Abstract
Neural development is a delicate process that can be disrupted by pollution that exerts detrimental impact on neural signaling. This commentary highlights recent discoveries in the arena of research at the interface of environmental toxicology and developmental neuroscience relating to toxicity mechanisms of bisphenol A (BPA), a ubiquitous chemical used in manufacturing of plastics and epoxy resins that is known to bind to and interfere with estrogen receptors, estrogen-receptor-related receptors and other receptors for gonadal steroids. It was recently observed that BPA disrupts the perinatal chloride shift, a key neurodevelopmental mechanism that brings down neuronal chloride from ~100 mM to ~20 mM within weeks. The chloride shift happens in all central nervous systems of vertebrates around parturition. High neuronal chloride supports neuron precursors’ migrations, low neuronal chloride is the prerequisite for inhibitory action of neurotransmitters GABA and glycine, and thus an absolute requisite for normal functioning of the mature CNS. One critical contributor to the neuronal chloride shift is the concomitant upregulation of expression of the chloride-extruding transporter molecule, KCC2. We highlight recent findings including our discovery that BPA disrupts the chloride shift in a sex-specific manner by recruiting epigenetics mechanisms. These could be relevant for childhood neuropsychiatric disorders as well as for liability to develop chronic neuropsychiatric diseases later in life.
Development of the central nervous system (CNS), in particular the brain, is driven by highly coordinated cascades of sequential and concurrent events that begin very early in human gestation and continue through adolescence. During early development, brain cells proliferate, migrate to the appropriate location, differentiate into the correct cell type, and establish synapses with nearby and distant cells, ultimately forming complex neuronal circuits.Citation1-Citation5 Disruption of this sequence of events at any point during brain development can have lasting consequences for brain function, which ultimately manifests as altered physiology, behavior and liability to respond to stress in a maladaptive manner and develop disease. Clear linkages between developmental disruption by environmental factors, including exposures to chemical pollution, and long-term neural and psychological outcomes, however, remain poorly characterized and thus a critical data gap toward improved understanding of neuropsychiatric disease.
Kcc2/KCC2 is a critical neuronal-specific gene of the CNS that codes for a chloride-extruding transporter molecule.Citation6-Citation10 Developmental upregulation of Kcc2 expression is conserved in vertebrates, essential for the chloride shift in CNS neurons, proper migration of precursor neurons to final locations, and synchronization of maturation at the cellular and neural network level.Citation11-Citation16 This is especially apparent for the delicate architecture of the mammalian cerebral cortex, where inhibitory precursor neurons originating from the median preganglionic eminence migrate considerable distances to their final destination in specific cortical layers.Citation11 This migration is critically dependent on the chloride shift and its appropriate developmental timing. Deceleration/attenuation of the ontogenetic upregulation of Kcc2 expression during the sensitive perinatal period can alter cortical organization and thus be regarded as an exemplar of developmental neurotoxicity.Citation11 In an earlier publication, we reported that a histone-deacetylase inhibitor, trichostatin-A (TSA) can accelerate the ontogenetic chloride shift by upregulating Kcc2 expression.Citation15 This demonstrated for the first time the relevance of epigenetic regulation for Kcc2 gene expression.
Bisphenol A (BPA) is an endocrine disrupting compound that has been primarily characterized regarding its interactions with estrogen signaling on a diversity of cell types, but is now recognized to also have other possible mechanisms of action.Citation17 BPA is a high production–volume plastic monomer produced at a rate of 3 million metric tons globally per year in the manufacturing of polycarbonate plastics, epoxy resins and plastic surface coatings, and is thus found in a wide variety of consumer items.Citation18 It is estimated that more than 90% of the US population have detectable levels of BPA in their urine.Citation19-Citation21 Concern has been expressed over the potential toxic and quasi-hormonal properties of BPA.
