Abstract
The ability to specifically reactivate epigenetically silenced genes would have great utility in experimental studies and potential therapeutic value. Here, we describe the specific targeting of thymidine DNA glycosylase (TDG), an enzyme involved in the mechanism of methylcytosine demethylation, to the promoter of Nos2, a gene silenced by methylation in fibroblasts, using artificial zinc finger DNA binding domains. Individual targeted TDG constructs had a small effect on Nos2 expression and methylation, but simultaneous targeting of a quartet of TDG constructs significantly restored responsiveness to LPS and IFN stimuli in association with marked cytosine demethylation at the promoter and CpG island; catalytically inactive TDG complexes had no effect. Whole-genome expression microarray and pathway analysis found only 42 genes that were affected by targeted TDG constructs; the majority are likely downstream of the effect on Nos2. This study therefore shows highly specific, directed reactivation of a single, silenced gene by targeting of a demethylase to the promoter.
Introduction
Virtually all cells in an organism contain all the genes of that organism's genome; it is the epigenetic silencing of “unused” genes that underlies the difference in structure and function of cellsCitation1,Citation2 This silencing is a powerful and heritable mechanism controlling gene function.Citation2 DNA methylation plays a pivotal role in epigenetic silencing of transcription:Citation3 conversely, demethylation of promoters or regulatory intragenic regions can enhance transcription.Citation4,Citation5 Gene-specific DNA demethylation would thus allow selective re-activation of many silenced genes for experimental or therapeutic purposes. However, existing approaches lack either specificity or efficiency in mammalian systems, as reviewed in.Citation6 Recent studies demonstrate that thymine-DNA-glycosylase (TDG)Citation7-Citation10 acts in concert with other factors to demethylate cytosine residues, although the mechanism is only partially understood. By using a specific DNA-binding domain of NF-κB we previously demonstratedCitation11 that tethering TDG molecules to gene promoters allows local demethylation and leads to transcriptional enhancement. TDG must be directly targeted to the DNA, since overexpressed, untethered TDG did not affect methylation or transcription. However, endogenous transcription factors target many genes simultaneously, limiting the utility of their DNA binding domains for specific regulation of individual genes. Because DNA-binding motifs may be highly sequence-specific, this approach has promising potential for precise demethylation of individual promoters. In this study, we therefore designed custom DNA-binding domains comprising artificial zinc finger (ZF) arrays constructed to specifically bind to 4 target sequences in the promoter of nitric oxide synthase 2 (Nos2). This gene is silenced by DNA methylation in 3T3 fibroblasts,Citation12 hence these cells have limited ability to upregulate production of nitric oxide (NO), a valuable defensive and regulatory factor.Citation13 However in our prior study, the Nos2 gene, among others, was responsive to demethylation,Citation11 hence we chose it as a sample target for the gene-specific assays reported here.
While it is not currently possible to design a single ZF to reliably target a unique promoter with no off-target binding, a difficulty in common for all existing DNA-binding domains, we postulated that simultaneously targeting multiple ZF-TDG constructs to the Nos2 promoter could allow a substantial improvement of the specificity and magnitude of the effect because of the cumulative activity of several TDG molecules. Off-target effects would be minimized, since, even if individual constructs may bind elsewhere in the genome, target sequences are clustered only at the Nos2 promoter. Here, we demonstrate that targeting the promoter with multiple ZF-TDG constructs indeed provided a cumulative effect and led to significant decreases in methylation in the promoter and CpG island of Nos2. This was associated with highly specific enhancement of transcriptional responsiveness of the gene to stimulation by LPS and IFNγ leading to ~3–4-fold increases in mRNA expression and to elevated production of NO (which is not a typical product of 3T3 fibroblasts). The controls included identical constructs with catalytically inactive TDG (single amino-acid mutation) and ZF-alone constructs.
