Abstract
Using a candidate gene approach we recently identified frequent methylation of the RASSF2 gene associated with poor overall survival in Ewing sarcoma (ES). To identify effective biomarkers in ES on a genome-wide scale, we used a functionally proven epigenetic approach, in which gene expression was induced in ES cell lines by treatment with a demethylating agent followed by hybridization onto high density gene expression microarrays. After following a strict selection criterion, 34 genes were selected for expression and methylation analysis in ES cell lines and primary ES. Eight genes (CTHRC1, DNAJA4, ECHDC2, NEFH, NPTX2, PHF11, RARRES2, TSGA14) showed methylation frequencies of > 20% in ES tumors (range 24–71%), these genes were expressed in human bone marrow derived mesenchymal stem cells (hBMSC) and hypermethylation was associated with transcriptional silencing. Methylation of NPTX2 or PHF11 was associated with poorer prognosis in ES. In addition, six of the above genes also showed methylation frequency of > 20% (range 36–50%) in osteosarcomas. Identification of these genes may provide insights into bone cancer tumorigenesis and development of epigenetic biomarkers for prognosis and detection of these rare tumor types.
Introduction
Ewing sarcoma is a rare malignant bone tumor occurring predominantly in children and adolescents, but is also seen in adults. Ewing sarcoma is the second most common primary bone tumor, characterized by EWS/ETS translocation and is thought to originate from mesenchymal stem cells.Citation1 The most common sites of involvement are the long bones, pelvis, ribs, and vertebral column. A combination of chemotherapy and excision, with or without radiotherapy, is used to manage the disease. Although prognosis for localized disease has improved greatly over the past decades, 20–30% of patients have metastasis at the time of diagnosis and prognosis for these patients is very poor. Hence, molecular markers that can help in diagnosis or prognosis are urgently required and may also provide novel targets for therapy.Citation2 Epigenetic changes, such as DNA methylation, can provide such biomarkers and have shown promise in other cancers.Citation3 Examples include predictive markers for cancer therapy: MGMT methylation predicts response to temozolomide treatment in glioblastoma patients;Citation4 prognostic biomarkers for cancer progression/outcome-promoter methylation of a panel of four genes (CDKN2A, CDH13, RASSF1A, and APC) predicts recurrence in early stage non-small cell lung cancer; and biomarkers for early detection of cancer—vimentin (VIM) gene methylation has been developed as the first commercially available DNA methylation-based diagnostic test for early detection of colorectal cancer.Citation5-Citation7
Previously, we undertook a candidate gene approach and demonstrated that RASSF2 methylation is associated with poor prognosis in ES patients.Citation8 Accumulation of promoter region DNA hypermethylation leading to silencing of cancer related genes are a frequent event in many types of cancers. In the current study, we used high density whole transcriptome gene expression arrays on ES cell lines after pharmacological unmasking of methylated DNA to identify novel genes epigenetically silenced in Ewing sarcoma.