We recently demonstrated that BPA enhanced repression of Kcc2 transcriptionCitation22 (). However, its effects did not result in an elimination of Kcc2 expression, which would be lethal, nor did it eliminate the upregulation of Kcc2. It did, however, decelerate and delay it, established in cultured neurons, organotypic brain slice culture and in vivo after gestational exposure. Therefore, BPA exposure could disrupt the layered architecture of the cerebral cortex because of over-migration of inhibitory precursor neurons. It is further anticipated that at this particularly sensitive time-point of neurodevelopment, BPA will disrupt dendritic spine formation and thereafter synaptic maturation, effectively compromising the functional set-up of inhibitory cortical neural circuits as well. This reasoning is consistent with studies showing disruption of cortical layer architecture in brains of mouse pups exposed to BPA through gestational feeding to their damsCitation23,Citation24 These neuroanatomical abnormalities in turn dovetail extremely well with behavioral deficits that have been observed in mice and rats gestationally exposed to BPA.Citation17,Citation25 Thus, we are describing a novel mechanism of neurodevelopmental toxicity by which altered neuronal chloride homeostasis and KCC2 expression subsequently compromise cortical neuronal and network structure. Because the chloride shift is important for development of the CNS elsewhere, by extension, other central neural structures may also be similarly impacted.
Figure 1. Exposure to BPA may disrupt development of the central nervous system by slowing down the removal of chloride from neurons. As an organism matures and the brain develops, chloride levels inside neurons decrease. However, when exposed to BPA, the chloride is removed more slowly from neurons. Female neurons appear to be more susceptible to the effects of BPA. In regards to the abscissa (left-hand), note a relevant ontological difference between rodents and humans: the chloride shift that coincides with the upregulation of KCC2/Kcc2 gene expression occurs between developmental stages E17 and P20 in rat neurons.Citation11,Citation62 In humans, KCC2 levels start increasing by the second trimester and reaches a peak at birth.Citation15,Citation22,Citation63
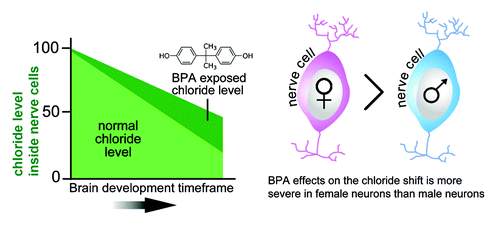
Links between perinatal BPA exposure and behavioral pathology are increasingly evident in laboratory animals and humans, and BPA effects frequently differ in males and females.Citation17 For instance, a number of studies reported anxiogenic effects of prenatal exposure to BPA in rodent species.Citation26-Citation28 For many behavioral outcomes, a consistent finding is that perinatal exposure to BPA disrupts the development of normal sexually-dimorphic behaviors, including anxiety, exploration, social interaction, aggression, reward sensitivity and spatial memory.Citation28-Citation32 In humans, sexually dimorphic disorders, such as attention-deficit/hyperactivity disorder and depression may be clinically correlated with such animal behaviors and may be related to disruption of the neuroendocrine system early in life.Citation33-Citation35 Interestingly, a recent epidemiological study reported that gestational BPA exposure is associated with increased hyperactivity and aggression scores in young girls, but not in boysCitation36,Citation37 (see also refs. Citation34 and Citation38 for recent data on human gestational exposure).