Results
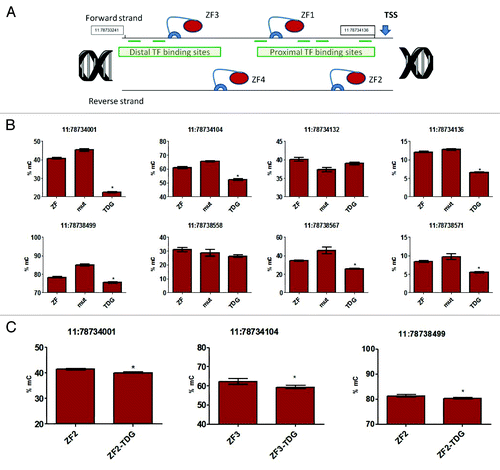
presents the schematic of targeting the Nos2 promoter. In the cells expressing all 4 ZF-TDG constructs DNA methylation has decreased significantly in multiple sites across the promoter and the nearest downstream CpG island by approximately 10–20% () compared with the inactive TDG or ZF-alone. Interestingly, even in the CpG sites with relatively low initial methylation we observed a significant reduction in mC content. Out of 8 sites tested, the drop in methylation was significant in 6. In contrast, in the cells expressing each single construct separately the decreases in methylation were comparatively small, although statistically significant. shows the three largest effects we observed were in the 2–5% range. This supports our hypothesis that multiple ZF-TDG constructs provide a cumulative effect when positioned in proximity to each other.
Figure 1. (A) Four ZF-TDG constructs (‘TDG’) were transduced into 3T3 fibroblasts. Controls included 4 constructs with mutant catalytically inactive TDG (‘mut’), ZF-only (‘ZF’-) and mock-transduced cells (not shown). (B) To analyze methylation changes induced by the constructs, we performed pyrosequencing assay to positions in the promoter (top, 11:78734001–78734136) and in the nearest CpG island (bottom, 11:78738499–78738571). We found statistically significant decrease in methylation at most sites in the ZF-TDG group (TDG) vs. mutant control (mut) or ZF-only control (ZF). Interestingly, even the 10–20% reduction in methylation at these sites was associated with a strong transcriptional response (), which emphasizes the potential of DNA demethylation for manipulation of gene expression. Of note, not all CpG sites (6 out of 8 tested) showed decreased methylation suggesting some CpG are causative for transcription and some are not. (C) To compare the magnitude of the effect we interrogated these sites in cells transduced with single ZF-TDG constructs and respective ZF-alone controls. Decreases in methylation in these cells were substantially less prominent, as shown in these three representative sites. All experiments were performed at least 3 times in triplicates, pyrosequencing measurements were performed in quadruplicates, *P < 0.05.
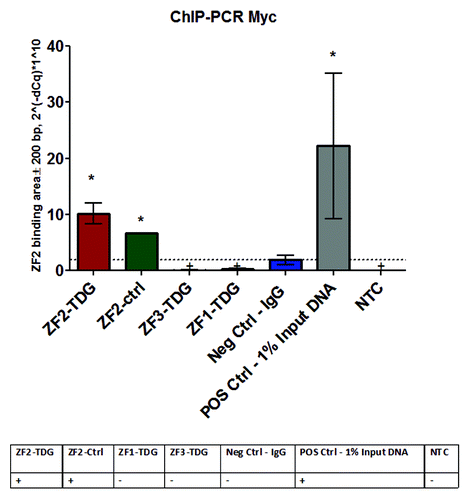
To confirm binding of the constructs to DNA in the promoter area we performed ChIP-PCR. In the example of ZF2 shown in we used anti-Myc Ab to precipitate sonicated fragments of DNA approximately 300–400 bp long followed by amplification of the DNA products by PCR against custom primers designed to flank the putative binding site of ZF2. In both ZF2-TDG and ZF2-alone provided similar signal, indicating that both constructs bound with similar affinity. Construct ZF1 includes Flag, not Myc, and therefore remained negative. Construct ZF3 includes Myc, but is positioned more than 700 bp away from ZF2 and therefore also remained negative. The table underneath the bar chart in summarizes the expectations from experimental and control groups in this assay. Similar testing for other constructs is not shown. In summary, these data indicate that binding of ZF constructs was sufficiently effective and specific to the desired target.
Figure 2. To confirm the binding of ZF constructs to desired DNA target areas in the promoter we performed ChIP-PCR using a standard sonication-based shearing protocol followed by Myc precipitation (ChIP kit, Sigma) and real-time PCR for a set of primers amplifying a small fragment of the Nos2 promoter. Both ZF2-TDG and the control ZF2 construct include Myc marker and, as evident, precipitated the desired fragment in Nos2 promoter indicating they were bound. ZF1-TDG uses FLAG instead of Myc, while ZF3-TDG is located ~700 bp away from ZF2 and is outside of the amplicon; they served as biological negative controls in the assay, together with control IgG they indicate that non-specific precipitation was minimal. Unprecipitated input DNA (diluted 1:100) served as positive control. *P < 0.05.
