Abstract
Genetic and epigenetic alterations are essential for the initiation and progression of human cancer. We previously reported that primary human medulloblastomas showed extensive cancer-specific CpG island DNA hypermethylation in critical developmental pathways. To determine whether genetically engineered mouse models (GEMMs) of medulloblastoma have comparable epigenetic changes, we assessed genome-wide DNA methylation in three mouse models of medulloblastoma. In contrast to human samples, very few loci with cancer-specific DNA hypermethylation were detected, and in almost all cases the degree of methylation was relatively modest compared with the dense hypermethylation in the human cancers. To determine if this finding was common to other GEMMs, we examined a Burkitt lymphoma and breast cancer model and did not detect promoter CpG island DNA hypermethylation, suggesting that human cancers and at least some GEMMs are fundamentally different with respect to this epigenetic modification. These findings provide an opportunity to both better understand the mechanism of aberrant DNA methylation in human cancer and construct better GEMMs to serve as preclinical platforms for therapy development.
Introduction
Cancer initiation and progression is a multistep process that involves a complex interplay between genetic and epigenetic alterations in a cell.Citation1 Recent whole genome and exome sequencing studies have identified many genetic alterations in cancer cells, a small subset of which that is frequently mutated across many different types of cancer (such as KRAS and TP53 - termed “mountains”), and a larger number of genetic perturbations that occur at low frequency (termed “hills”).Citation2 Similarly, genome-wide studies of human tumors have demonstrated very large numbers of epigenetic changes, such as aberrant DNA methylation, that contribute to the cancer phenotype. Examples of epigenetic “mountains” that are frequently altered in many cancers include CDKN2A and VHL inactivation by promoter CpG island DNA hypermethylation.Citation3 Genes classified as “hills,” regardless of whether their function is altered by genetic or epigenetic means, most likely define pathways that require multiple perturbations to drive cancer development.Citation4
Genetically engineered mouse models (GEMMs) of cancer have been extensively used to study human cancer biology. Using sophisticated targeting and expression techniques, mouse models have been used as platforms to study drug resistance, early detection, metastasis, cancer prevention and for the preclinical development of novel targeted therapeutics.Citation5 While most cancer GEMMs have either overexpressed oncogenes or targeted deletions of tumor suppressor genes, very few of these models have examined how epigenetic changes contribute to the cancer phenotype.
We previously found common regions of cancer-specific DNA methylation changes in critical developmental regulatory pathways in primary medulloblastoma patient samples.Citation6 To determine whether GEMMs of medulloblastoma share a similar pattern of epigenetic dysregulation, we examined genome-wide DNA methylation in three different GEMMs of medulloblastoma.
Results
Mouse models of medulloblastoma lack extensive promoter CpG island DNA hypermethylation
We initially chose a Shh-driven mouse model of medulloblastoma in which a constitutively activated form of the Smoothened gene (SmoA1) is expressed within cerebellar granule neuron precursors through the regulation of the NeuroD2 promoter. Medulloblastomas form in approximately 50% of SmoA1 mice by approximately 6 mo of age and show molecular profiles that resemble Shh-driven human medulloblastomas.Citation7 To detect differential DNA methylation, we performed the denaturation analysis of methylation differences (DAMD) assay, a technique that identifies DNA methylation differences in CpG islands based on the increased melting temperature of cytosine methylated DNA.Citation6,Citation8 In contrast to the extensive promoter CpG island DNA hypermethylation in human medulloblastoma patient samples observed previously,Citation6 no DAMD-positive hypermethylated loci were observed in tumors from SmoA1 mice. Cerebella from wild-type mice served as the comparator. To determine if this finding was specific to the SmoA1 allele, we analyzed a different allele of Smoothened (SmoA2) that drives medulloblastoma in ~75% of animals by 6 mo of age, and also displays aberrant cerebellar development.Citation9 Assaying tumors from SmoA2 mice, we again observed no DAMD-positive loci (DAMD data for SmoA1 and SmoA2 accessible via Gene Expression Omnibus database, accession number GSE45342). These results suggest that the extensive promoter CpG island DNA hypermethylation previously observed in patient samples is not present in these two Shh-driven mouse models.
