been described to play essential roles during placental development and in pathologies of the placenta (reviewed in ref. Citation9). Several miRNAs are specific of the trophoblast and may be able to regulate invasion, proliferation, and iron metabolism, especially when this regulation involves hypoxia, to which miR-210, for instance, seems to be very responsive.Citation10 Therefore, miRNAs may function in fail-safe mechanisms, assuring fidelity of expression domains crucial for patterning/development. They could therefore be important for placentation, which is a finely regulated step for a harmonious pregnancy. Herein, we wished to specifically study miR-34a putative effects on SERPINA3 expression. This is based upon the observation that miR-34a overexpression in HTC116 cells (a colon cancer cell line) leads to a decrease of SERPINA3 mRNA.Citation11 miR-34a involvement in placental physiology is also substantiated by observations showing that it downregulates the Notch and Jagged pathways in choriocarcinoma, thus suppressing their invasive propertiesCitation12 and by its possible involvement in the enhanced invasive capacities of trophoblasts in the context of placenta accreta.Citation13 The miR-34a gene is located on chromosome 1p36, it has two exons, the mature miRNA being encoded by the second one, like all miRNAs from the miR-34 family. miR-34a has the particularity to have a very large intron of about 30 kb. It was shown that miR-34a is regulated by P53, via P53 binding sites in its promoter, in the context of cancer.Citation14 Several direct miR-34a targets implicated in different biological pathways have been validated. For example, miR-34a inhibits the apoptosis regulator BCL2. It is also involved in the inhibition of CyclinD1 and E2, as well as cyclin dependent kinases CDK4 and CDK6, MET, the hepatocyte growth factor receptor, and the proto-oncogene N-MYC, all involved in cell cycle, leading to G1 arrest and E2F3 transcription factor inhibition, which leads to proliferation inhibition. Delta-like protein DLL1, a notch ligand, is also inhibited by miR-34a, which means that Notch signaling, important for placenta vascularization and development, as mentioned above,Citation15,Citation16 can be disturbed by miR-34a. Lastly, SIRT1, a NAD-dependent deacetylase, is a miR-34a target; since it has also been shown to inhibit several pro-apoptotic proteins including P53, SIRT1 inhibition may initiate a positive feedback loop leading to P53 overexpressionCitation17,Citation18
Here, we hypothesized that abnormal miR-34a expression could contribute to placental diseases, for instance, by deregulating SERPINA3. In this study, we show for the first time that pri-miR-34a and mature miR-34a expression, as well as the promoter methylation of the gene, is affected in pathological placentas. P53, known as a key regulator of miR-34a expression, is reduced in all pathological placentas. We also show that miR-34a is induced by hypoxia in choriocarcinoma cells (24–48h), but not after 72h, time at which there is an important increase of miR-34a promoter methylation. Finally, we show in this cell model that miR-34a is indeed able to inhibit SERPINA3 expression, presumably indirectly.
Results
Abnormal expression of pri-miR-34a in pathological placentas
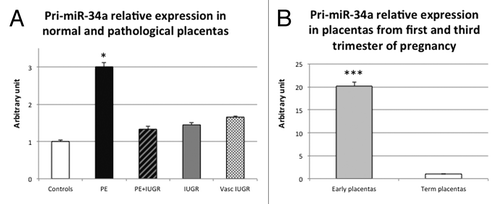
First, we assessed pri-miR-34a expression in control and pathological placentas. miRNAs quantification is not straightforward due to their small size, so we first estimated miR-34a expression by the quantification of the spliced primary miRNA (pri-miR-34a), as previously described.Citation18 When the complete data set was analyzed by ANOVA, no significant values were reached. However, in preeclamptic placentas, pri-miR-34a was increased (3-fold) compared with control placentas (, P = 1.9 × 10−2 by the Student test).
Figure 1. pri-miR-34a quantity is significantly higher in PE and first trimester placentas. (A) Expression was assessed by qRT-PCR in 10 term control placentas (Controls), 10 preeclamptic placentas (PE), 5 preeclamptic placentas associated with IUGR (PE+IUGR), 7 IUGR placentas (IUGR) and 6 vascular IUGR (vasc IUGR). The Ct values of each sample were normalized by those obtained for SDHA. The expression level obtained for the control placentas was arbitrary set to one. *P< 0.05 by the Student t-test relative to the control samples. (B) Expression was assessed by qRT-PCR in 10 term control placentas (Term placentas) and 26 placentas from first trimester of pregnancy (Early placentas). The Ct values of each sample were normalized by those obtained for SDHA. The expression level obtained for the control term placentas was arbitrary set to one. ***P < 0.001.
Differential expression of pri-miR-34a according to term
We then investigated whether there was a difference in pri-miR-34a expression in early placentas (at the first trimester of pregnancy) vs. term placentas and showed a striking elevation (20-fold) of pri-miR-34a RNA at early terms (, P = 1.3 × 10−7 by the Student test).
We then wondered whether the increase observed in preeclamptic placentas was due to placental term rather than to pathological state. Due to obvious limited access to control samples, preeclamptic placentas are somewhat younger than control placentas in the large majority of studies. Transcriptome analysis demonstrated nevertheless that expression variations are quite limited during the last trimester of gestation.Citation19 In the present studies, gestational ages were 33 weeks and 4 d ± 5 d of amenorrhea (preeclampsia) vs. 39 weeks and 3 d ± 1 d (controls). To assess whether miR-34a expression diminishes according to placental term, we performed correlation analyses, using all the values obtained for term placentas (controls and pathological) and we could not find any significant correlation. The observed increase of pri-miR-34a in preeclamptic placentas is thus not linked to the gestational age.
Putative links between P53 and miR-34a expression in the placental context
As miR-34a is regulated by P53,Citation17 we analyzed the expression of this factor in control and pathological placentas (). There was a systematic decrease of P53 mRNA level in all the pathological placentas studied. In preeclamptic placentas, a 3.2- to 3.7-fold decrease was observed (P = 5.5 × 10−6 and P = 2.2 × 10−3 for PE and PE+IUGR placentas, respectively). In IUGR placentas, we observed a 2.2- to 2.7-fold decrease (P = 1.3 × 10−3 and P = 4.1 × 10−3 for IUGR and vascular IUGR placentas, respectively). We also assessed P53 expression in first trimester placentas () and observed a slight not significant increase.