Results
Identification of silenced and hypermethylated genes in ES patient cohort
Three Ewing sarcoma cell lines were treated with the demethylating agent 5-aza-2′-deoxycytidine (5 μM) for 5 d to re-activate epigenetically silenced/downregulated genes and changes in gene expression were measured on Illumina’s HumanHT-12 v4 gene expression arrays that contain probes for > 47,000 transcripts across the human transcriptome. Gene expression was also measured for human bone marrow derived mesenchymal stem cells (hBMSC) as the control tissue on the same gene expression arrays as the ES cell lines. In order to prioritize genes for methylation analysis, we initially focused on genes which showed a 2-fold or greater increase in expression following demethylation in two or more cell lines. Transcripts that represented hypothetical proteins and imprinted genes were also removed as well as genes located on X and Y chromosomes. Next, we excluded genes that did not have a predicted promoter region CpG island, as predicted by the human genome browser (www.genome.ucsc.edu). Finally, 34 genes were selected for methylation analysis (Fig. S1; Table S1). These genes showed no (or low) basal expression before 5-aza-2'-deoxycytidine treatment, were upregulated after treatment with the demethylating agent and were expressed in hBMSC. Next, 22 genes from the above list of 34 genes were selected for confirmation of the gene expression microarray data. RT-PCR analyses demonstrated a good correlation (17 out of 22 confirmed; 77%) with microarray based estimates of changes in gene expression and were expressed in hBMSC (). No further analysis of the 5 false positive genes was performed. Among the remaining 29 genes analyzed for methylation in ES cell lines using COBRA (Combined Bisulfite Restriction Analysis), 21 showed methylation in two or more cell lines (72%). In the majority of the cases, there was a good correlation between gene expression and gene methylation status (). We next analyzed primary ES tumors for methylation of the above 21 genes found to be frequently methylated in ES cell lines. In primary ES sample cohort (n = 46), 8 genes showed methylation frequency of > 20% in ES tumors (CTHRC1 [29%], DNAJA4 [28%], ECHDC2 [71%], NEFH [47%], NPTX2 [61%], PHF11 [24%], RARRES2 [28%], TSGA14 [60%] ( and ; Table S2). These genes were expressed (and unmethylated) in hBMSC and showed upregulation of gene expression in methylated ES cell lines after 5-azaDC treatment (). The remaining genes were either unmethylated or showed low methylation frequency in primary ES tumors. COBRA results for the frequently methylated genes were confirmed by clone sequencing of bisulfite modified DNA and, as expected, showed heterogeneous pattern of methylation in primary tumors ().
Figure 1. Expression and methylation analysis in ES cell lines. (A) Expression results for 4 selected genes in ES cell lines and hBMSC (CTHRC1, RARRES2, TSGA14, DNAJA4, and GAPDH as control). Three ES cell lines are shown for each gene with (+) indicating the samples after 5-azaDC treatment and (-) indicating the samples without 5-azaDC treatment. (B) COBRA results for the selected genes. Undigested (U) samples were loaded next to the BstU1 digested samples (D). Three ES cell lines are shown for each gene with (*) indicating that the cell line is methylated and the positive control (C) is in vitro methylated DNA.
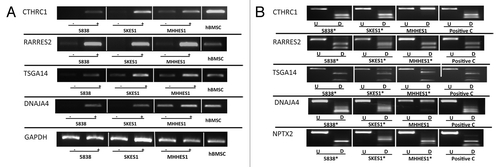
Figure 2. COBRA analysis for selected gene in primary ES samples. Results of COBRA analysis for 5 genes (CTHRC1, DNAJA4, ECHDC2, NPTX2 and RARRES2) with methylation frequency of > 20% in ES samples. COBRA results are shown for primary ES samples with in vitro methylated DNA as positive control (C). The Undigested (U) samples are loaded next to the BstU1 digested samples (D). (*) indicates methylated samples.
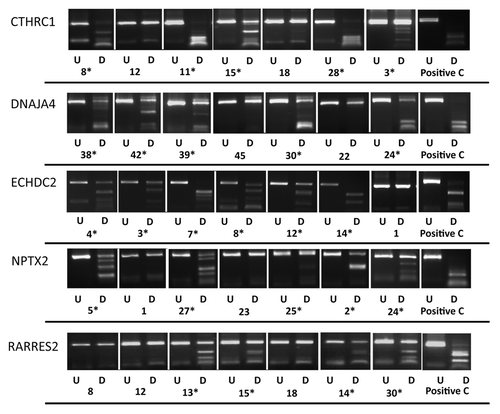
Figure 3. Frequently methylated genes in ES. Graph showing methylation frequency for each gene in primary ES tumors.