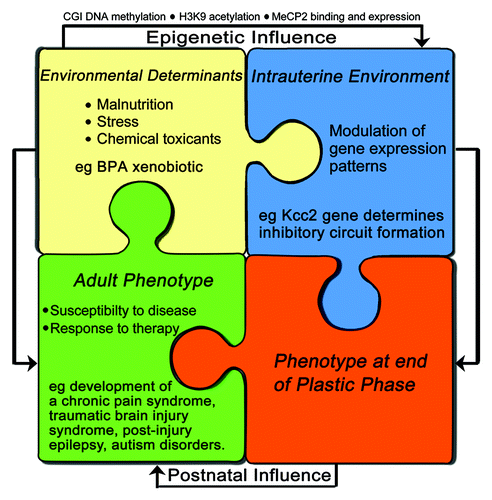
Understanding the mechanisms by which BPA alters CNS development via epigenetic effects on Kcc2 broadens our insight into how the environment can influence gene expression patterns and subsequent phenotype. In view of the possibly long-lasting duration of epigenetic changes it is reasonable to assume that such changes could be relevant for disease susceptibility later in life (). This reasoning is especially applicable to stresses and injuries that can potentially downregulate Kcc2 expression, such as traumatic brain injury, epilepsy or chronic pain states.Citation9,Citation39-Citation43 Subsequent upregulation would re-normalize chloride homeostasis in the CNS and lead to clinical improvement. However, re-normalization of KCC2/Kcc2 expression after stress- or injury-mediated downregulation of KCC2/Kcc2 could be delayed by perinatal epigenetic changes of the KCC2/Kcc2 promoter, thus contributing to disease chronicity for diseases in which KCC2/Kcc2 downregulation has been demonstrated to be a part of the molecular pathology in the CNS.Citation39,Citation44
Figure 2. A model for neurodevelopmental disorders and susceptibility to suffer from maladaptive neural plasticity later in life: perinatal epigenetic changes of key genes for nervous system functioning, such as Kcc2, plasticity and repair mechanisms counteract. The degree and effectiveness of these regulatory (i.e., damage-limiting) mechanisms is further affected by how much they are affected by BPA exposure. In case these mechanisms are severely impaired themselves, then epigenetic dysregulation is more likely to lead to manifest neurodevelopmental disorders. In case counter-regulatory repair mechanisms are rather effective, it can lead to complete restitution, or it can lead to normal development, but a predisposition to show maladaptive changes in response to stress/ injury/ hormonal changes later in life. For example, development of a chronic pain syndrome, a post-traumatic brain injury syndrome, post-injury epilepsy and autism disorders.
Against this background, our discovery of the epigenetic effects of BPA on chloride regulation in neurons during the perinatal periodCitation22 raises several points of discussion that we are addressing here.
1) The methylating effects of BPA in in vivo and in vitro experiments appear to be different. Perinatal exposure to BPA may activate a specific series of biochemical pathways involving DNA synthesis and chromatin organization within maternal and fetal tissues that ultimately lead to activation of DNA-methyl-transferases and other components of the epigenetic machinery that target the Kcc2 gene promoter in cortical neurons and very likely other CNS neurons. In contrast, these biochemical pathways may not be similarly affected in a homogenous culture of cortical neurons. The critical difference could be rooted in non-neuronal cells in the developing brain, such as developing astrocytes. For example, developing glial cells, in response to BPA exposure, could facilitate neuronal epigenetic changes.Citation45 Additionally, differences could result from the function of the placenta as it is exposed to BPA. The developing brain could be affected by maternal humoral factors via the placenta so that, ultimately, epigenetic changes in developing neurons result.