Figure 2. P53 mRNAs is significantly decreased in all the pathological placentas. (A) Expression was assessed by qRT-PCR in 11 control placentas (Controls), 13 preeclamptic placentas (PE), 4 preeclamptic placentas associated with IUGR (PER), 7 IUGR placentas (IUGR) and 7 vascular IUGR (vasc IUGR). The Ct values of each sample were normalized by those obtained for SDHA. The expression level obtained for the control placentas was arbitrary set to one. P = 1.3 × 10−5 by ANOVA. **P< 0.01, ***P< 0 0.001. (B) Expression was assessed by qRT-PCR in 8 control term placentas (Term placentas) and 8 placentas from first trimester of pregnancy (Early placentas). The Ct values of each sample were normalized by those obtained for SDHA. The expression level obtained for the control term placentas was arbitrary set to one.
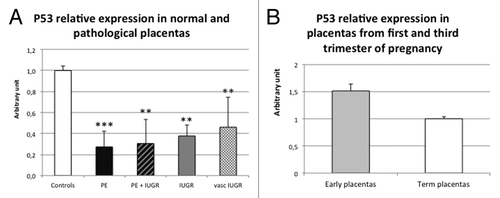
Consequently, P53 does not seem responsible for pri-miR-34a increase in preeclamptic placentas, since it is reduced in this pathology. It could however participate in the pri-miR-34a increase observed in first trimester placentas.
Epigenetic analysis (DNA methylation) of the miR-34a promoter in placentas
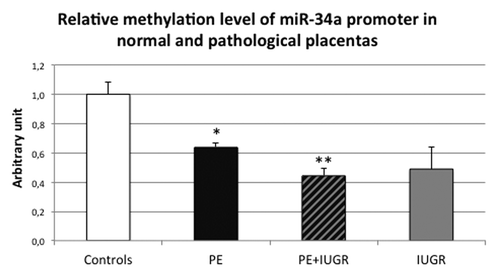
Other mechanisms could explain miR-34a regulation, such as promoter methylation. Indeed, the miR-34a promoter contains a CpG island, which could constitute a target for DNA methylation. We assessed the methylation level of the promoter by McrBC digestion followed by quantitative PCR in normal and pathological placentas. In this experiment, a systematic hypomethylation of the promoters was observed in all preeclamptic placentas, associated or not with IUGR, compared with control placentas (, P = 4.6 × 10−2 and P = 6.6 × 10−3 by post hoc Student test, respectively, for PE and PE+IUGR group compared with control group). This could contribute to the elevated expression of pri-miR-34a in preeclamptic placentas.
Figure 3. miR-34a promoter methylation is significantly decreased in PE placentas (associated or not with IUGR). Methylation was assessed by McrBC digestion followed by quantitative PCR in 5 term control placentas (Controls), 5 preeclamptic placentas (PE), 5 preeclamptic placentas associated with IUGR (PE+IUGR) and 5 IUGR placentas (IUGR). The Ct values of each sample were normalized by those obtained for non-digested DNA. The methylation level obtained for the control placentas was arbitrary set to one. *P < 0.05 and **P < 0.01.
In addition, we analyzed the methylation level in first trimester placentas, but could not find any significant difference compared with term placentas (Fig. S1A).
Oxygen pressure is an important regulator of pri-miR-34a expression in choriocarcinoma cells
In order to better understand how the pri-miR-34a is increased in pathophysiological placentas, we assessed the hypothesis of a regulation by hypoxia, since it is known that pathological placentas have an increased level of hypoxia inducible factor (HIF).Citation20 Therefore, we used JEG-3 cells as a proxy of trophoblasts, cultivated them in hypoxia (2% O2) and kinetically evaluated pri-miR-34a expression (). This revealed a strong induction of pri-miR-34a expression after 24 and 36 h of hypoxia (23 fold, P = 2.2 × 10−2 and 6 fold, P = 2.4 × 10−2 by Student test comparing normoxia and hypoxia ΔCt values, for 24 h and 36 h of hypoxia respectively). Quite interestingly, however, at 72 h the level of miR-34a was back to normal (oxygen atmospheric pressure) values.
Figure 4. Hypoxia induces the expression of pri-miR-34a following this of P53. (A) Pri-miR-34a level was assessed by qRT-PCR in cells submitted to 3, 6, 24, 36, or 72 h of hypoxia (2% O2) or under classical normoxic culture condition (20% O2). The Ct values of each sample were normalized by those obtained for SDHA. The graphical representation shows the relative expression in hypoxia compared with normoxia. P = 2.2 × 10−4 by two-factor ANOVA. *P < 0.05. (B) P53 expression was assessed by qRT-PCR in cells submitted to 3, 6, 24, 36, 48, or 72 h of hypoxia (2% O2) or under classical normoxic culture condition (20% O2). The Ct values of each sample were normalized by those obtained for CyclophilinA. The graphical representation shows the relative expression in hypoxia compared with normoxia. P = 4.3 × 10−3 by two-factor ANOVA. *P < 0.05.