The DAMD assay detects differential DNA methylation based on the increased melting temperature (Tm) of methylated DNA.Citation6 Because DNA sequence composition also strongly affects the Tm of DNA, the denaturation conditions used in the DAMD assay do not detect differential DNA methylation in low-density CpG regions of the genome. Thus, the lack of DAMD-positive loci in tumor specimens from mice could formally be due to differences in sequence composition between the mouse and human genomes.
To address this possibility, we performed Reduced Representation Bisulfite Sequencing (RRBS), a high throughput sequencing-based method for quantitative, single-nucleotide resolution of cytosine methylation that provides good coverage of CpG islands and promoter regions across the genome.Citation10 RRBS was performed on a representative human medulloblastoma patient sample, and tumors from three different mouse models of medulloblastoma: the two Shh-driven medulloblastoma mouse models (SmoA1 and SmoA2) previously examined using the DAMD assay, and a MYCN-driven mouse model of medulloblastoma (Glt1-tTA:TRE-MYCN/Luc) that recapitulates the histology of human classic and large cell anaplastic medulloblastoma.Citation11 For the identification of differentially methylated regions in tumors, human and mouse strain-specific normal cerebella were used as controls. RRBS analysis of the human medulloblastoma patient sample revealed strong promoter CpG island DNA hypermethylation (>60% more methylated than normal cerebellum) of 121 loci (; Table S1), while the mouse models of medulloblastoma displayed far fewer loci, ranging from 0 to 16 loci (; Table S2). If the stringency for methylation in the tumors was reduced to identify loci with > 33% more methylation than the control, the patient sample had 315 additional loci, while tumors from the mouse models of medulloblastoma still demonstrated fewer loci, ranging from 7 to 70 (; Tables S1 and S2).
Table 1. DNA methylation of promoter CpG islands in human and mouse medulloblastoma
RRBS library characteristics between the human and mouse samples were similar, with the mouse samples having greater CpG coverage (Table S3). Metaplots of the RRBS data from both human and mouse promoters containing a CpG island demonstrate the expected pattern of relative protection from CpG DNA methylation of the transcriptional start site (TSS) in both tumor and normal cerebellum (Fig. S1). When the subset of promoters displaying CpG island DNA hypermethylation or hypomethylation are similarly analyzed, the TSS continues to show relative hypomethylation (Figs. S1B and C).
Traditional bisulfite sequencing of representative loci confirmed the results obtained by RRBS. In the human sample, loci that had >60% methylation in the tumor displayed extensive CpG methylation, while normal cerebellum was largely unmethylated at those loci (GPR75, MIR155HG, and TMEM176B_A; ). Good agreement between RRBS and traditional bisulfite sequencing was also obtained for loci that displayed 33–59% methylation in the human samples (SALL4; ), as well as the SmoA2 tumor and normal cerebellum (). However, the mouse samples did not show the same degree of dense methylation as was present in the human samples with the exception of one locus (1810013D10Rik) from SmoA2 that demonstrated a pattern consistent with one allele of the gene being densely methylated, while the other allele was completely unmethylated ().
Figure 1. Bisulfite sequence analysis of loci identified by Reduced Representation Bisulfite Sequencing from human and SmoA2 medulloblastoma. (A) Bisulfite sequence analysis of GPR75, MIR155HG, TMEM176B_A, and SALL4 from human medulloblastoma (MB) and normal cerebellum (Cbl). The black rectangle shows the genomic region subjected to bisulfite sequence analysis; the mRNA structure (exon, large rectangle; intron, thin line; untranslated region (UTR), small rectangle; arrow, direction of transcription) is shown in blue; any associated CpG island is shown using a green rectangle. Solid circles represent CpG methylation, and open circles depict unmodified CpG dinucleotides. (B) Bisulfite sequence analysis of 5730507C01Rik, 1810013D10Rik, and Vmn1r90 from SmoA2 and wild-type cerebellum (WT Cbl).