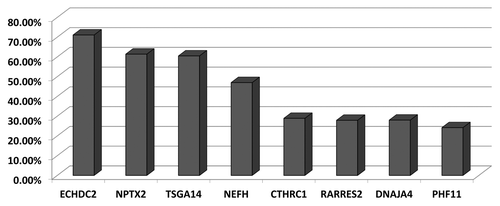
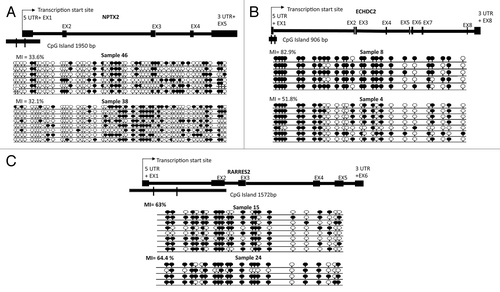
Figure 4. Bisulfite sequencing analysis in primary ES samples and schematics representing sequenced region for three genes (NPTX2, ECHDC2, and RARRES2). (A) Single clone bisulfite sequencing analysis of NPTX2 promoter region CpG island shows methylation index of 33.6% in ES sample 46 and 32.1% in ES sample 38. (B) Single clone and bisulfite sequencing analysis of ECHDC2 CpG island region shows methylation index of 82.9% and 51.8% in ES samples 8 and 4 respectively. (C) Sequencing analysis of RARRES2 CpG island region shows methylation index of 63% and 64.4% in ES samples 15 and 24 respectively. Individual alleles are represented by a horizontal line, black and white circles indicate methylated and unmethylated CpG dinucleotides respectively. The percentage of methylation indexes were calculated as follows: (number of methylated CpG dinucleotide/number of CpG dinucleotide sequenced) x 100.
Methylation frequency in osteosarcomas
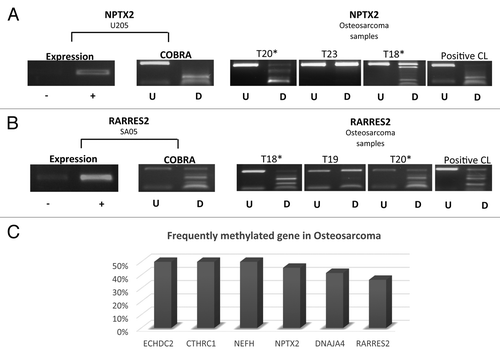
Osteosarcoma is the most frequently occurring primary bone tumor. Analysis of 6 of the above 8 genes in osteosarcomas demonstrated that these genes were also frequently methylated in these tumors (n = 12) (range of frequency 36–50%) (). The frequently methylated genes demonstrated upregulation of gene expression in methylated osteosarcoma cell lines after treatment with 5-azaDC. Hence, some of these genes may have biological and or clinical relevance for development of osteosarcomas.
Figure 5. Expression and methylation analysis for NPTX2 and RARRES2 in osteosarcoma cell lines and primary tumors. (A) NPTX2 (B) RARRES2 expression and methylation (COBRA) results in osteosarcoma cell line and tumors. (C) Frequently methylated genes in osteosarcoma with the methylation frequency for each gene. -, gene expression before 5-azaDC treatment; +, gene expression after 5-azaDC; U, undigested samples; D, BstU1 digested samples; *, methylated samples; CL, positive control.
ES patient survival and gene methylation
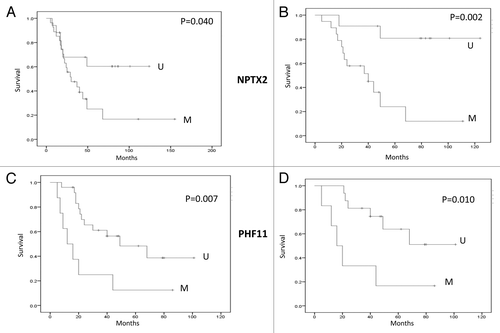
We analyzed whether methylation in ES patient samples for the 8 frequently methylated genes was associated with patient survival. There was no significant association between methylation status and overall survival for 6 of the 8 genes as demonstrated by Kaplan-Meier survival curves. However, NPTX2 methylation (p = 0.04; p = 0.002 for ≤ 18 y) or PHF11 (p = 0.007; p = 0.01 for ≤ 18 y) methylation was associated with significantly poorer prognosis ().