2) Pertaining to relevance of our findings for human exposures, the mice in our experiments were fed chow containing 50 mg BPA/kg pellet; therefore, in our experiments, mouse exposure to BPA was effectively 10 mg/kg body weight/d through diet (≈250 µg). The amount fed to the mice, though on the high side, may still be physiologically relevant. A recent study measured epigenetic responses following gestational exposure to a range of BPA levels (ng-µg-mg). They showed overall global methylation across all BPA exposure groups in comparison to controls; however, there were no statistically significant differences in global methylation among the three BPA exposed groups.Citation46 Another group exposed rats to BPA in drinking water, leading to exposures of 35–55 µg per animal per day; by this method serum BPA levels were assessed to be equivalent to those reported in humans. They reported that BPA exposure decreased overall expression of genes known to affect socio-sexual behavior by about 1.5- to 2-fold. Interestingly, the sex differences of two sexually dimorphic genes (Kiss-1 and Bdnf) were eliminated with BPA exposure.Citation27 However, it remains to be seen if disappearance of sexual dimorphism of gene expression is mediated by epigenetic mechanisms. Yet, another group recently reported epigenetic effects in the form of DNA methylation and altered gene regulation in the brain in response to very low-dose gestational BPA exposures, applying 2, 2,0 and 200 µg BPA/kg/d.Citation47 Genes of the epigenetic machinery were affected as well as sex-hormone nuclear receptors, some of them in a sex-specific manner, also in the cerebral cortex and the hypothalamus. Offspring’s behavior was significantly altered at all three doses. This study documents epigenetic effects on gene regulation in the brain at the low end of the dose, thus providing counter-argument to previously published positions.Citation48
3) The sexual dimorphism effects of BPA that have been observed in our experiments point toward up-stream signaling mechanisms mediated by sex-hormone receptors. BPA is a documented estrogen disruptor.Citation49-Citation52 Sex-specific estrogen receptor expression is present in perinatal brains of rodents and primates (including humans). For example, in the perinatal rat brain, estrogen is masculinizing. Perinatal exposure to BPA has also been shown to affect the organization of numerous estrogen-sensitive structures including the neocortex, amygdala, preoptic area, and hypothalamic regions important for sex-specific reproductive physiology and behavior.Citation24,Citation30,Citation47,Citation49,Citation53-Citation55 Viewing these results in concert, there is cause for concern that BPA exposure in humans may potentially result in adverse neuropsychiatric and other health outcomes.Citation56-Citation58 For example, BPA exposure can also increase expression of leptin-receptors, thus predisposing affected subjects to future metabolic dysregulation, possibly obesity, if this dysregulation were affecting the brain.Citation59,Citation60
Finally, our own results show that BPA led to increased CpG methylation and MECP2 occupancy in the upstream repressor-element 1 (RE1) site, a region ~1.8 kb upstream of the transcription start site (TSS) of the KCC2/Kcc2 gene, also known to harbor a CpG shore. Future experiments will be performed to ascertain if epigenetic alteration of the KCC2/Kcc2 shore region is causal for increased repression of transcription. For instance, using cultured cortical neurons, we can make use of the CRISPR-Cas systemCitation61 to engineer a deletion of the 500 bp promoter shore region containing the upstream RE1 site, then assess Kcc2 mRNA abundance, in the presence and absence of BPA.
In conclusion, our data revealing that BPA can delay the perinatal chloride shift in cortical neurons via epigenetic mechanisms unveils a novel mechanism by which environmental factors can shape neural development. Perturbation of the chloride shift likely affects neuronal migration and synaptic maturation in the developing brain’s cortex. Therefore, we have identified a novel example of neurodevelopmental toxicity, mediated by BPA. Our study evokes pertinent follow-up questions related to molecular and cellular mechanisms of DNA-methylation and other epigenetic mechanisms that affect Kcc2 expression. In addition, critical involvement of sex-hormone receptors upstream of epigenetic effects will be assessed.