Although the RRBS identified loci in the mouse models of medulloblastoma showed only modest changes in CpG island methylation, a few common methylated loci (>33% hypermethylation) were identified among the different mouse tumor samples. One region showed increased CpG island methylation in all of the mouse tumor samples, and this locus encompasses two genes (Cbln1 and Gm2694) that are divergently transcribed and share a common promoter CpG island (Fig. S2). Cbln1 encodes for Cerebellin precursor protein 1, a secreted glycoprotein essential for normal synaptic structure and function in the cerebellum,Citation12 while Gm2694 encodes a protein of unknown function. As shown in Fig. S3A, the RRBS data demonstrated minor differences in DNA methylation between the tumors and normal cerebellum control at this locus. Common to SmoA1, SmoA2, and one of the MYCN derived tumors was Grm4, encoding a type 4 metabotropic glutamate (mGlu4) receptor. mGlu4 receptors are expressed by cerebellar granule precursor cells (CGPs), and activation of these receptors reduces proliferation and enhances differentiation of CGPs.Citation13 A previous study also demonstrated that activation of mGlu4 receptors reduces medulloblastoma cell proliferation by inhibiting the PI-3-K pathway.Citation14 Lastly, common to SmoA2 and one of the MYCN tumors was Map3k6, a proapoptotic gene that has been shown to be a tumor suppressor in a keratinocyte tumorigenesis model.Citation15 There was only one methylated (>33%) locus shared between any of the mouse medulloblastoma tumors and the patient sample. Common to SmoA2 and the patient sample was FAM83F (family with sequence similarity 83, member F), a gene of unknown function. This locus showed a modest increase in DNA methylation in the mouse tumors compared with the DNA hypermethylation observed in the patient sample (Fig. S3B). Therefore, although minor methylation differences can be found at a small number of mostly non-overlapping loci in mouse models of medulloblastomas, the mouse models do not display the strong promoter CpG island hypermethylation of a large set of shared loci present in human tumors.Citation6 However, the limited number of affected genes in the mouse models may be of interest for future studies in medulloblastoma biology.
Mouse models of Burkitt lymphoma and breast cancer do not display extensive promoter CpG island DNA hypermethylation
Given our findings examining promoter CpG island DNA hypermethylation in mouse models of medulloblastoma, we wanted to determine whether this phenomenon was common to other GEMMs. We performed the DAMD assay on tumors from the Eµ-Myc mouse model of Burkitt lymphomaCitation16 and the MMTV-rtTA/TetO-NeuNT transgenic mammary carcinoma model.Citation17 Similar to our findings examining mouse models of medulloblastoma, we did not detect any DAMD-positive loci (DAMD data available via accession number GSE45342), suggesting that the extensive promoter CpG island DNA hypermethylation observed in human cancers is not a common feature of mouse models of cancer.
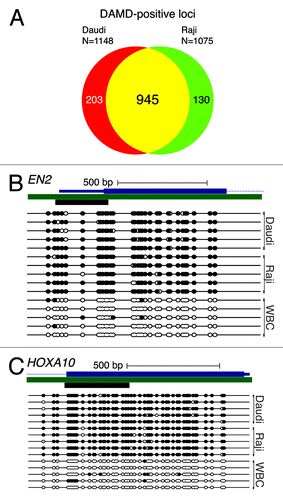
Previously published studies of breast cancer patient samples have demonstrated extensive promoter CpG island DNA hypermethylation,Citation18-Citation21 For Burkitt lymphoma, however, data are more limited. Candidate gene studiesCitation22,Citation23 and an array-based examination of a small subset of the genomeCitation24 suggested that promoter CpG island DNA hypermethylation can occur in Burkitt lymphoma. Since we did not have access to Burkitt lymphoma patient samples, we performed the DAMD assay on two human Burkitt lymphoma cell lines, Daudi and Raji. While the long-term propagation of cancer cell lines in culture can lead to increased promoter CpG island DNA hypermethylation, genome-wide DNA methylation studies in cell lines can still inform on cancer biology.Citation25 In contrast to Eµ-Myc mice, extensive promoter CpG island DNA hypermethylation was observed for Daudi (1148 loci) and Raji (1075) with a high degree of overlap (945 loci; ; Table S4). Traditional bisulfite sequence analysis confirmed that the DAMD-positive loci represented regions of DNA hypermethylation (). Taken together with the previously published reports of Burkitt lymphoma and breast cancer patient samples, these results suggest that human medulloblastomas, Burkitt lymphomas, and breast cancers display aberrant promoter CpG island DNA hypermethylation, whereas this epigenetic change is greatly diminished, or absent, in their corresponding genetically engineered mouse models, indicating a fundamental epigenetic difference between the mouse models and human disease.