Figure 6. Survival analysis. (AandB) Kaplan-Meier Survival curves for NPTX2 methylation status and overall survival in ES sample cohort (M; Methylated and U; Unmethylated). (CandD) Kaplan-Meier Survival curves for PHF11 methylation status and overall survival in the ES sample cohort (M, Methylated; U, Unmethylated). A and C all ages; B and D patients 18 y or below.
Discussion
Cancer specific epigenetic alterations are a hallmark of tumor suppressor gene inactivation in human cancers. In recent years, various high throughput genome-wide screening approaches have been developed to identify DNA methylation in human cancers, including MEDIP, MIRA/MBD, methylation arrays, next generation sequencing, and functional epigenomics. In the current study, we used pharmacological inhibition of DNA methylation (functional epigenomics) combined with gene expression microarrays to identify genome-wide functional methylation changes in Ewing sarcoma. We have previously utilized this approach to successfully identify novel hypermethylated tumor suppressor genes in clear cell renal cell carcinoma.Citation9,Citation10
We identified 8 genes (CTHRC1, DNAJA4, ECHDC2, NEFH, NPTX2, PHF11, RARRES2, TSGA14) that were hypermethylated in > 20% of primary ES tumors, for which gene expression was reactivated or upregulated in methylated ES cell lines after treatment with a demethylating agent. Six of the above genes (CTHRC1, DNAJA4, ECHDC2, NEFH, NPTX2, RARRES2) were also frequently methylated in osteosarcoma.
Collagen Triple Helix Repeat Containing 1 (CTHRC1) is a soluble protein involved in bone formation, morphogenesis, and vascular remodeling. CTHRC1 gene has been reported to be linked with different types of solid cancers and associated with metastasis and cancer invasion.Citation11 Wang et al., reported that CTHRC1 was re-expressed after treating gastric cancer cell lines with 5-azaDC.Citation11
DnaJ (Hsp40) homolog (DNAJA4) is involved in oncogenesis, cell differentiation, and tissue development. Mahoney et al. reported promoter hypermethylation in both rhabdomyosarcoma cell lines and primary patient samples. Moreover, they also used RT-qPCR to confirm the upregulation of gene expression after treating cell line with 5-azaDC.Citation12 In a genome-wide screen, Dnaja4 was recently identified as a target of c-Myc mediated DNA methylation dependent transcriptional silencing.Citation13
ECHDC2, located at 1p32.3, showed the highest methylation frequency in ES samples (71%); ECHDC2 was expressed and unmethylated in hBMSC. Interestingly, microsatellite instability and allelic loss at 1p was demonstrated in 35% of Ewing sarcomas.Citation14
Neurofilament heavy polypeptide (NEFH) was previously shown to regulate Akt-β-catenin pathway and was frequently methylated in esophageal cancer.Citation15 Hence, cancer cells with methylated NEFH could be targeted with specific inhibitors of the deregulated pathways.
The association between neuronal pentraxin II (NPTX2) gene methylation and pancreatic cancer was reported in many different studies.Citation16,Citation17 NPTX2 is known to inhibit proliferation and invasion of pancreatic cancer cells.Citation18 In our present study, NPTX2 was frequently methylated in ES patient’s samples (27/44), with methylation frequency of 61%, and was unmethylated but expressed in control hBMSC. In addition, we found that NPTX2 methylation was linked to poor prognosis in ES patients.