Acknowledgments
Supported by funding from Duke University, the Klingenstein Fund, New York, NY, and the NIH (R21NS066307) to Liedtke W.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Hanganu-Opatz IL. Between molecules and experience: role of early patterns of coordinated activity for the development of cortical maps and sensory abilities. Brain Res Rev 2010; 64:160 - 76; http://dx.doi.org/10.1016/j.brainresrev.2010.03.005; PMID: 20381527
- Uhlhaas PJ, Roux F, Rodriguez E, Rotarska-Jagiela A, Singer W. Neural synchrony and the development of cortical networks. Trends Cogn Sci 2010; 14:72 - 80; http://dx.doi.org/10.1016/j.tics.2009.12.002; PMID: 20080054
- Heng JI, Chariot A, Nguyen L. Molecular layers underlying cytoskeletal remodelling during cortical development. Trends Neurosci 2010; 33:38 - 47; http://dx.doi.org/10.1016/j.tins.2009.09.003; PMID: 19837469
- Whitford KL, Dijkhuizen P, Polleux F, Ghosh A. Molecular control of cortical dendrite development. Annu Rev Neurosci 2002; 25:127 - 49; http://dx.doi.org/10.1146/annurev.neuro.25.112701.142932; PMID: 12052906
- Caviness VS Jr., Rakic P. Mechanisms of cortical development: a view from mutations in mice. Annu Rev Neurosci 1978; 1:297 - 326; http://dx.doi.org/10.1146/annurev.ne.01.030178.001501; PMID: 386903
- Song L, Mercado A, Vázquez N, Xie Q, Desai R, George AL Jr., Gamba G, Mount DB. Molecular, functional, and genomic characterization of human KCC2, the neuronal K-Cl cotransporter. Brain Res Mol Brain Res 2002; 103:91 - 105; http://dx.doi.org/10.1016/S0169-328X(02)00190-0; PMID: 12106695
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol 2004; 468:57 - 64; http://dx.doi.org/10.1002/cne.10983; PMID: 14648690
- Andäng M, Lendahl U. Ion fluxes and neurotransmitters signaling in neural development. Curr Opin Neurobiol 2008; 18:232 - 6; http://dx.doi.org/10.1016/j.conb.2008.06.001; PMID: 18638551
- Price TJ, Cervero F, Gold MS, Hammond DL, Prescott SA. Chloride regulation in the pain pathway. Brain Res Rev 2009; 60:149 - 70; http://dx.doi.org/10.1016/j.brainresrev.2008.12.015; PMID: 19167425
- Rivera C, Voipio J, Kaila K. Two developmental switches in GABAergic signalling: the K+-Cl- cotransporter KCC2 and carbonic anhydrase CAVII. J Physiol 2005; 562:27 - 36; http://dx.doi.org/10.1113/jphysiol.2004.077495; PMID: 15528236
- Bortone D, Polleux F. KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 2009; 62:53 - 71; http://dx.doi.org/10.1016/j.neuron.2009.01.034; PMID: 19376067
- Fiumelli H, Woodin MA. Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr Opin Neurobiol 2007; 17:81 - 6; http://dx.doi.org/10.1016/j.conb.2007.01.002; PMID: 17234400
- Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 2001; 105:521 - 32; http://dx.doi.org/10.1016/S0092-8674(01)00341-5; PMID: 11371348
- Uvarov P, Ludwig A, Markkanen M, Rivera C, Airaksinen MS. Upregulation of the neuron-specific K+/Cl- cotransporter expression by transcription factor early growth response 4. J Neurosci 2006; 26:13463 - 73; http://dx.doi.org/10.1523/JNEUROSCI.4731-06.2006; PMID: 17192429
- Yeo M, Berglund K, Augustine G, Liedtke W. Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J Neurosci 2009; 29:14652 - 62; http://dx.doi.org/10.1523/JNEUROSCI.2934-09.2009; PMID: 19923298
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999; 397:251 - 5; http://dx.doi.org/10.1038/16697; PMID: 9930699