Figure 2. Human Burkitt lymphoma cell lines demonstrate extensive promoter CpG island DNA hypermethylation. (A) DAMD-positive loci from the human Burkitt lymphoma cell lines Daudi and Raji are depicted, and common loci are indicated with a Venn diagram. (B and C) Bisulfite sequence analysis of engrailed homeobox 2 (EN2) and homeobox A10 (HOXA10) from Daudi, Raji, and peripheral white blood cells (WBC). See legend for labeling schematic.
Mouse fibroblasts passaged through crisis obtain extensive genome-wide promoter CpG island DNA hypermethylation
Previous work over 20 y ago demonstrated that immortalizing mouse fibroblasts could lead to an increase in DNA methylation of the CpG island encompassing the MYOD1 gene.Citation26 Experiments using human cell lines have shown that genome-wide DNA methylation of promoter CpG islands increases during the process of immortalization,Citation27,Citation28 and most recently, this result was confirmed in mouse embryonic fibroblasts.Citation29
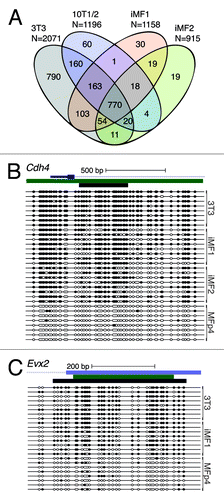
To determine whether the genome-wide DNA hypermethylation of promoter CpG islands that occurs in mouse cells during the process of immortalization is more similar to human cancers than the modest methylation changes observed in GEMMs, we created newly immortalized mouse fibroblasts by serially passaging primary cells in vitro and selecting clones that spontaneously acquired the ability to bypass replicative senescence.Citation30 The DAMD assay was performed on two independent newly immortalized mouse fibroblast cell lines (iMF1 and iMF2), and two well-established mouse fibroblast immortalized cell lines (3T3 and 10T1/2); an early passage culture of non-immortalized mouse primary fibroblasts served as a control. When compared with the early passage mouse primary fibroblast culture, all of the immortalized mouse fibroblasts displayed extensive promoter CpG island DNA hypermethylation of a large number of loci (3T3, 2071 loci; 10T1/2, 1196; iMF1, 1158; iMF2, 915), similar to the dense hypermethylation in human cancers. Many of these loci were common to all four of the immortalized mouse cell lines (770 loci; ; Table S4), and bisulfite sequence analysis confirmed that these loci displayed DNA hypermethylation (). These results demonstrate that mouse cells are capable of undergoing extensive promoter CpG island DNA hypermethylation, similar to what is observed in human cancers, when they are spontaneously immortalized. Furthermore, this finding implies that GEMMs of cancer do not activate a similar process of immortalization-associated CpG island hypermethylation.
Figure 3. Mouse fibroblast cells obtain global promoter CpG island DNA hypermethylation during the process of spontaneous immortalization. (A) DAMD-positive loci from the established mouse fibroblast cell lines 3T3 and 10T1/2, and two independent newly immortalized mouse fibroblast cell lines (iMF1 and iMF2), are depicted. Common loci are indicated on a Venn diagram. (B and C) Bisulfite sequence analysis of Cdh4 and Evx2 from 3T3, iMF1, iMF2, and an early passage primary mouse fibroblast culture (MFp4). See legend for labeling schematic.