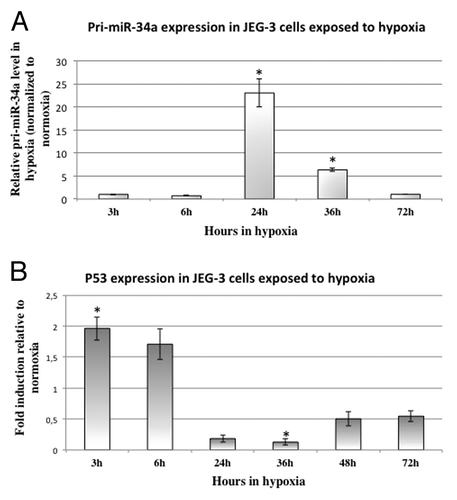
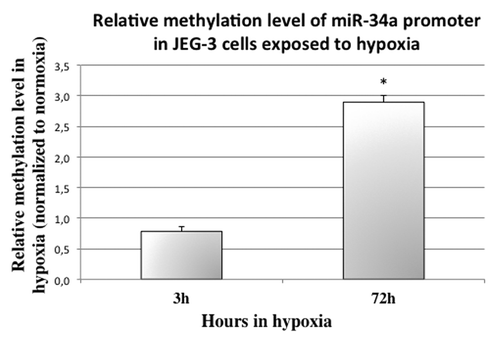
To understand how hypoxia regulates pri-miR-34a, we analyzed its effect on P53 () and on miR-34a promoter methylation (). P53 is rapidly induced by hypoxia: around 2-fold after 3h (P = 1.1 × 10−2 by Student test comparing normoxia and hypoxia). However, after 36h, P53 mRNA is significantly decreased (around 8-fold, P = 3.4 × 10−2, by Student test comparing normoxia and hypoxia). Then, we checked whether hypoxia could influence miR-34a promoter methylation (). Indeed, at 72 h of hypoxia, methylation of the promoter was increased by ~3-fold in choriocarcinoma cells (P = 2.4 × 10−2 by Post hoc Student test compared with 72 h in normoxic conditions). This hypermethylation was not present at 24 h (Fig. S1B).
Figure 5. miR-34a promoter methylation is significantly increased in cells submitted to 72 h of hypoxia. McrBC digestion followed by quantitative PCR in cells submitted to 3 and 72 h of normoxia or hypoxia was performed to assess methylation status. The Ct values of each sample were normalized by those obtained for non-digested DNA. P = 3.9 × 10−3 by two-factor ANOVA. The graphical representation shows the relative methylation status in hypoxia compared with normoxia. *P < 0.05.
Mature miR-34a expression in placentas and choriocarcinoma cells exposed to hypoxia
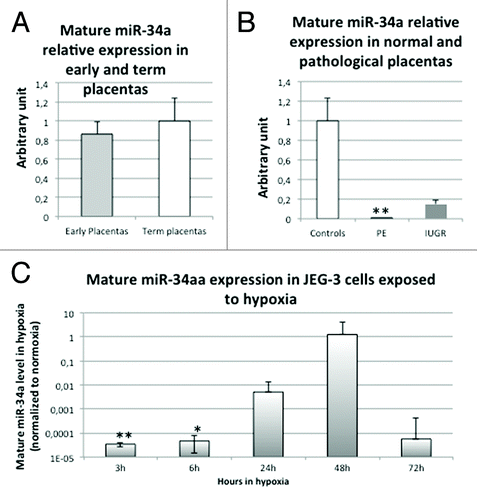
In order to check whether the mature miR-34a displays the same profile than the pri-miR-34a, we performed qPCR following a polyadenylation step on extracted RNA (), as described in reference Citation21. In first trimester placentas, we did not observe an increase of mature miR-34a (), despite the 20-fold increase of pri-miR-34a. This probably indicates that the maturation of the pri-miRNA is occurring with a very low efficiency during the first trimester. When we looked at the pathological placentas (), we observed a striking reduction of the mature miR-34a in PE placentas (more than 250-fold, P = 6.6 × 10−3, by Student test comparing control and PE placentas), and a 7-fold decrease in IUGR placentas (non significant). This is in opposition with the observed increase of pri-miR-34a in PE placentas. Thus, the maturation process seems defective in pathological placentas. The pri-miR34a increase that we observed could be a failed attempt to compensate the almost absence of mature miRNA. In this sense, we showed that in cells, the mature miR-34a is also strikingly decreased by 3 h and 6 h of hypoxia (), compared with normoxia (P = 1.0 × 10−3 and P = 3.8 × 10−2, for 3 h and 6 h, respectively, by Student test comparing normoxia and hypoxia). After 24 h and 48 h of hypoxia, this reduction is not present anymore, suggesting that, upon induction of the pri-miR-34a, the amount available could compensate for the low maturation of the pri-miRNA. Finally, after 72 h of hypoxia, when the pri-miR-34a is not anymore induced, the mature miR-34a seems, once again, decreased, compared with normoxia (non-significant).
Figure 6. Mature miR-34a in first trimester placentas, terms placentas (control and pathological) and in cells submitted to hypoxia. (A) Mature miR-34a quantity was assessed by qRT-PCR preceded by a polyadenylation step in 8 Term control placentas (Term placentas) and 8 placentas from first trimester of pregnancy (Early placentas). The Ct values of each sample were normalized by a geometric means of those obtained for 5S RNA and SDHA. The expression level obtained for the control placentas was arbitrary set to one. (B) Mature miR-34a quantity was assessed by qRT-PCR preceded by a polyadenylation step in 8 control placentas (Controls), 5 preeclamptic placentas (PE) and 4 IUGR placentas (IUGR). The Ct values of each sample were normalized by a geometric means of those obtained for 5S RNA and SDHA. The expression level obtained for the control placentas was arbitrary set to one. **P < 0.01 by the Student t-test relative to the control samples. (C) Mature miR-34a level was assessed by qRT-PCR preceded by a polyadenylation step in cells submitted to 3, 6, 24, 48, or 72 h of hypoxia (2% O2) or under classical normoxic culture condition (20% O2). The Ct values of each sample were normalized by a geometric means of those obtained for 5S RNA and SDHA.. The graphical representation shows the relative expression in hypoxia compared with normoxia. P = 1.1 × 10−4 by two-factor ANOVA. *P < 0.05, **P < 0.01.
Effects of miR-34a expression on SERPINA3 regulation
In order to know whether miR-34a regulates SERPINA3, we overexpressed it in JEG-3 cells (). As a positive control, we assessed BCL2 expression in these cells, as it is a known target of miR-34a, and we could indeed show that its mRNA level was decreased 3-fold compared with cells transfected with an empty vector (P = 1.6 × 10−2 by Student test). In the same conditions, we showed a 2.2-fold decrease of SERPINA3 mRNA in cells transfected with miR-34a compared with cells transfected with an empty vector (P = 1.0 × 10−2 by Student test), thus confirming the ability of miR-34a to downregulate SERPINA3 expression.