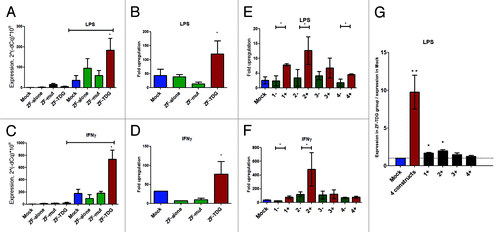
As shown in , responsiveness to both LPS and IFNγ was increased by several fold in cells expressing all 4 constructs, compared with any of the controls, which showed minimal upregulation (). In a separate series of experiments we tested the effect of only one construct per cell and found that a single construct provides, on average, a weaker response (). In summary, we postulate that transcriptional effect of ZF-TDG constructs is also cumulative (). Interestingly, ZF #2 was the closest to TSS and the proved the most effective construct to enhance responsiveness to both LPS and IFN. Construct #1 was slightly farther and provided less prominent responses; #4 was still farther and led to a significant enhancement of response only to LPS but not IFN, while #3 being the most remote was minimally effective. This suggests that proximity to TSS may play a role in targeted demethylation-based gene function enhancement, but the effect can also be explained by confinement of the differentially methylated site to the proximal Nos2 promoter.Citation12
Figure 3. (A–D) Four simultaneous ZF-TDG constructs provided a strong transcriptional enhancement of Nos2 in response to stimuli without affecting background expression. Cells were stimulated either by LPS (0.5 ng/mL for 2 h) (A and B); or by IFNγ (200 U/mL 24 h) (C and D). Expression is normalized to β-actin. In B and D fold upregulation was obtained by dividing expression after stimulation to background unstimulated expression in that group. (E and F) To identify the efficiency of each construct separately, we transduced cells with each ZF-TDG or its ZF-only control (1+ vs. 1-, etc.) and tested effects on upregulation of Nos2 in identical conditions. ZF-TDG #2 led to the greatest increase in Nos2 transcription > #1 > > ##s 4 and 3. Interestingly, ZF #2 was the closest to TSS, followed by #1 and #4, with #3 being the most remote, which suggests that proximity to TSS may play a role. Notably, the fold change by each single construct was substantially lower than the cumulative effect of all 4, most prominently for the case of LPS, as seen in (G) where the fold upregulation is normalized to the level in Mock. All experiments were performed at least 3 times in triplicates, *P < 0.05; **P < 0.01.
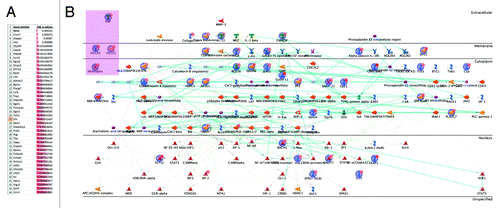
To determine whether the effect was gene-specific we performed gene chip microarray analysis on ZF-TDG and control ZF -mutant inactive TDG samples from the cells expressing 4 constructs at once. While LPS exposure induced multiple expression changes in both groups, factorial ANOVA shows that only 42 genes (out of 14000 genes over 22690 probesets in the Affymetrix 430A 2.0 array) were differentially upregulated by LPS in ZF-TDG compared with ZF-Mut (), with Nos2 showing the most significant P value (P = 0.0002). Pathway analysis (Metacore) showed that 38 of these 42 genes were linked to Nos2 in a Dijkstra’s algorithm “shortest paths” network over ≤ 2 steps in the path, indicating that differential elevation of these genes may be secondary to upregulation of Nos2. While more than just Nos2 transcription was affected, we conclude that the results of transcriptional enhancement by ZF-TDG are highly gene-specific.
Figure 4. To determine gene-specificity of the effect microarray analysis was performed over LPS-stimulated and control RNA samples from cells carrying ZF-TDG and ZF-mutant control TDG. (A) RMA values were extracted using RMA Express; after quality control and outlier removal the resultant RMA matrix was analyzed in TIGR MeV software by factorial ANOVA to identify genes upregulated after LPS exposure only in the TDG, and not in the Mut groups with at least 1.1 fold. (B) Pathway analysis of the list in 4A using GeneGo MetaCore indicates that all except 4 of the genes are linked to Nos2 in a “shortest paths” network over < 2 intermediaries, indicating that differential elevation of these genes may be secondary to upregulation of Nos2.