Discussion
We examined genome-wide DNA methylation in GEMMs, with a primary focus on examining promoter CpG island DNA hypermethylation in primary tumor samples from mouse models of medulloblastoma. In contrast to the extensive DNA hypermethylation of these regions of the genome observed in patient samples,Citation6 relatively few promoter CpG islands in three different mouse models of medulloblastoma displayed this epigenetic modification, and those loci did not show the same degree of hypermethylation seen in human cancers.
A very limited number of genome-wide DNA methylation studies of GEMMs of cancer have been reported. One of these examined genome-wide DNA methylation in the same genetically driven Eµ-Myc mouse model of Burkitt lymphoma used in our current study.Citation31 Robust promoter CpG island DNA hypermethylation of individual genes was not described, but rather only a trend toward increasing promoter CpG island DNA hypermethylation could be discerned when data for all genes were analyzed in aggregate. A separate study in Eµ-Myc mice demonstrated a modest increase in DNA methylation by transgenic expression of DNMT3B7, a truncated isoform of DNA methyltransferase 3B that has been found in human cancer cells.Citation32 This finding is not cancer-specific, however, since an increase in promoter CpG island DNA methylation was observed by ectopic expression of mouse Dnmt3b in normal mouse colon cells.Citation33
Mouse cells are capable of obtaining extensive promoter CpG island DNA hypermethylation when they are immortalized, as recently shownCitation29 and in this study. It is possible that this phenomenon is a result of culturing cells, as it has been observed that progressive promoter CpG island DNA hypermethylation occurs after extensive passaging of cancer cells in vitro. However, while differential CpG methylation has also been observed upon culturing non-transformed cells, these methylation differences occur predominately as unmethylated CpGs in the bodies of genes involved in regulating cellular proliferation, rather then promoter CpG island DNA hypermethylation.Citation25 Given that extensive promoter CpG island DNA hypermethylation occurs as an early step in the process of immortalization in both mouse and human fibroblasts, these findings suggest that this phenomenon may contribute to the bypass of senescence, an important barrier to cancer development.
While many GEMMs use an oncogenic driver to initiate cancer, chronic inflammatory states have been shown to lead to tumor formation in mice. Patients with inflammatory bowel disease (IBD) are at an increased risk of developing colorectal cancer,Citation34 and mouse models of this disease recapitulate this phenomenon. Using glutathione peroxidase Gpx1 and Gpx2 double-knockout (Gpx1/2-KO) mice, genome-wide DNA methylation studies of tumors and pre-cancerous lesions demonstrated a limited number of loci with moderate degree of promoter CpG DNA methylation.Citation35 This result, along with our findings in spontaneously immortalized mouse fibroblasts, suggests that the molecular machinery responsible for promoter CpG island DNA hypermethylation is present in mouse cells, but this global epigenetic event is not initiated nor required for tumorigenesis in GEMMs driven by an oncogene, at least not in the five GEMMs we examined. Cancer arising from inflammatory states, however, may require the initiation of global epigenetic dysregulation in pre-neoplastic tissues to allow for the selection of clones that harbor the prerequisite epigenetic and genetic changes found in a malignant cell. In support of the hypothesis that a global change in DNA methylation may be an early event in inflammatory cancer, it was demonstrated that oxidative damage could cause rapid redistribution of a silencing complex containing DNA methyltransferases and Polycomb members to promoter CpG islands.Citation36 Future studies to elucidate the differences between GEMMs and human cancers will hopefully give insight into what changes are necessary for human carcinogenesis as well as aid in developing more faithful GEMMs as platforms for preclinical drug testing, especially for those drugs that target the epigenome.
Material and Methods
Detailed Material and Methods may be found in Supplemental Materials and Methods.
Ethics statement
Pre-existing patient de-identified samples were obtained in accordance with IRB protocol.