Figure 7. SERPINA3 expression is downregulated in cells transfected by a plasmid encoding miR-34a, but this effect is not mediated by SERPINA3 3′UTR. (A) Expression of BCL2 and SERPINA3 was assessed by qRT-PCR in cells transfected by an empty vector (Empty) or a plasmid encoding miR-34a (miR-34a). The Ct values of each sample were normalized by those obtained for SDHA. The expression level obtained for the control cells (empty vector) was arbitrary set to one. *P < 0.05. (B) Luciferase activity was measured in JEG-3 cells transfected with two different firefly luciferase vectors (CMV-luc to measure the basal level, and CMV-luc-3′UTR-SERPINA3, which contains the 3′UTR sequence of SERPINA3 downstream of the luciferase gene) and with an empty vector or a plasmid encoding miR-34a. The values of each sample were normalized by those obtained for Renilla luciferase. The graphical representation shows the fold induction following miR-34a expression. NS, Non Significant.
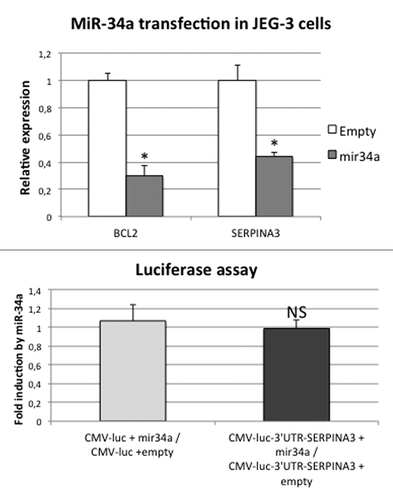
Since the negative regulatory effect of miR-34a on SERPINA3 expression can be direct or indirect, we tested the possibility of a direct interaction between the miRNA and SERPINA3 mRNA as a target. First, in silico, we scanned SERPINA3 for identifying a miR-34a target using predicting algorithms (such as TargetScan and miRanda), but without evincing a specific sequence. Second, we explored SERPINA3 3′UTR manually for sequence complementarity to the seed region of miR-34a. The seed region is located in position 2 to 7 in the mature RNA, where it seems necessary to have an almost perfect match to this part to have an inhibitory effect. In the SERPINA3 3′UTR, we found only an imperfect match. Third, to eventually determine if this may be enough to induce silencing, we cloned the complete 3′UTR of SERPINA3 downstream of the luciferase gene and performed transfections of this constructions in JEG-3 cells, in parallel with either the miR-34a plasmid or an empty vector (). We could not prove any difference in luciferase activity following miR-34a transfection, suggesting that, apparently, miR-34a does not target directly the 3′UTR of SERPINA3.
Discussion
Previously, several placental miRNAs have been shown to be deregulated in the context of placental disease or trophoblast differentiation.Citation22-Citation28 Here, we show that pri-miR-34a is overexpressed in preeclamptic placentas and that its placental expression is much higher during the first trimester of pregnancy than at term, suggesting a possible developmental role for this miRNA. However, these differences are not translated into a mature miR-34a increase, and, in fact, for PE placentas, the opposite profile is observed. This could be explained by a very low maturation of the pri-miRNA in early pregnancy, and a completely defective maturation in PE placentas. The maturation of pri-miR-34a consists in several steps: splicing, a first cleavage into pre-miRNA and a second cleavage into mature miRNA; all these steps can be finely regulated. In preeclampsia, the higher level of pri-miR-34a could be a compensation for a defective maturation (negative feedback?).
Concerning miR-34a regulation, on the one hand, we showed that P53, a known activator of miR-34a, is reduced in all pathological placentas and, on the other hand, that hypoxia induced pri-miR-34a expression in JEG-3 cells. We observed that, consistently, this induction is preceded by this of P53. Hypoxia also seems to influence the maturation process, as the level of mature miR-34a is dramatically reduced after 3 h of hypoxia compared with normoxia. Later on, at 24 h and 36 h of hypoxia exposure, the quantity of pri-miR-34a increases, the level of mature miRNA is progressively brought back to that observed in normoxia, suggesting an efficient compensation of the altered maturation process.
We also studied the methylation status of the miR-34a promoter (around the P53 putative binding sites present in this promoter). We found hypomethylation in all placentas from preeclamptic pregnancies (associated or not with IUGR). On the other hand, hypoxia induced a hypermethylation in JEG-3 cells at 72 h.
Finally, we show that SERPINA3 is downregulated by miR-34a overexpression in JEG-3, this regulation being independent of the 3′UTR of SERPINA3. Previously, we have demonstrated that SERPINA3 is upregulated in placental diseases, in association with the abnormal methylation of its promoter.Citation6 Here, we show that another mechanism could be at work in regulating this gene: the quasi-absence of mature miR-34a in pathological placentas compared with controls could partly be responsible for SERPINA3 upregulation in placental diseases.
It is generally admitted that the placental defects leading to preeclampsia occur early in pregnancy, when pri-miR-34a is the most expressed, suggesting that it could play a role in early stages of preeclampsia. As we did not observe a high level of mature miR-34a in early placentas, maturation probably has low efficiency. However, having high levels of pri-miR-34a in early pregnancies (probably stored within the cells) could be a physiological strategy to allow a rapid response when the mature miRNA becomes necessary in the placenta.