To establish biological effect of the transcriptional enhancement of Nos2 we performed analysis of NO production based on the total nitrate levels in culture supernatants. demonstrates significant (P < 0.05) elevation of nitrate after 48 h of LPS incubation. Effectively we obtained an almost 2-fold enhancement of NO production in the ZF-TDG cells compared with controls.
Figure 5. In cells expressing all 4 constructs, NO production was measured in the supernatants (phenol-free DMEM complete) after 48 h of 1 ng/mL LPS stimulation using the colorimetric Griess method. (A) The values are total nitrate after nitrite-to-nitrate conversion. (B) Total nitrate normalized to cell counts per well. All experiments were performed at least 3 times in triplicates, *P < 0.05; **P < 0.01.
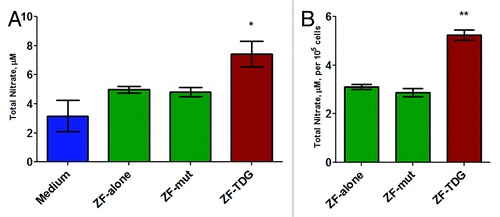
We conclude that direct tethering of multiple TDG molecules via custom designed ZFs to the Nos2 promoter led to decreased methylation and highly specific transcriptional enhancement of Nos2 allowing elevated NO production in fibroblasts.
Discussion
Two key attributes that distinguish epigenetic methods from other approaches to modify gene expression are heritability and regulation. An epigenetic mark may persist across cell division, even after the causative stimulus has ceased. It may not affect constitutive gene expression, but rather enhance (or decrease) a gene’s susceptibility to environmental or developmental stimulus. Thus, the ability to specifically target epigenetically silenced genes for reactivation would have powerful and diverse applications. Experimentally, it would enable study of the biological role of individual genes and their silencing in different developmental and pathological states while, in contrast to conventional overexpression studies, preserving the regulatory context and subtleties. The approach might also provide novel therapeutic avenues for a multitude of diseases where aberrant gene silencing contributes to the pathology.Citation6,Citation14-Citation16 In this work we are the first to address the cutting edge questions of epigenetic manipulation with our focus on demonstrating the gene specificity and magnitude of transcriptional enhancement that can be accomplished via targeted tethering of an epigenetically active enzyme.
We have targeted the Nos2 gene, important for signaling and host defense, and whose promoter is silenced by methylation in fibroblasts.Citation12,Citation13 We have previously shown that TDG can demethylate the silenced Nos2 promoter when artificially targeted, and that this demethylation is associated with increased transcriptional responsiveness to stimuli.Citation11 However, the use of endogenous DNA-binding domains, as in our earlier study, limits flexibility for targeting, increases the likelihood of off-target effects, and raises issues of competition between the demethylation construct and endogenous transcription factors. For the potential widespread utility of the approach the off-target effects must be minimal. In this report, we address these issues by using multiple artificial DNA-binding domains, targeted to neighboring sites in the Nos2 promoter. Artificial zinc finger domains have been used successfully by others to tether nucleasesCitation17 or methylasesCitation18 to DNA, thus enabling editing of gene structure or inhibition of gene function, respectively. Other artificial targeting approaches include transcription activator-like effectors (TALEs). Our reasons for choosing ZF in this case include: (1) TALEs have difficulty binding to highly methylated sitesCitation19; and (2) ZF proteins can naturally internalize into cells and nuclei, without the need for encoding in lentivirus, which increases the future potential of this approach.Citation17 In addition to the role of TDG, mechanistic studies suggest ten-eleven translocation proteins (Tet) are important in the demethylation process,Citation20 likely in collaboration with TDG. However, unlike TDG,Citation11 untethered Tet overexpression induces genome-wide demethylation,Citation7 making TDG more suitable for targeted editing.