PHF finger protein 11 (PHF11) is one of the PHD (plant homeodomain)-containing transcription factors located at chromosome 13q14.2. These transcription factors are expressed in the nucleus and play important roles in cell proliferation and differentiation in human disease. In a functional screen using monochromosome transfer followed by gene expression microarray analysis, a critical tumor-suppressive region was mapped to 13q14 in esophageal carcinoma containing 9 candidate tumor suppressor genes, including PHF11. PHF11 expression was downregulated or absent in 100% of Esophageal Carcinoma cell lines, confirmed by RT-PCR.Citation19 This gene was frequently methylated in primary ES patient samples (24%) and methylation was associated with poorer prognosis. PHF11 had not been reported previously to be methylated in any human cancer.
Retinoic Acid Receptor Responder 2 (RARRES2) encodes for a recently identified secreted chemotactic protein (Chemerin) that initiates chemotaxis via the G protein-coupled receptor CMKLR1 and plays a role in adaptive and innate immunity. RARRES2 was reported to be downregulated and frequently hypermethylated in primary adrenocortical tumors.Citation20 In a genome-wide gene expression screen, RARRES2 was differentially expressed between malignant and benign adrenocortical carcinomas and could accurately distinguish between the two entities.Citation21 In the present study, RARRES2 showed cancer specific frequent hypermethylation in ES.
TSGA14 (CEP41) encodes a centrosomal and microtubule-binding protein. The gene is located adjacent to the imprinted gene MEST on 7q32 but escapes genomic imprinting.Citation22 This is the first report of DNA methylation of TSGA14 in human cancer.
In summary, we performed a genome-wide screen for functional methylation changes in Ewing sarcoma and identified a number of novel genes with diverse functions that are hypermethylated in these tumors. The identification of these genes may provide a basis for developing biomarkers for diagnostic and prognostic purposes and for understanding pathways deregulated in this rare cancer sub-type.
Materials and Methods
Cell culture, 5-Aza-2'-deoxycytidine treatment, and microarray analysis
Ewing sarcoma cell lines were maintained in RPMI medium for MHHES-1 and 5838 and McCoy’s 5A medium for SKES-1 (Sigma-Aldrich), supplemented with 15% FBS, glutamine and antibiotics, at 37°C, 5% CO2.Citation23 After a 24 h seeding period, cells were treated with 5µM 5-aza-2'-deoxycitidine (5-azaDC) freshly prepared in sterile ddH2O, to achieve genome demethylation. Treatment was performed over 5 d with daily media changes and addition of fresh 5-azaDC. RNA was prepared as described in Gharanei et al.Citation8 Total RNA from the three ES cell lines (before and after treatment) and RNA from hBMSC was sent to Cambridge Genomic Services (http://www.cgs.path.cam.ac.uk/). Briefly, the total RNA was amplified and converted to cDNA before hybridization to the HumanHT-12 v4 BeadChips (Illumina) and data production according to the manufacturer protocols.
Patient samples
Clinical information for the ES patient samples and isolation of hBMSC samples has been described previously (n = 46).Citation8 The study was approved by the relevant institutional review board and was conducted according to the principles outlined in the Declaration of Helsinki.
RT-PCR, bisulfite modification and promoter methylation analysis
RT-PCR was performed as described previously,Citation24 with annealing temperatures ranging from 56–60 °C. Bisulfite modification, COBRA analysis, cloning, and bisulfite DNA sequencing was performed as described previouslyCitation24 (methylation and expression primers are provided in Tables S3 and S4). CpG islands in the putative promoter regions were identified using the human genome browser. PCR primers (semi-nested or fully-nested) for methylation analysis were designed within the appropriate CpG island close to the transcription start site.