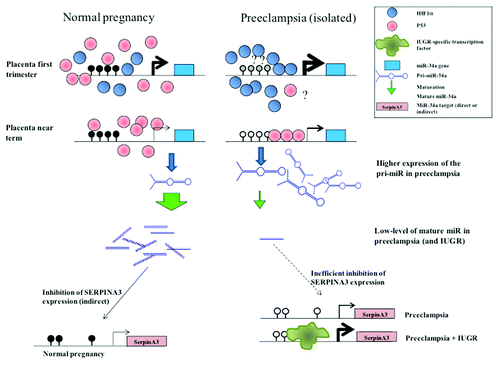
Based on our present results, we propose here the following model (): during normal pregnancies, the miR-34a promoter is methylated; in these cases, even if P53 is expressed at a relatively high level, it is not able to induce pri-miR-34a expression; reciprocally, in preeclampsia, there is a smaller amount of P53, but the promoter is hypomethylated and may have a better responsiveness to the inducer. In these cases, pri-miR-34a expression is increased, but this is not sufficient to completely compensate the defective maturation, and the mature miR-34a is much reduced compared with normal placentas, contributing probably to the observed increase of SERPINA3 expression in placental diseases. At the beginning of pregnancy, the higher expression of pri-miR-34a could be explained by the abundance of HIF1α in physiological hypoxia, and probably even more so in preeclampsia.Citation20 Indeed, several HIF binding sites (RCGTG) are presents in the miR-34a promoter (near the P53-box), and we showed in JEG-3 cells that pri-miR-34a is robustly induced by hypoxia. However, this activation by hypoxia is certainly mediated by a cofactor present in trophoblast cells since miR-34a has been previously found to be decreased by hypoxia in another cell type (renal tubular epithelial cell).Citation29 Furthermore, pri-miR-34a induction by hypoxia in the cell did not last more than 36 h, probably due to progressive promoter methylation. While the miR34a level was considerably reduced at 3 and 6 h of hypoxia, its level tended to be restored at 36–48 h. Overexpression of the pri-miRNA can therefore be considered as an efficient compensatory mechanism to the defective maturation triggered by hypoxia.
Figure 8. miR-34a regulation along pregnancy if physiological and pathological cases. In this model, we propose that miR-34a regulation is mediated by the combined effect of methylation status and transcription factors binding. At the end of pregnancy, P53 could bind the hypomethylated promoter of pathological placentas and induce a higher expression of the pri-miR. At the beginning of pregnancy, when the placenta is rather hypoxic, HIF1α would induce a high expression level of miR-34a in physiological cases and a higher one in pathological cases, leading to an enhanced downregulation of target genes. We have shown previously that the SERPINA3 promoter is demethylated in pathological placentas and that this is associated with an increased expression. In IUGR, we have shown that specific transcription factors (ZBTB7B) are specific mediators of this high induction. The induction of SERPINA3 in pathology could be partly explained by the low level of the mature miR-34a, resulting of an inefficient activity of the miRNA maturation machinery, even though the precursor is present at a high level, may be through the absence of a feedback regulation.
At the beginning of pregnancy, a pathological long hypoxia could induce a low level of pri-miR-34a by promoter hypermethylation (such as observed in the cells at 72 h of hypoxia), associated to a lessened level of mature miR, aggravated by hypoxia-dependent defective maturation. This could contribute to the physiopathology of preeclampsia by inducing higher levels of SERPINA3, with deleterious effect on invasion.Citation6 Indeed, we already proposed a role of this protein in placental diseases.Citation4-Citation6 However, how miR-34a regulates SERPINA3 remains elusive. We show here that it is probably indirect, hypothetically by downregulating an activator of SERPINA3 expression. Nevertheless, it could be through another part of SERPINA3 mRNA, as it is now demonstrated that miRNA can act on coding regions.Citation30
In conclusion, we bring new evidences about an involvement of miR-34a in the pathophysiology of placental disease, thus reinforcing previous studies showing that miR-34a regulates the invasive capacity of trophoblast cells,Citation12,Citation31 consistently with its involvement in preeclampsia and placental development.
Material and Methods
Human material samples and ethics
Placentas from normal and pathological pregnancies (PE and IUGR) were collected after caesarean delivery from 3 maternities (St Antoine, Port-Royal, and Institut de Puériculture) following the indications of the Cochin Hospital ethical committee. We had access to complete clinical data for each individual. Placental fragments from voluntary termination of pregnancy were also collected and formed the “early placenta” group.
The inclusion criteria used for PE patients were the following clinical thresholds: systolic pressure > 140 mmHg, diastolic pressure > 90 mmHg, and proteinuria > 0.3 g per 24 h. The inclusion criteria used for IUGR was weight at birth < 10th percentile, and it was characterized as vascular if a notch was observed during uterine Doppler at the second or/and the third trimester assessment.
We classified the patients in 3 groups: isolated PE (PE), PE associated with IUGR (PE+IUGR), isolated IUGR (IUGR), and vascular IUGR (vasc IUGR). Women who underwent caesarean surgery without suffering any disease during pregnancy formed the control group.
Fragments (10 to 20 mm) from the placental subchorial zone were dissected as described previously.Citation32,Citation33 Maternal membranes were eliminated, and floating villi were washed in Hanks’ balanced salt solution (HBSS). A fraction of these samples was kept frozen and another kept in TRIzol (5 mL/g, Invitrogen).
RNA extraction, poly-adenylation, and quantitative RT-PCR conditions
Total RNA from collected tissues was extracted using TRIzol Reagent (Invitrogen) in accordance with the manufacturer’s instructions, treated with RNase-free DNase, and quantified by spectrophotometry.
For mature miRNA quantification, a polyadenylation step was performed as described in.Citation21 Briefly, 4 µg of total RNA were treated with poly(A) polymerase (Ambion) according to manufacturer’s instructions. Then a phenol-chloroform extraction followed by an isopropanol precipitation was performed in order to eliminate all reagents (enzymes, ions). A specific reverse transcription was then made with a specific universal primer (containing a poly(T) part followed by a universal sequence, which will be used for the qPCR amplification, Table S1).
For mRNA and pri-miR-34a quantification, reverse transcription (RT) was performed according to a standardized protocol. Briefly, 4 µg of total DNase-treated RNA was reverse transcribed in a volume of 25 µL at 39 °C using the M-MLV Reverse Transcriptase (Invitrogen) and random primers during 1 h.
Quantitative RT-PCR was performed using the LightCycler® 480 SYBR Green I Master Mix (Roche Applied Science) in accordance with the manufacturer’s instructions. The reaction was performed in a Light-Cycler 480 Thermocycler (Roche Applied Science). Primers were designed for the coding sequences of the different genes using the PRIMER3 software (http://frodo.wi.mit.edu/cgi-bin/primer3) and aligned with basic local alignment search tool software (BLAST) to avoid nonspecific annealing. Samples were submitted to cycling according to the following PCR program: 95 °C for 5 min followed by 45 cycles of 3 temperature steps (94 °C for 10 s, 58 °C for 15 s, and 72 °C for 15 s). Finally, samples were submitted to a progressive temperature elevation (from 65 to 99 °C at 0.1 °C/s), resulting in a melting curve, enabling to check the PCR products homogeneity. In addition, amplification products were systematically controlled by agarose gel electrophoresis.