Cell culture, mouse strains, and genomic DNA isolation
The human Burkitt lymphoma cell lines DaudiCitation37 and RajiCitation38 were maintained in RPMI-1640; mouse fibroblast cells were all propagated in DMEM. Media in all cases was supplemented with 10% FBS and 1% Pen/Strep. Newly immortalized mouse fibroblast cell lines were obtained by serially passaging cells until cell division ceased (approximately at passage 16), and then spontaneously immortalized clones were subsequently isolated.Citation30 Genomic DNA for RRBS analysis was obtained by growing an isolated clone immediately after immortalization. Mouse medulloblastoma tumors were obtained from SmoA1,Citation7 SmoA2,Citation9 and Glt1-tTA:TRE-MYCN/Luc mice.Citation11 All mice were maintained in accordance with the NIH Guide for the Care and Use of Experimental Animals with approval from the Institutional Animal Care and Use Committee from each institution. Mouse tumor or normal tissue was dissected and snap frozen prior to genomic DNA isolation. Genomic DNA from all sources was isolated using the DNeasy Blood and Tissue Kit (Qiagen, cat# 69504). Genomic DNA from human peripheral white blood cells was obtained from Promega (cat# G3041).
DNA methylation and bioinformatics analysis
The Denaturation Analysis of Methylation Differences (DAMD) assay was performed and analyzed as previously described.Citation8 Sample library preparation for Reduced Representation Bisulfite Sequencing (RRBS) was performed by service provider (Zymo Research). Briefly, mapping of RRBS reads was performed using BSMAP (v2.74).Citation39 Methylation analysis was performed using R. See Supplemental Material and Methods for detailed description of the data analysis. Bisulfite DNA conversion, PCR, and sequence analysis was performed as described.Citation8 Primer sequences can be found in Table S5. The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession number GSE45342).
Additional material
Download Zip (5.6 MB)Acknowledgments
We thank M Parker for the gift of primary mouse fibroblast cells. SJD was supported as a St Baldrick’s Foundation Scholar, a Hyundai Hope on Wheels Scholar, and by the American Brain Tumor Association/Emily Dorfman Foundation for Children Basic Research Fellowship in memory of Emily Ann Dorfman, Alex’s Lemonade Stand Foundation, and University of Washington Child Health Research Center (NIH grant K12 HD43376). This study was also supported by NIAMS AR045113 (SJT).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/26486
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science 2013; 339:1546 - 58; http://dx.doi.org/10.1126/science.1235122; PMID: 23539594
- Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 2012; 13:679 - 92; http://dx.doi.org/10.1038/nrg3270; PMID: 22945394
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 2011; 11:726 - 34; http://dx.doi.org/10.1038/nrc3130; PMID: 21941284
- Politi K, Pao W. How genetically engineered mouse tumor models provide insights into human cancers. J Clin Oncol 2011; 29:2273 - 81; http://dx.doi.org/10.1200/JCO.2010.30.8304; PMID: 21263096
- Diede SJ, Guenthoer J, Geng LN, Mahoney SE, Marotta M, Olson JM, Tanaka H, Tapscott SJ. DNA methylation of developmental genes in pediatric medulloblastomas identified by denaturation analysis of methylation differences. Proc Natl Acad Sci U S A 2010; 107:234 - 9; http://dx.doi.org/10.1073/pnas.0907606106; PMID: 19966297
- Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res 2004; 64:7794 - 800; http://dx.doi.org/10.1158/0008-5472.CAN-04-1813; PMID: 15520185
- Mahoney SE, Yao Z, Keyes CC, Tapscott SJ, Diede SJ. Genome-wide DNA methylation studies suggest distinct DNA methylation patterns in pediatric embryonal and alveolar rhabdomyosarcomas. Epigenetics 2012; 7:400 - 8; http://dx.doi.org/10.4161/epi.19463; PMID: 22419069