Additional material
Download Zip (268 KB)Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
Abdullah Alholle was supported in part by Kuwait Medical Genetics Centre (KMGC), Ministry of Health, Kuwait and funding from University of Birmingham, UK. We would also like to thank Bone Cancer Research Trust and Ministero della Salute of Italy, Ricerca Corrente L2013 for financial support.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/26266
References
- Toomey EC, Schiffman JD, Lessnick SL. Recent advances in the molecular pathogenesis of Ewing’s sarcoma. Oncogene 2010; 29:4504 - 16; http://dx.doi.org/10.1038/onc.2010.205; PMID: 20543858
- Shukla N, Schiffman J, Reed D, Davis IJ, Womer RB, Lessnick SL, Lawlor ER, COG Ewing Sarcoma Biology Committee. Biomarkers in Ewing Sarcoma: The Promise and Challenge of Personalized Medicine. A Report from the Children’s Oncology Group. Front Oncol 2013; 3:141; PMID: 23761859
- Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 2012; 13:679 - 92; http://dx.doi.org/10.1038/nrg3270; PMID: 22945394
- Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343:1350 - 4; http://dx.doi.org/10.1056/NEJM200011093431901; PMID: 11070098
- Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S, Glöckner S, Piantadosi S, Gabrielson E, Pridham G, et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med 2008; 358:1118 - 28; http://dx.doi.org/10.1056/NEJMoa0706550; PMID: 18337602
- Chen WD, Han ZJ, Skoletsky J, Olson J, Sah J, Myeroff L, Platzer P, Lu S, Dawson D, Willis J, et al. Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed vimentin gene. J Natl Cancer Inst 2005; 97:1124 - 32; http://dx.doi.org/10.1093/jnci/dji204; PMID: 16077070
- Itzkowitz S, Brand R, Jandorf L, Durkee K, Millholland J, Rabeneck L, Schroy PC 3rd, Sontag S, Johnson D, Markowitz S, et al. A simplified, noninvasive stool DNA test for colorectal cancer detection. Am J Gastroenterol 2008; 103:2862 - 70; http://dx.doi.org/10.1111/j.1572-0241.2008.02088.x; PMID: 18759824
- Gharanei S, Brini AT, Vaiyapuri S, Alholle A, Dallol A, Arrigoni E, Kishida T, Hiruma T, Avigad S, Grimer R, et al. RASSF2 methylation is a strong prognostic marker in younger age patients with Ewing sarcoma. Epigenetics 2013; 8; In press http://dx.doi.org/10.4161/epi.25617; PMID: 23887284
- Morris MR, Gentle D, Abdulrahman M, Maina EN, Gupta K, Banks RE, Wiesener MS, Kishida T, Yao M, Teh B, et al. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res 2005; 65:4598 - 606; http://dx.doi.org/10.1158/0008-5472.CAN-04-3371; PMID: 15930277
- Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Latif F, Maher ER. Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene 2010; 29:2104 - 17; http://dx.doi.org/10.1038/onc.2009.493; PMID: 20154727
- Wang P, Wang YC, Chen XY, Shen ZY, Cao H, Zhang YJ, Yu J, Zhu JD, Lu YY, Fang JY. CTHRC1 is upregulated by promoter demethylation and transforming growth factor-β1 and may be associated with metastasis in human gastric cancer. Cancer Sci 2012; 103:1327 - 33; http://dx.doi.org/10.1111/j.1349-7006.2012.02292.x; PMID: 22590977
- Mahoney SE, Yao Z, Keyes CC, Tapscott SJ, Diede SJ. Genome-wide DNA methylation studies suggest distinct DNA methylation patterns in pediatric embryonal and alveolar rhabdomyosarcomas. Epigenetics 2012; 7:400 - 8; http://dx.doi.org/10.4161/epi.19463; PMID: 22419069