The threshold cycle number (Ct) values were collected with the LightCycler480 software (Roche Applied Science) in the exponential phase of the PCR reaction. These Cts were normalized by the Ct values obtained for the succinate dehydrogenase subunit A (SDHA) for mRNA and by those of 5S RNA for miRNA used as normalizing genes.
All primers are presented as Table S1.
Plasmid construction
To produce the pcDNA-miR-34a vector, a 1025 bp fragment encompassing the miR-34a hairpin was amplified from placental genomic DNA and cloned into pcDNA3.1 vector (Life Technologies) digested by EcoRV (Fermentas). It was already described that such construction could allow the expression and maturation of miRNA.Citation34
For Luc-3′UTR-SERPINA3, a 353 bp fragment corresponding to the 3′UTR of SERPINA3 was amplified from placental genomic DNA and cloned into a modified pGL3 basic vector (Promega, modified by the integration of a minimal CMV upstream of the luciferase gene) digested by XbaI (Fermentas).
Cell culture, hypoxia, and transfection
The JEG-3 cell line is a conventionally used model of trophoblast cells.Citation35 JEG-3 cells were grown in Dulbecco’s modified Eagle’s medium Glutamax (Life Technologies) supplemented with 10% of heat inactivated fetal calf serum (Life Technologies) and 1% penicillin/streptomycin, at 37 °C in the presence of 5% CO2.
To analyze the effect of hypoxia on gene expression, cells were seeded in 60 mm diameter glass dishes, placed in a Lwoff chamber at 37 °C and exposed to an oxygen-depleted atmosphere (2% O2, 5% CO2) or maintained as control at 37 °C in humidified normal atmosphere (20% O2 – 5% CO2). Cells were then harvested for RNA extraction using TRIzol Reagent (Invitrogen) at 3, 24, 36 and 72 h. The kinetics was done twice independently. For 3, 24, and 72 h, a part of the cells was kept for genomic DNA extraction.
To assess BCL2 and SERPINA3 expression following miR-34a transfection, cells were seeded the day before transfection at 80% of confluence, and then transfected using lipofectamine 2000 (Life technologies) according to manufacturer’s instruction. Three independent experiments each including three replicates for both conditions (empty vector and pcDNA-miR-34a) were performed.
For luciferase assay, cells were seeded the day before transfection at 30% of confluence, and then transfected using the calcium phosphate method as previously described.Citation6 For luciferase assays (see below), cells were seeded in 24-well plates and harvested after 48 h. Quantities of plasmid DNA used for transfections were, respectively, 1 μg per well total for luciferase assays (i.e., 590 ng of reporter luciferase, 400 ng of miR-34a-expressing vector and 10 ng of renilla, pRL-RSV, Promega). DNA quantifications were performed using a Nanodrop spectrophotometer. Each experiment included three to five replicates for each condition and was repeated thrice independently.
Luciferase assay
miR-34a activity on the 3′UTR of SERPINA3 was assessed by the Dual-Luciferase Reporter Assay System (Promega). A Renilla luciferase vector (pRL-RSV, Promega) was co-transfected in all experiments to monitor transfection efficiency. Luminescence was measured using a Berthold Lumat LB9507 luminometer (EG&G Berthold). The experiment was performed twice independently with three to six replicates per condition. The observed firefly activity was divided by the activity recorded from Renilla luciferase vector (pRL-RSV, Promega), and the mean values of the replicates were calculated. These mean values were then divided by the mean values of the corresponding condition to obtain the fold induction by miR-34a expression compared with the empty vector.
Genomic DNA extraction and methylation analysis with McrBC digestion
DNA was isolated following the protocol of Jeanpierre.Citation36 DNA was purified by phenol chloroform extraction followed by ethanol precipitation and subsequently dissolved in water overnight. The DNA concentration and quality were determined by measuring the absorbance at 260 nm and 280 nm in a spectrophotometer (NanoDrop 1000; Thermo Scientific).
The methylation status of the miR-34a promoter was determined by methylation-sensitive McrBC-PCR assay, as previously described.Citation37-Citation39
Genomic DNA (1μg) was treated either with 25 U of McrBC endonuclease (New England Biolabs, Beverly, MA) or mock treated with an equivalent volume of water in a 50-μl reaction mixture containing 1× NEB2 buffer, 0.1 mg/ml BSA, and 2 mm GTP. Treated and mock-treated reactions were incubated at 37 °C during 6 h. Then, the enzyme was inactivated (65°C, 20 min), and the samples were precipitated, washed, and resuspended in 30 μl of H2O. After the McrBC treatment, subsequent fluorescence-based quantitative PCR was used, starting from 2 μl of resuspended DNAs. Quantitative PCR was performed using the LightCycler® 480 SYBR Green I Master Mix (Roche Applied Science) in accordance with the manufacturer’s instructions. The reaction was performed in a Light-Cycler 480 Thermocycler (Roche Applied Science).
Specific PCR primers for miR-34a promoter and a region without McrBC restriction site (as a reference for normalizing) were used. Samples were submitted to cycling according to the following PCR program: 95 °C for 5 min followed by 45 cycles of 3 temperature steps (94 °C for 10 s, 58 °C for 15 s, and 72 °C for 15 s). Finally, samples were submitted to a progressive temperature elevation (from 65 to 99 °C at 0.1 °C/s), resulting in a melting curve, enabling to check the PCR products homogeneity. In addition, amplification products were systematically controlled by agarose gel electrophoresis.
The Ct (threshold cycle number) values were collected with the LightCycler software (Roche) in the exponential phase of the PCR. Cleavage of methylated DNA by McrBC induces DNA strand breaks and abrogates PCR amplification. Conversely, the presence of unmethylated cytosines in DNA prevents enzyme cleavage and can be detected by PCR amplification. Mock-treated DNA (undigested) served as a control and was considered as the maximal PCR product recovery. Methylated DNA has decreased amounts of PCR product after McrBC digestion. Thus, the Ct values of treated samples were normalized by the Ct values of the mock-treated DNA (ΔCt). The results are presented as fold induction of PCR product recovery after the digestion of DNA with McrBC relative to undigested DNA (computed by 2−ΔCt).