- Wolstenholme JT, Rissman EF, Connelly JJ. The role of Bisphenol A in shaping the brain, epigenome and behavior. Horm Behav 2011; 59:296 - 305; http://dx.doi.org/10.1016/j.yhbeh.2010.10.001; PMID: 21029734
- Geens T, Aerts D, Berthot C, Bourguignon JP, Goeyens L, Lecomte P, Maghuin-Rogister G, Pironnet AM, Pussemier L, Scippo ML, et al. A review of dietary and non-dietary exposure to bisphenol-A. Food Chem Toxicol 2012; 50:3725 - 40; http://dx.doi.org/10.1016/j.fct.2012.07.059; PMID: 22889897
- Bushnik T, Haines D, Levallois P, Levesque J, Van Oostdam J, Viau C. Lead and bisphenol A concentrations in the Canadian population. Health Rep 2010; 21:7 - 18; PMID: 20973429
- Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003-2004. Environ Health Perspect 2008; 116:39 - 44; http://dx.doi.org/10.1289/ehp.10753; PMID: 18197297
- Galloway T, Cipelli R, Guralnik J, Ferrucci L, Bandinelli S, Corsi AM, Money C, McCormack P, Melzer D. Daily bisphenol A excretion and associations with sex hormone concentrations: results from the InCHIANTI adult population study. Environ Health Perspect 2010; 118:1603 - 8; http://dx.doi.org/10.1289/ehp.1002367; PMID: 20797929
- Yeo M, Berglund K, Hanna M, Guo JU, Kittur J, Torres MD, Abramowitz J, Busciglio J, Gao Y, Birnbaumer L, et al. Bisphenol A delays the perinatal chloride shift in cortical neurons by epigenetic effects on the Kcc2 promoter. Proc Natl Acad Sci U S A 2013; 110:4315 - 20; http://dx.doi.org/10.1073/pnas.1300959110; PMID: 23440186
- Nakamura K, Itoh K, Sugimoto T, Fushiki S. Prenatal exposure to bisphenol A affects adult murine neocortical structure. Neurosci Lett 2007; 420:100 - 5; http://dx.doi.org/10.1016/j.neulet.2007.02.093; PMID: 17532137
- Komada M, Asai Y, Morii M, Matsuki M, Sato M, Nagao T. Maternal bisphenol A oral dosing relates to the acceleration of neurogenesis in the developing neocortex of mouse fetuses. Toxicology 2012; 295:31 - 8; http://dx.doi.org/10.1016/j.tox.2012.02.013; PMID: 22426297
- Masuo Y, Ishido M. Neurotoxicity of endocrine disruptors: possible involvement in brain development and neurodegeneration. J Toxicol Environ Health B Crit Rev 2011; 14:346 - 69; http://dx.doi.org/10.1080/10937404.2011.578557; PMID: 21790316
- Cox KH, Gatewood JD, Howeth C, Rissman EF. Gestational exposure to bisphenol A and cross-fostering affect behaviors in juvenile mice. Horm Behav 2010; 58:754 - 61; http://dx.doi.org/10.1016/j.yhbeh.2010.07.008; PMID: 20691692
- Patisaul HB, Sullivan AW, Radford ME, Walker DM, Adewale HB, Winnik B, Coughlin JL, Buckley B, Gore AC. Anxiogenic effects of developmental bisphenol A exposure are associated with gene expression changes in the juvenile rat amygdala and mitigated by soy. PLoS One 2012; 7:e43890; http://dx.doi.org/10.1371/journal.pone.0043890; PMID: 22957036
- Gioiosa L, Fissore E, Ghirardelli G, Parmigiani S, Palanza P. Developmental exposure to low-dose estrogenic endocrine disruptors alters sex differences in exploration and emotional responses in mice. Horm Behav 2007; 52:307 - 16; http://dx.doi.org/10.1016/j.yhbeh.2007.05.006; PMID: 17568585
- Palanza P, Gioiosa L, vom Saal FS, Parmigiani S. Effects of developmental exposure to bisphenol A on brain and behavior in mice. Environ Res 2008; 108:150 - 7; http://dx.doi.org/10.1016/j.envres.2008.07.023; PMID: 18949834
- Wolstenholme JT, Taylor JA, Shetty SR, Edwards M, Connelly JJ, Rissman EF. Gestational exposure to low dose bisphenol A alters social behavior in juvenile mice. PLoS One 2011; 6:e25448; http://dx.doi.org/10.1371/journal.pone.0025448; PMID: 21980460
- Jašarević E, Williams SA, Vandas GM, Ellersieck MR, Liao C, Kannan K, Roberts RM, Geary DC, Rosenfeld CS. Sex and dose-dependent effects of developmental exposure to bisphenol A on anxiety and spatial learning in deer mice (Peromyscus maniculatus bairdii) offspring. Horm Behav 2013; 63:180 - 9; http://dx.doi.org/10.1016/j.yhbeh.2012.09.009; PMID: 23051835