- Licchesi JD, Van Neste L, Tiwari VK, Cope L, Lin X, Baylin SB, Herman JG. Transcriptional regulation of Wnt inhibitory factor-1 by Miz-1/c-Myc. Oncogene 2010; 29:5923 - 34; http://dx.doi.org/10.1038/onc.2010.322; PMID: 20697356
- Ohali A, Avigad S, Cohen IJ, Meller I, Kollender Y, Issakov J, Goshen Y, Yaniv I, Zaizov R. High frequency of genomic instability in Ewing family of tumors. Cancer Genet Cytogenet 2004; 150:50 - 6; http://dx.doi.org/10.1016/j.cancergencyto.2003.08.014; PMID: 15041223
- Kim MS, Chang X, LeBron C, Nagpal JK, Lee J, Huang Y, Yamashita K, Trink B, Ratovitski EA, Sidransky D. Neurofilament heavy polypeptide regulates the Akt-beta-catenin pathway in human esophageal squamous cell carcinoma. PLoS One 2010; 5:e9003; http://dx.doi.org/10.1371/journal.pone.0009003; PMID: 20140245
- Sato N, Fukushima N, Maitra A, Matsubayashi H, Yeo CJ, Cameron JL, Hruban RH, Goggins M. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res 2003; 63:3735 - 42; PMID: 12839967
- Park JK, Ryu JK, Yoon WJ, Lee SH, Lee GY, Jeong KS, Kim YT, Yoon YB. The role of quantitative NPTX2 hypermethylation as a novel serum diagnostic marker in pancreatic cancer. Pancreas 2012; 41:95 - 101; http://dx.doi.org/10.1097/MPA.0b013e318221c903; PMID: 21778928
- Zhang L, Gao J, Li L, Li Z, Du Y, Gong Y. The neuronal pentraxin II gene (NPTX2) inhibit proliferation and invasion of pancreatic cancer cells in vitro. Mol Biol Rep 2011; 38:4903 - 11; http://dx.doi.org/10.1007/s11033-010-0632-y; PMID: 21161403
- Ko JM, Chan PL, Yau WL, Chan HK, Chan KC, Yu ZY, Kwong FM, Miller LD, Liu ET, Yang LC, et al. Monochromosome transfer and microarray analysis identify a critical tumor-suppressive region mapping to chromosome 13q14 and THSD1 in esophageal carcinoma. Mol Cancer Res 2008; 6:592 - 603; http://dx.doi.org/10.1158/1541-7786.MCR-07-0154; PMID: 18403638
- Rechache NS, Wang Y, Stevenson HS, Killian JK, Edelman DC, Merino M, Zhang L, Nilubol N, Stratakis CA, Meltzer PS, et al. DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J Clin Endocrinol Metab 2012; 97:E1004 - 13; http://dx.doi.org/10.1210/jc.2011-3298; PMID: 22472567
- Fernandez-Ranvier GG, Weng J, Yeh RF, Khanafshar E, Suh I, Barker C, Duh QY, Clark OH, Kebebew E. Identification of biomarkers of adrenocortical carcinoma using genomewide gene expression profiling. Arch Surg 2008; 143:841 - 6, discussion 846; http://dx.doi.org/10.1001/archsurg.143.9.841; PMID: 18794420
- Yamada T, Kayashima T, Yamasaki K, Ohta T, Yoshiura Ki, Matsumoto N, Fujimoto S, Niikawa N, Kishino T. The gene TSGA14, adjacent to the imprinted gene MEST, escapes genomic imprinting. Gene 2002; 288:57 - 63; http://dx.doi.org/10.1016/S0378-1119(02)00428-6; PMID: 12034494
- Avigad S, Shukla S, Naumov I, Cohen IJ, Ash S, Meller I, Kollender Y, Issakov J, Yaniv I. Aberrant methylation and reduced expression of RASSF1A in Ewing sarcoma. Pediatr Blood Cancer 2009; 53:1023 - 8; http://dx.doi.org/10.1002/pbc.22115; PMID: 19637319
- Ricketts CJ, Morris MR, Gentle D, Brown M, Wake N, Woodward ER, Clarke N, Latif F, Maher ER. Genome-wide CpG island methylation analysis implicates novel genes in the pathogenesis of renal cell carcinoma. Epigenetics 2012; 7:278 - 90; http://dx.doi.org/10.4161/epi.7.3.19103; PMID: 22430804