- Dey J, Ditzler S, Knoblaugh SE, Hatton BA, Schelter JM, Cleary MA, Mecham B, Rorke-Adams LB, Olson JM. A distinct Smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol Cell Biol 2012; 32:4104 - 15; http://dx.doi.org/10.1128/MCB.00862-12; PMID: 22869526
- Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc 2011; 6:468 - 81; http://dx.doi.org/10.1038/nprot.2010.190; PMID: 21412275
- Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan Q-W, Goldenberg DD, Lau J, Masic S, Nguyen K, Yakovenko S, et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev 2010; 24:1059 - 72; http://dx.doi.org/10.1101/gad.1907510; PMID: 20478998
- Hirai H, Pang Z, Bao D, Miyazaki T, Li L, Miura E, Parris J, Rong Y, Watanabe M, Yuzaki M, et al. Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat Neurosci 2005; 8:1534 - 41; http://dx.doi.org/10.1038/nn1576; PMID: 16234806
- Canudas AM, Di Giorgi-Gerevini V, Iacovelli L, Nano G, D’Onofrio M, Arcella A, Giangaspero F, Busceti C, Ricci-Vitiani L, Battaglia G, et al. PHCCC, a specific enhancer of type 4 metabotropic glutamate receptors, reduces proliferation and promotes differentiation of cerebellar granule cell neuroprecursors. J Neurosci 2004; 24:10343 - 52; http://dx.doi.org/10.1523/JNEUROSCI.3229-04.2004; PMID: 15548648
- Iacovelli L, Arcella A, Battaglia G, Pazzaglia S, Aronica E, Spinsanti P, Caruso A, De Smaele E, Saran A, Gulino A, et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors inhibits the growth of medulloblastomas. J Neurosci 2006; 26:8388 - 97; http://dx.doi.org/10.1523/JNEUROSCI.2285-06.2006; PMID: 16899734
- Iriyama T, Takeda K, Nakamura H, Morimoto Y, Kuroiwa T, Mizukami J, Umeda T, Noguchi T, Naguro I, Nishitoh H, et al. ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. EMBO J 2009; 28:843 - 53; http://dx.doi.org/10.1038/emboj.2009.32; PMID: 19214184
- Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med 1988; 167:353 - 71; http://dx.doi.org/10.1084/jem.167.2.353; PMID: 3258007
- Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, Dugan KD, Innocent N, Cardiff RD, Schnall MD, Chodosh LA. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell 2002; 2:451 - 61; http://dx.doi.org/10.1016/S1535-6108(02)00212-X; PMID: 12498714
- Hill VK, Ricketts C, Bieche I, Vacher S, Gentle D, Lewis C, Maher ER, Latif F. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res 2011; 71:2988 - 99; http://dx.doi.org/10.1158/0008-5472.CAN-10-4026; PMID: 21363912
- Novak P, Jensen T, Oshiro MM, Watts GS, Kim CJ, Futscher BW. Agglomerative epigenetic aberrations are a common event in human breast cancer. Cancer Res 2008; 68:8616 - 25; http://dx.doi.org/10.1158/0008-5472.CAN-08-1419; PMID: 18922938
- Rodenhiser DI, Andrews J, Kennette W, Sadikovic B, Mendlowitz A, Tuck AB, Chambers AF. Epigenetic mapping and functional analysis in a breast cancer metastasis model using whole-genome promoter tiling microarrays. Breast Cancer Res 2008; 10:R62; http://dx.doi.org/10.1186/bcr2121; PMID: 18638373
- Ruike Y, Imanaka Y, Sato F, Shimizu K, Tsujimoto G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genomics 2010; 11:137; http://dx.doi.org/10.1186/1471-2164-11-137; PMID: 20181289
- Lee S, Syed N, Taylor J, Smith P, Griffin B, Baens M, Bai M, Bourantas K, Stebbing J, Naresh K, et al. DUSP16 is an epigenetically regulated determinant of JNK signalling in Burkitt’s lymphoma. Br J Cancer 2010; 103:265 - 74; http://dx.doi.org/10.1038/sj.bjc.6605711; PMID: 20551953
- Murai M, Toyota M, Satoh A, Suzuki H, Akino K, Mita H, Sasaki Y, Ishida T, Shen L, Garcia-Manero G, et al. Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br J Cancer 2005; 92:1165 - 72; http://dx.doi.org/10.1038/sj.bjc.6602422; PMID: 15756280