- Jašarević E, Sieli PT, Twellman EE, Welsh TH Jr., Schachtman TR, Roberts RM, Geary DC, Rosenfeld CS. Disruption of adult expression of sexually selected traits by developmental exposure to bisphenol A. Proc Natl Acad Sci U S A 2011; 108:11715 - 20; http://dx.doi.org/10.1073/pnas.1107958108; PMID: 21709224
- Harley KG, Gunier RB, Kogut K, Johnson C, Bradman A, Calafat AM, Eskenazi B. Prenatal and early childhood bisphenol A concentrations and behavior in school-aged children. Environ Res 2013; Forthcoming http://dx.doi.org/10.1016/j.envres.2013.06.004; PMID: 23870093
- Braun JM, Hauser R. Bisphenol A and children’s health. Curr Opin Pediatr 2011; 23:233 - 9; http://dx.doi.org/10.1097/MOP.0b013e3283445675; PMID: 21293273
- Martel MM, Klump K, Nigg JT, Breedlove SM, Sisk CL. Potential hormonal mechanisms of attention-deficit/hyperactivity disorder and major depressive disorder: a new perspective. Horm Behav 2009; 55:465 - 79; http://dx.doi.org/10.1016/j.yhbeh.2009.02.004; PMID: 19265696
- Braun JM, Yolton K, Dietrich KN, Hornung R, Ye X, Calafat AM, Lanphear BP. Prenatal bisphenol A exposure and early childhood behavior. Environ Health Perspect 2009; 117:1945 - 52; PMID: 20049216
- Braun JM, Kalkbrenner AE, Calafat AM, Yolton K, Ye X, Dietrich KN, Lanphear BP. Impact of early-life bisphenol A exposure on behavior and executive function in children. Pediatrics 2011; 128:873 - 82; http://dx.doi.org/10.1542/peds.2011-1335; PMID: 22025598
- Gerona RR, Woodruff TJ, Dickenson CA, Pan J, Schwartz JM, Sen S, Friesen M, Fujimoto VY, Hunt PA. BPA, BPA glucuronide, and BPA sulfate in mid-gestation umbilical cord serum in a northern California cohort. Environ Sci Technol 2013; Forthcoming http://dx.doi.org/10.1021/es402764d; PMID: 23941471
- Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol 2008; 4:490 - 503; http://dx.doi.org/10.1038/ncpneuro0883; PMID: 18769373
- Lu Y, Zheng J, Xiong L, Zimmermann M, Yang J. Spinal cord injury-induced attenuation of GABAergic inhibition in spinal dorsal horn circuits is associated with downregulation of the chloride transporter KCC2 in rat. J Physiol 2008; 586:5701 - 15; http://dx.doi.org/10.1113/jphysiol.2008.152348
- Palma E, Amici M, Sobrero F, Spinelli G, Di Angelantonio S, Ragozzino D, Mascia A, Scoppetta C, Esposito V, Miledi R, et al. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci U S A 2006; 103:8465 - 8; http://dx.doi.org/10.1073/pnas.0602979103; PMID: 16709666
- Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus 2002; 12:258 - 68; http://dx.doi.org/10.1002/hipo.10014; PMID: 12000122
- Zhang H, Mei X, Wang W, Sun X. Blocking conversion of GABAergic inhibition as a potential mechanism of propofol-mediated brain protection following resuscitation. Drug News Perspect 2009; 22:525 - 9; http://dx.doi.org/10.1358/dnp.2009.22.9.1437960; PMID: 20072729
- Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 2012; 18:467 - 86; http://dx.doi.org/10.1177/1073858412438697; PMID: 22547529
- Miyatake M, Miyagawa K, Mizuo K, Narita M, Suzuki T. Dynamic changes in dopaminergic neurotransmission induced by a low concentration of bisphenol-A in neurones and astrocytes. J Neuroendocrinol 2006; 18:434 - 44; http://dx.doi.org/10.1111/j.1365-2826.2006.01434.x; PMID: 16684133
- Anderson OS, Nahar MS, Faulk C, Jones TR, Liao C, Kannan K, Weinhouse C, Rozek LS, Dolinoy DC. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ Mol Mutagen 2012; 53:334 - 42; http://dx.doi.org/10.1002/em.21692; PMID: 22467340
- Kundakovic M, Gudsnuk K, Franks B, Madrid J, Miller RL, Perera FP, Champagne FA. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc Natl Acad Sci U S A 2013; 110:9956 - 61; http://dx.doi.org/10.1073/pnas.1214056110; PMID: 23716699