Statistical analysis
Statistical significance was evaluated using one or two-ways ANOVA followed by post-hoc tests (Student-Neuman Keuls) on the Ct differences (ΔCt) or normalized Relative Light Units. The calculations were made using the Excel® Add-in StatistiXL package. For the correlation analysis, a linear regression was performed on Excel and the significance of the observed value of R was calculated with VassarStats website (http://www.vassarstats.net). The threshold for significance was fixed at 0.05.
Additional material
Download Zip (160.7 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/26196
References
- Sibai BM. Thrombophilia and severe preeclampsia: time to screen and treat in future pregnancies?. Hypertension 2005; 46:1252 - 3; http://dx.doi.org/10.1161/01.HYP.0000188904.47575.7e; PMID: 16286582
- Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science 2005; 308:1592 - 4; http://dx.doi.org/10.1126/science.1111726; PMID: 15947178
- Astedt B, Lindoff C, Lecander I. Significance of the plasminogen activator inhibitor of placental type (PAI-2) in pregnancy. Semin Thromb Hemost 1998; 24:431 - 5; http://dx.doi.org/10.1055/s-2007-996035; PMID: 9834009
- Chelbi ST, Mondon F, Jammes H, Buffat C, Mignot TM, Tost J, Busato F, Gut I, Rebourcet R, Laissue P, et al. Expressional and epigenetic alterations of placental serine protease inhibitors: SERPINA3 is a potential marker of preeclampsia. Hypertension 2007; 49:76 - 83; http://dx.doi.org/10.1161/01.HYP.0000250831.52876.cb; PMID: 17088445
- Auer J, Camoin L, Guillonneau F, Rigourd V, Chelbi ST, Leduc M, Laparre J, Mignot TM, Vaiman D. Serum profile in preeclampsia and intra-uterine growth restriction revealed by iTRAQ technology. J Proteomics 2010; 73:1004 - 17; http://dx.doi.org/10.1016/j.jprot.2009.12.014; PMID: 20079470
- Chelbi ST, Wilson ML, Veillard AC, Ingles SA, Zhang J, Mondon F, Gascoin-Lachambre G, Doridot L, Mignot TM, Rebourcet R, et al. Genetic and epigenetic mechanisms collaborate to control SERPINA3 expression and its association with placental diseases. Hum Mol Genet 2012; 21:1968 - 78; http://dx.doi.org/10.1093/hmg/dds006; PMID: 22246292
- Davis BN, Hata A. Regulation of MicroRNA Biogenesis: A miRiad of mechanisms. Cell Commun Signal 2009; 7:18; http://dx.doi.org/10.1186/1478-811X-7-18; PMID: 19664273
- van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest 2007; 117:2369 - 76; http://dx.doi.org/10.1172/JCI33099; PMID: 17786230
- Cherblanc FL, Davidson RW, Di Fruscia P, Srimongkolpithak N, Fuchter MJ. Perspectives on natural product epigenetic modulators in chemical biology and medicine. Nat Prod Rep 2013; 30:605 - 24; http://dx.doi.org/10.1039/c3np20097c; PMID: 23396528
- Lee DC, Romero R, Kim JS, Tarca AL, Montenegro D, Pineles BL, Kim E, Lee J, Kim SY, Draghici S, et al. miR-210 targets iron-sulfur cluster scaffold homologue in human trophoblast cell lines: siderosis of interstitial trophoblasts as a novel pathology of preterm preeclampsia and small-for-gestational-age pregnancies. Am J Pathol 2011; 179:590 - 602; http://dx.doi.org/10.1016/j.ajpath.2011.04.035; PMID: 21801864
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007; 129:1311 - 23; http://dx.doi.org/10.1016/j.cell.2007.05.022; PMID: 17604720
- Pang RT, Leung CO, Ye TM, Liu W, Chiu PC, Lam KK, Lee KF, Yeung WS. MicroRNA-34a suppresses invasion through downregulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells. Carcinogenesis 2010; 31:1037 - 44; http://dx.doi.org/10.1093/carcin/bgq066; PMID: 20351093
- Umemura K, Ishioka S, Endo T, Ezaka Y, Takahashi M, Saito T. Roles of microRNA-34a in the pathogenesis of placenta accreta. J Obstet Gynaecol Res 2013; 39:67 - 74; http://dx.doi.org/10.1111/j.1447-0756.2012.01898.x; PMID: 22672425
- Kumamoto K, Spillare EA, Fujita K, Horikawa I, Yamashita T, Appella E, Nagashima M, Takenoshita S, Yokota J, Harris CC. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res 2008; 68:3193 - 203; http://dx.doi.org/10.1158/0008-5472.CAN-07-2780; PMID: 18451145
- Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev 1994; 8:707 - 19; http://dx.doi.org/10.1101/gad.8.6.707; PMID: 7926761
- Zhao WX, Lin JH. Notch signaling pathway and human placenta. Int J Med Sci 2012; 9:447 - 52; http://dx.doi.org/10.7150/ijms.4593
- Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ 2010; 17:193 - 9; http://dx.doi.org/10.1038/cdd.2009.56; PMID: 19461653
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007; 26:731 - 43; http://dx.doi.org/10.1016/j.molcel.2007.05.017; PMID: 17540598
- Winn VD, Haimov-Kochman R, Paquet AC, Yang YJ, Madhusudhan MS, Gormley M, Feng KT, Bernlohr DA, McDonagh S, Pereira L, et al. Gene expression profiling of the human maternal-fetal interface reveals dramatic changes between midgestation and term. Endocrinology 2007; 148:1059 - 79; http://dx.doi.org/10.1210/en.2006-0683; PMID: 17170095