- Martín-Subero JI, Kreuz M, Bibikova M, Bentink S, Ammerpohl O, Wickham-Garcia E, Rosolowski M, Richter J, Lopez-Serra L, Ballestar E, et al, Molecular Mechanisms in Malignant Lymphomas Network Project of the Deutsche Krebshilfe. New insights into the biology and origin of mature aggressive B-cell lymphomas by combined epigenomic, genomic, and transcriptional profiling. Blood 2009; 113:2488 - 97; http://dx.doi.org/10.1182/blood-2008-04-152900; PMID: 19075189
- Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 2013; 23:555 - 67; http://dx.doi.org/10.1101/gr.147942.112; PMID: 23325432
- Jones PA, Wolkowicz MJ, Rideout WM 3rd, Gonzales FA, Marziasz CM, Coetzee GA, Tapscott SJ. De novo methylation of the MyoD1 CpG island during the establishment of immortal cell lines. Proc Natl Acad Sci U S A 1990; 87:6117 - 21; http://dx.doi.org/10.1073/pnas.87.16.6117; PMID: 2385586
- Landan G, Cohen NM, Mukamel Z, Bar A, Molchadsky A, Brosh R, Horn-Saban S, Zalcenstein DA, Goldfinger N, Zundelevich A, et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet 2012; 44:1207 - 14; http://dx.doi.org/10.1038/ng.2442; PMID: 23064413
- Novak P, Jensen TJ, Garbe JC, Stampfer MR, Futscher BW. Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res 2009; 69:5251 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-08-4977; PMID: 19509227
- Tommasi S, Zheng A, Weninger A, Bates SE, Li XA, Wu X, Hollstein M, Besaratinia A. Mammalian cells acquire epigenetic hallmarks of human cancer during immortalization. Nucleic Acids Res 2013; 41:182 - 95; http://dx.doi.org/10.1093/nar/gks1051; PMID: 23143272
- Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 1963; 17:299 - 313; http://dx.doi.org/10.1083/jcb.17.2.299; PMID: 13985244
- Conerly ML, Teves SS, Diolaiti D, Ulrich M, Eisenman RN, Henikoff S. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res 2010; 20:1383 - 90; http://dx.doi.org/10.1101/gr.106542.110; PMID: 20709945
- Shah MY, Vasanthakumar A, Barnes NY, Figueroa ME, Kamp A, Hendrick C, Ostler KR, Davis EM, Lin S, Anastasi J, et al. DNMT3B7, a truncated DNMT3B isoform expressed in human tumors, disrupts embryonic development and accelerates lymphomagenesis. Cancer Res 2010; 70:5840 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-10-0847; PMID: 20587527
- Steine EJ, Ehrich M, Bell GW, Raj A, Reddy S, van Oudenaarden A, Jaenisch R, Linhart HG. Genes methylated by DNA methyltransferase 3b are similar in mouse intestine and human colon cancer. J Clin Invest 2011; 121:1748 - 52; http://dx.doi.org/10.1172/JCI43169; PMID: 21490393
- Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, Williams C, Price A, Talbot I, Forbes A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004; 126:451 - 9; http://dx.doi.org/10.1053/j.gastro.2003.11.010; PMID: 14762782
- Hahn MA, Hahn T, Lee D-H, Esworthy RS, Kim B-W, Riggs AD, Chu FF, Pfeifer GP. Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res 2008; 68:10280 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-08-1957; PMID: 19074896
- O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 2011; 20:606 - 19; http://dx.doi.org/10.1016/j.ccr.2011.09.012; PMID: 22094255
- Klein E, Klein G, Nadkarni JS, Nadkarni JJ, Wigzell H, Clifford P. Surface IgM-kappa specificity on a Burkitt lymphoma cell in vivo and in derived culture lines. Cancer Res 1968; 28:1300 - 10; PMID: 4174339
- Pulvertaft JV. Cytology of Burkitt's tumour (African lymphoma). Lancet 1964; 1:238 - 40; http://dx.doi.org/10.1016/S0140-6736(64)92345-1; PMID: 14086209
- Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 2009; 10:232; http://dx.doi.org/10.1186/1471-2105-10-232; PMID: 19635165