We created four constructs, in which TDG was tethered to a different ZF array, targeting separate sites in the Nos2 promoter. Each individual ZF-TDG construct produced a modest reduction in methylation (). This may be expected: an artificial DNA binding domain designed in silico is unlikely to bind with the affinity of a natural transcription factor, refined over many generations of evolution. However, simultaneous targeting of four different ZF-TDG constructs had a cumulative effect, with prevalence of methylation being reduced by over a third at some sites (). This reduced methylation correlated with increased responsiveness to LPS and IFNγ (classical inducers of Nos2 in cell types where the gene is not silenced) at the level of mRNA (), and led to significant elevation of NO production (). We reasoned that the requirement for multiple TDG activities may increase specificity: while a single 9 bp binding motif (for 3 ZFs in each ZF array) might occur at random every 260 kB, the probability of multiple sites occurring in proximity will be many times lower. Even if clusters of off-target binding sites do occur, they may not be close enough to a silenced promoter to have an effect. This hypothesis is supported by genome-wide transcriptional profiling: targeting catalytically active TDG to the Nos2 promoter of 3T3 cells conferred inducibility to only forty-two (42) additional genes (0.3%), of which 38 were linked to the NOS2 activity, and so are likely downstream effects of Nos2 activation rather than true off-target effects. We chose to screen for secondary effects at the level of gene expression using microarray rather than global methylcytosine analysis because (1) off-target modulation of gene expression is the primary practical concern, rather than the underlying changes in methylation; and (2) the changes in methylation we found when a single construct bound the Nos2 promoter were too small to be reliably detected by existing global epigenomic techniques, indeed they required repeated pyrosequencing runs for reliable detection; hence it would be difficult to rely on epigenome-wide data
We point out that we have not determined the mechanism of the small number of off-target outcomes. We postulate that they reflect a combination of off-target effects of the construct and (largely) secondary effects of elevated NOS2 levels. Further studies are necessary to rigorously address these possibilities. Nevertheless, the results show a > 99% specificity in the observed transcriptional enhancement.
Notably, the background trace levels of Nos2 expression and NO production were not affected by ZF-TDG and control constructs. Only the stimulated transcription was enhanced, which is a desirable effect of epigenetic manipulation for future applications.
Our analysis of the target area of Nos2 was limited to its promoter and the nearest downstream CpG island; other CpG sites in this region were not analyzed. We note that some of the CpG’s we interrogated did not change their methylation status when the majority did. We also note that the CpG island is located approximately 1000 bp into the gene, and we detect similar mC% changes in the island as we do in the promoter. In contrast, however, we observe an apparent reduction in transcriptional efficacy as the distance between the TDG tethering site and TSS increases. Further study will be required to describe the range of effect of tethered TDG, how this relates to the looping and packaging of chromosome in the nucleus, and to what extent the spatial considerations are applicable between different loci.
In conclusion, in this report we show that tethering of TDG to a tetrad of artificial DNA binding domains allows highly specific restoration of the responsiveness of an epigenetically silenced gene to external stimuli.
Methods
Construct design and delivery
Zinc finger arrays were designed using Zifit (zifit.partners.org), a free utility that provides both context-dependent assembly (CoDA) and oligomerized pool engineering (OPEN) algorithms.Citation21 For added reliability we selected only the ZFs that were suggested by both of the algorithms. We designed 4 arrays, two of them for the coding strand, and two for the reverse strand of the Nos2 promoter, with the aim of delivering 4 tethered fusion molecules at once. Polynucleotides encoding each array were custom synthesized as “mini-genes” (IDT, Inc.). Murine TDG was tethered to each ZF via a poly-glycine linker, and transduced into NIH-3T3 cells as previously descibed.Citation11 Control cells were identically transduced, except with a mutant, inactive TDG (a single mutation in catalytic Asn151→Ala151 [N151A]),Citation22 or with ZF alone. The fusion constructs also included a selection marker (LNGFR) and an epitope flag (Myc, HA, or FLAG) to enable ChIP and protein studies. All eight ZF-TDG and mutant catalytically inactive ZF-Mut (TDG N151A) genes were expressed at similar levels (data not shown).
Packaging of lentivirus into 293T cells, transduction into NIH-3T3 cells (ATCC) and selection using paramagnetic anti-LNGFR antibody coated nanoparticles (Miltenyi) were performed as described previously.Citation11 Transduction efficiency was typically greater than 90%. We performed 4 consecutive transductions in order to deliver each of the 4 constructs.