- Teeguarden JG, Hanson-Drury S. A systematic review of Bisphenol A “Low Dose” studies in the context of human exposure: A case for establishing standards for reporting “low-dose” effects of chemicals. Food Chem Toxicol 2013; Forthcoming PMID: 23867546
- Kundakovic M, Champagne FA. Epigenetic perspective on the developmental effects of bisphenol A. Brain Behav Immun 2011; 25:1084 - 93; http://dx.doi.org/10.1016/j.bbi.2011.02.005; PMID: 21333735
- Takayanagi S, Tokunaga T, Liu X, Okada H, Matsushima A, Shimohigashi Y. Endocrine disruptor bisphenol A strongly binds to human estrogen-related receptor gamma (ERRgamma) with high constitutive activity. Toxicol Lett 2006; 167:95 - 105; http://dx.doi.org/10.1016/j.toxlet.2006.08.012; PMID: 17049190
- Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol 2006; 102:175 - 9; http://dx.doi.org/10.1016/j.jsbmb.2006.09.017; PMID: 17088055
- Vom Saal FS, Nagel SC, Coe BL, Angle BM, Taylor JA. The estrogenic endocrine disrupting chemical bisphenol A (BPA) and obesity. Mol Cell Endocrinol 2012; 354:74 - 84; http://dx.doi.org/10.1016/j.mce.2012.01.001; PMID: 22249005
- Cao J, Mickens JA, McCaffrey KA, Leyrer SM, Patisaul HB. Neonatal Bisphenol A exposure alters sexually dimorphic gene expression in the postnatal rat hypothalamus. Neurotoxicology 2012; 33:23 - 36; http://dx.doi.org/10.1016/j.neuro.2011.11.002; PMID: 22101008
- Cao J, Rebuli ME, Rogers J, Todd KL, Leyrer SM, Ferguson SA, Patisaul HB. Prenatal bisphenol a exposure alters sex-specific estrogen receptor expression in the neonatal rat hypothalamus and amygdala. Toxicol Sci 2013; 133:157 - 73; http://dx.doi.org/10.1093/toxsci/kft035; PMID: 23457122
- He Z, Paule MG, Ferguson SA. Low oral doses of bisphenol A increase volume of the sexually dimorphic nucleus of the preoptic area in male, but not female, rats at postnatal day 21. Neurotoxicol Teratol 2012; 34:331 - 7; http://dx.doi.org/10.1016/j.ntt.2012.03.004; PMID: 22507915
- Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocr Rev 2009; 30:75 - 95; http://dx.doi.org/10.1210/er.2008-0021; PMID: 19074586
- Hengstler JG, Foth H, Gebel T, Kramer PJ, Lilienblum W, Schweinfurth H, Völkel W, Wollin KM, Gundert-Remy U. Critical evaluation of key evidence on the human health hazards of exposure to bisphenol A. Crit Rev Toxicol 2011; 41:263 - 91; http://dx.doi.org/10.3109/10408444.2011.558487; PMID: 21438738
- Machtinger R, Combelles CM, Missmer SA, Correia KF, Williams P, Hauser R, Racowsky C. Bisphenol-A and human oocyte maturation in vitro. Hum Reprod 2013; 28:2735 - 45; http://dx.doi.org/10.1093/humrep/det312; PMID: 23904465
- Mackay H, Patterson ZR, Khazall R, Patel S, Tsirlin D, Abizaid A. Organizational effects of perinatal exposure to bisphenol-A and diethylstilbestrol on arcuate nucleus circuitry controlling food intake and energy expenditure in male and female CD-1 mice. Endocrinology 2013; 154:1465 - 75; http://dx.doi.org/10.1210/en.2012-2044; PMID: 23493373
- Ptak A, Gregoraszczuk EL. Bisphenol A induces leptin receptor expression, creating more binding sites for leptin, and activates the JAK/Stat, MAPK/ERK and PI3K/Akt signalling pathways in human ovarian cancer cell. Toxicol Lett 2012; 210:332 - 7; http://dx.doi.org/10.1016/j.toxlet.2012.02.003; PMID: 22343039
- Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol 2013; 31:681 - 3; http://dx.doi.org/10.1038/nbt.2661; PMID: 23929336
- Li H, Tornberg J, Kaila K, Airaksinen MS, Rivera C. Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur J Neurosci 2002; 16:2358 - 70; http://dx.doi.org/10.1046/j.1460-9568.2002.02419.x; PMID: 12492431
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci 2007; 27:9866 - 73; http://dx.doi.org/10.1523/JNEUROSCI.2761-07.2007; PMID: 17855601