- Caniggia I, Mostachfi H, Winter J, Gassmann M, Lye SJ, Kuliszewski M, Post M. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J Clin Invest 2000; 105:577 - 87; http://dx.doi.org/10.1172/JCI8316; PMID: 10712429
- Sdassi N, Silveri L, Laubier J, Tilly G, Costa J, Layani S, Vilotte JL, Le Provost F. Identification and characterization of new miRNAs cloned from normal mouse mammary gland. BMC Genomics 2009; 10:149; http://dx.doi.org/10.1186/1471-2164-10-149; PMID: 19351399
- Yan T, Liu Y, Cui K, Hu B, Wang F, Zou L. MicroRNA-126 regulates EPCs function: Implications for a role of miR-126 in preeclampsia. J Cell Biochem 2013; 114:2148 - 59; http://dx.doi.org/10.1002/jcb.24563; PMID: 23553946
- Kumar P, Luo Y, Tudela C, Alexander JM, Mendelson CR. The c-Myc-regulated microRNA-17~92 (miR-17~92) and miR-106a~363 clusters target hCYP19A1 and hGCM1 to inhibit human trophoblast differentiation. Mol Cell Biol 2013; 33:1782 - 96; http://dx.doi.org/10.1128/MCB.01228-12; PMID: 23438603
- Fu G, Brkić J, Hayder H, Peng C. MicroRNAs in Human Placental Development and Pregnancy Complications. Int J Mol Sci 2013; 14:5519 - 44; http://dx.doi.org/10.3390/ijms14035519; PMID: 23528856
- Wang W, Feng L, Zhang H, Hachy S, Satohisa S, Laurent LC, Parast M, Zheng J, Chen DB. Preeclampsia up-regulates angiogenesis-associated microRNA (i.e., miR-17, -20a, and -20b) that target ephrin-B2 and EPHB4 in human placenta. J Clin Endocrinol Metab 2012; 97:E1051 - 9; http://dx.doi.org/10.1210/jc.2011-3131; PMID: 22438230
- Muralimanoharan S, Maloyan A, Mele J, Guo C, Myatt LG, Myatt L. MIR-210 modulates mitochondrial respiration in placenta with preeclampsia. Placenta 2012; 33:816 - 23; http://dx.doi.org/10.1016/j.placenta.2012.07.002; PMID: 22840297
- Gao WL, Liu M, Yang Y, Yang H, Liao Q, Bai Y, Li YX, Li D, Peng C, Wang YL. The imprinted H19 gene regulates human placental trophoblast cell proliferation via encoding miR-675 that targets Nodal Modulator 1 (NOMO1). RNA Biol 2012; 9:1002 - 10; http://dx.doi.org/10.4161/rna.20807; PMID: 22832245
- Lázár L, Nagy B, Molvarec A, Szarka A, Rigó J Jr.. Role of hsa-miR-325 in the etiopathology of preeclampsia. Mol Med Rep 2012; 6:597 - 600; PMID: 22710575
- Du R, Sun W, Xia L, Zhao A, Yu Y, Zhao L, Wang H, Huang C, Sun S. Hypoxia-induced down-regulation of microRNA-34a promotes EMT by targeting the Notch signaling pathway in tubular epithelial cells. PLoS One 2012; 7:e30771; http://dx.doi.org/10.1371/journal.pone.0030771; PMID: 22363487
- Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci U S A 2008; 105:14879 - 84; http://dx.doi.org/10.1073/pnas.0803230105; PMID: 18812516
- Pang RT, Leung CO, Lee CL, Lam KK, Ye TM, Chiu PC, Yeung WS. MicroRNA-34a is a tumor suppressor in choriocarcinoma via regulation of Delta-like1. BMC Cancer 2013; 13:25; http://dx.doi.org/10.1186/1471-2407-13-25; PMID: 23327670
- Mondon F, Mignot TM, Rebourcet R, Jammes H, Danan JL, Ferré F, Vaiman D. Profiling of oxygen-modulated gene expression in early human placenta by systematic sequencing of suppressive subtractive hybridization products. Physiol Genomics 2005; 22:99 - 107; http://dx.doi.org/10.1152/physiolgenomics.00276.2004; PMID: 15797968
- Vaiman D, Mondon F, Garcès-Duran A, Mignot TM, Robert B, Rebourcet R, Jammes H, Chelbi ST, Quetin F, Marceau G, et al. Hypoxia-activated genes from early placenta are elevated in preeclampsia, but not in Intra-Uterine Growth Retardation. BMC Genomics 2005; 6:111; http://dx.doi.org/10.1186/1471-2164-6-111; PMID: 16129025
- Rao DS, O'Connell RM, Chaudhuri AA, Garcia-Flores Y, Geiger TL, Baltimore D. MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity 2010; 33:48 - 59
- Kohler PO, Bridson WE, Hammond JM, Weintraub B, Kirschner MA, Van Thiel DH. Clonal lines of human choriocarcinoma cells in culture. Acta Endocrinol Suppl (Copenh) 1971; 153:137 - 53; PMID: 5314233
- Jeanpierre M. A rapid method for the purification of DNA from blood. Nucleic Acids Res 1987; 15:9611; http://dx.doi.org/10.1093/nar/15.22.9611; PMID: 3684611
- Holemon H, Korshunova Y, Ordway JM, Bedell JA, Citek RW, Lakey N, Leon J, Finney M, McPherson JD, Jeddeloh JA. MethylScreen: DNA methylation density monitoring using quantitative PCR. Biotechniques 2007; 43:683 - 93; http://dx.doi.org/10.2144/000112597; PMID: 18072598
- Ordway JM, Bedell JA, Citek RW, Nunberg A, Garrido A, Kendall R, Stevens JR, Cao D, Doerge RW, Korshunova Y, et al. Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis 2006; 27:2409 - 23; http://dx.doi.org/10.1093/carcin/bgl161; PMID: 16952911
- Borghese B, Barbaux S, Mondon F, Santulli P, Pierre G, Vinci G, Chapron C, Vaiman D. Research resource: genome-wide profiling of methylated promoters in endometriosis reveals a subtelomeric location of hypermethylation. Mol Endocrinol 2010; 24:1872 - 85; http://dx.doi.org/10.1210/me.2010-0160; PMID: 20685852