We performed flow cytometry using FacsCanto II cytometer (BD Biosciences). We used anti-LNGFR antibody (Miltenyi) to confirm success of transduction and selection, and a cell viability kit (Invitrogen) with Cytox Green and Annexin V stains for necrotic and apoptotic cells. Presence of ZF-based constructs did not compromise cell viability (data not shown).
Cell culture
NIH-3T3 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with fetal bovine serum, glutamine, penicillin, streptomycin and HEPES (VWR) in tissue-culture treated 6-well plates (BD). For colorimetric NO assay the cells were cultured in the Phenol Red-free version of DMEM supplemented similarly. We performed nitric oxide assay in the supernatants after 48hrs of exposure to 1 ng/mL LPS using the colorimetric nitrate kit based on the modified Griess assay (Cayman, Inc.); the procedure was performed in full adherence to manufacturer's recommendations including the nitrite to nitrate conversion step.
Except where stated otherwise, cells were stimulated with 250 μg/ml of murine recombinant IFNγ (Peprotech) or with 0.5 ng/mL of LPS (Sigma).
Gene expression analysis
Cells were harvested by trypsinisation followed by immediate isolation of RNA and DNA using Qiagen RNEasy and DNEasy kits, in complete adherence to the manufacturer’s instructions. RNA was reverse transcribed into cDNA using iScript cDNA Synthesis Kit (Bio-Rad) and amplified in a CFX96 real-time PCR system (Bio-Rad) with respective primers (IDT, Inc.), see for sequences, using the SsoAdvanced SYBRGreen Master Mix (Bio-Rad) with a typical protocol including a hot start of 95 °C for 1 min, 40 cycles of amplification of 95 °C, 60 °C, and 72 °C, and a melt curve from 65 °C to 95 °C in 1 s. increments. Expression was normalized to β-actin, expression values were calculated as 2(-dCq) multiplied by an appropriate quotient to remove decimals and zeros. Fold upregulation was calculated by normalizing the stimulated expression to background (delta delta Cq). DNA (500 ng) was converted in a bisulfite reaction using Qiagen EpiTect kit following the manufacturer recommended procedure. PCR amplification with respective primers (IDT DNA, see for sequences) continued for 40–49 cycles, depending on the target. PCR product was verified by gel electrophoresis. Remaining product was loaded into a pyrosequencing reaction and analyzed using Pyromark Q96 MD pyrosequencer (Qiagen) in compliance with manufacturer recommended procedure.
Table 1. Primer sequences
Microarray expression assays were performed at the Dana-Farber Cancer Institute core facility using the Affymetrix 430A 2.0 murine arrays (n = 3 per group). Microarrays were validated by testing several random genes via real-time PCR. We used RMA Express and TIGR MeV software to identify the genes upregulated after LPS exposure only in the ZF-TDG, not in the ZF-mut group with at least 1.1-fold change. Specifically, RMA values were extracted without log transformation or filtering and assembled into a single matrix, which was loaded into MeV, annotated and analyzed by factorial ANOVA with the threshold P value of 0.05 (without Bonferroni correction). The resultant list was processed to exclude the genes downregulated by LPS exposure, poorly annotated targets and targets with < 1.1-fold upregulation to LPS and with < 1.1-fold difference between ZF-TDG and ZF-mut.
Pathway analysis was performed using Metacore online pathway portal (GeneGo) which allows building networks and maps of interacting factors based on extensive proprietary curated database. The list of 42 gene names was processed to evaluate any direct and indirect interactions allowing a maximum of 2 intermediary factors that are not on the list (Dijkstra algorithm); the analysis was performed with default settings.
Statistical analysis
Experiments were repeated 3 times. In each of the biological repeats, each RNA sample was analyzed at least in triplicates; each DNA and protein sample was analyzed at least in duplicates. Pyrosequencing assays were repeated 3 times in quadruplicates. Data other than microarrays were analyzed in Prism 5.0 (GraphPad Software, Inc.) using ANOVA with post-tests or Mann-Whitney test, as appropriate based on normality and variance requirements. Statistical significance was acceptable when P < 0.05. Data in the bar charts are expressed as MEAN ± SEM.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The study was supported in part by NIEHS grants ES015425 to Fedulov A and ES017588 to Kobzik L.
References
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008; 454:766 - 70; PMID: 18600261
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33:Suppl 245 - 54; http://dx.doi.org/10.1038/ng1089; PMID: 12610534
- Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy?. Genes Cancer 2011; 2:607 - 17; http://dx.doi.org/10.1177/1947601910393957; PMID: 21941617
- Flintoft L. DNA methylation: Looking beyond promoters. Nat Rev Genet 2010; 11:596; PMID: 20680023
- Majid S, Dar AA, Ahmad AE, Hirata H, Kawakami K, Shahryari V, Saini S, Tanaka Y, Dahiya AV, Khatri G, et al. BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 2009; 30:662 - 70; http://dx.doi.org/10.1093/carcin/bgp042; PMID: 19221000
- de Groote ML, Verschure PJ, Rots MG. Epigenetic Editing: targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic Acids Res 2012; 40:10596 - 613; http://dx.doi.org/10.1093/nar/gks863; PMID: 23002135
- Zhu B, Benjamin D, Zheng Y, Angliker H, Thiry S, Siegmann M, Jost JP. Overexpression of 5-methylcytosine DNA glycosylase in human embryonic kidney cells EcR293 demethylates the promoter of a hormone-regulated reporter gene. Proc Natl Acad Sci U S A 2001; 98:5031 - 6; http://dx.doi.org/10.1073/pnas.091097298; PMID: 11296268
- Zhu B, Zheng Y, Hess D, Angliker H, Schwarz S, Siegmann M, Thiry S, Jost JP. 5-methylcytosine-DNA glycosylase activity is present in a cloned G/T mismatch DNA glycosylase associated with the chicken embryo DNA demethylation complex. Proc Natl Acad Sci U S A 2000; 97:5135 - 9; http://dx.doi.org/10.1073/pnas.100107597; PMID: 10779566
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011; 146:67 - 79; http://dx.doi.org/10.1016/j.cell.2011.06.020; PMID: 21722948
- Zhu J-K. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet 2009; 43:143 - 66; http://dx.doi.org/10.1146/annurev-genet-102108-134205; PMID: 19659441
- Gregory DJ, Mikhaylova L, Fedulov AV. Selective DNA demethylation by fusion of TDG with a sequence-specific DNA-binding domain. Epigenetics 2012; 7:344 - 9; http://dx.doi.org/10.4161/epi.19509; PMID: 22419066
- Chan GC, Fish JE, Mawji IA, Leung DD, Rachlis AC, Marsden PA. Epigenetic basis for the transcriptional hyporesponsiveness of the human inducible nitric oxide synthase gene in vascular endothelial cells. J Immunol 2005; 175:3846 - 61; PMID: 16148131
- Bogdan C. Nitric oxide and the immune response. Nat Immunol 2001; 2:907 - 16; http://dx.doi.org/10.1038/ni1001-907; PMID: 11577346
- Tsai H-C, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res 2011; 21:502 - 17; http://dx.doi.org/10.1038/cr.2011.24; PMID: 21321605
- Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet 2013; 132:359 - 83; http://dx.doi.org/10.1007/s00439-013-1271-x; PMID: 23370504
- Bierne H, Hamon M, Cossart P. Epigenetics and bacterial infections. Cold Spring Harb Perspect Med 2012; 2:a010272; http://dx.doi.org/10.1101/cshperspect.a010272; PMID: 23209181
- Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF 3rd. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods 2012; 9:805 - 7; http://dx.doi.org/10.1038/nmeth.2030; PMID: 22751204
- Rivenbark AG, Stolzenburg S, Beltran AS, Yuan X, Rots MG, Strahl BD, Blancafort P. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 2012; 7:350 - 60; http://dx.doi.org/10.4161/epi.19507; PMID: 22419067
- Bultmann S, Morbitzer R, Schmidt CS, Thanisch K, Spada F, Elsaesser J, Lahaye T, Leonhardt H. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Res 2012; 40:5368 - 77; http://dx.doi.org/10.1093/nar/gks199; PMID: 22387464
- He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011; 333:1303 - 7; http://dx.doi.org/10.1126/science.1210944; PMID: 21817016
- Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, Curtin SJ, Blackburn JS, Thibodeau-Beganny S, Qi Y, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods 2011; 8:67 - 9; http://dx.doi.org/10.1038/nmeth.1542; PMID: 21151135
- Hardeland U, Bentele M, Jiricny J, Schär P. Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J Biol Chem 2000; 275:33449 - 56; http://dx.doi.org/10.1074/jbc.M005095200; PMID: 10938281