
Abstract
Maternal diet can affect fetal gene expression through epigenetic mechanisms. Wilson disease (WD), which is caused by autosomal recessive mutations in ATP7B encoding a biliary copper transporter, is characterized by excessive hepatic copper accumulation, but variability in disease severity. We tested the hypothesis that gestational supply of dietary methyl groups modifies fetal DNA methylation and expression of genes involved in methionine and lipid metabolism that are impaired prior to hepatic steatosis in the toxic milk (tx-j) mouse model of WD. Female C3H control and tx-j mice were fed control (choline 8 mmol/Kg of diet) or choline-supplemented (choline 36 mmol/Kg of diet) diets for 2 weeks throughout mating and pregnancy to gestation day 17. A second group of C3H females, half of which were used to cross foster tx-j pups, received the same diet treatments that extended during lactation to 21 d postpartum. Compared with C3H, fetal tx-j livers had significantly lower copper concentrations and significantly lower transcript levels of Cyclin D1 and genes related to methionine and lipid metabolism. Maternal choline supplementation prevented the transcriptional deficits in fetal tx-j liver for multiple genes related to cell growth and metabolism. Global DNA methylation was increased by 17% in tx-j fetal livers after maternal choline treatment (P < 0.05). Maternal dietary choline rescued the lower body weight of 21 d tx-j mice. Our results suggest that WD pathogenesis is modified by maternal in utero factors, including dietary choline.
Introduction
Wilson disease (WD) is an autosomal recessive disease caused by excessive hepatic copper (Cu) accumulation that results from mutations in the ATP7B biliary Cu transporter.Citation1 Although considered a monogenic disease, WD is characterized by a variety of clinical hepatic presentations ranging from fatty liver to cirrhosis and fulminant hepatic failure and can occur from 3 to 70 y of age.Citation2,Citation3 Despite numerous clinical studies, a convincing phenotype-genotype correlation for WD has not been established.Citation4-Citation6 Indeed, several case reports describe twins with identical WD genotypes but different phenotypes.Citation7-Citation9
Using the tx-j mouse that is characterized by a mutation in the ATP7B Cu transporter and consequent hepatic Cu accumulation similar to WD patients,Citation10,Citation11 we previously demonstrated altered hepatic methionine metabolism and global DNA hypomethylation in association with Cu accumulation and altered gene expression.Citation12 The importance of epigenetic regulation through DNA methylation for variability in the onset, severity, and progression of many human diseases is increasingly recognized.Citation13,Citation14 Methyl donors are particularly needed during pregnancy and lactation as they are critical for fetal and postnatal development.Citation14,Citation15 Studies in various animal models have shown that maternal supplementation of dietary methyl donors can affect DNA methylation, gene expression, and phenotype in the offspring.Citation16-Citation18 Methionine metabolism provides a supply of methyl groups through its metabolite S-adenosylmethionine (SAM), which is derived from methionine; SAM provides methyl groups for methylation reactions catalyzed by DNA and histone methyltransferases. As diagrammed in , methionine originates from both the transmethylation reaction of homocysteine and 5-methyltetrahydrofolate, or from betaine, which is an alternate methyl donor derived from choline, that is largely provided by dietary sources such as egg yolk, potatoes, cauliflower, lentils, and oats. Choline is required for neural tube closure during embryonic life and significant amounts of maternal choline are needed to ensure an adequate fetal supply.Citation15 In addition, phosphatidylcholine, the principal metabolite of choline, is a major component of cell membrane phospholipids and is essential for cell integrity and signaling. Choline supplementation of pregnant rats can increase levels of histone and DNA methylation in fetal liver.Citation19 In addition, choline deficiency is associated with fatty liver in adult life.Citation20 While multiple benefits of choline supplementation have been reported, no previous study has addressed the influence of maternal nutritional and methylation status as potential factors affecting WD presentation and progression. We hypothesized that gestational supply of dietary methyl groups modifies fetal DNA methylation and expression of genes involved in methionine and lipid metabolism that are impaired prior to hepatic steatosis in the toxic milk (tx-j) mouse model of WD. To test this hypothesis, we fed tx-j and control dams with control or choline supplemented diets before mating, throughout pregnancy, and lactation during which newborn tx-j mice were cross-fostered after postpartum day 1–3 to C3H dams that have normal milk copper content and were fed either control or choline-supplemented diets to improve their viability. To control for cross fostering effects, another two groups of each genotype were suckled from their birth mothers for 6 d. Fetal and 21-d-old (21d) pup livers were collected to study the expression of selected genes relevant to liver damage and DNA methylation. Our studies showed that maternal choline supplementation corrected several growth, methionine metabolism, global DNA methylation, and gene expression deficits of the mutant tx-j mouse model of WD without changing DNA methylation of selected genes. The data suggest that maternal choline supplementation of tx-j mice created an environment for adaptation of gene expression to growth stimuli.
Figure 1. Representation of methionine metabolism. MS, methionine synthase; MAT, methionine adenosyl transferase; SAHH, S-adenosylhomocysteine transferase; DNMT, DNA methyltransferase.
Results
Reproductive success and weanling growth
The sex distributions among C3H and tx-j mice at 6d (not shown) and 21d were similar. C3H (control) and tx-j mice had similar reproductive success and choline supplementation did not affect the number of viable fetuses and pups (). The body weights of weanling 21d tx-j pups were significantly lower than those of C3H mice, but maternal choline supplementation of tx-j mice corrected this phenotype (P < 0.001) ().
Table 1. Effects of choline supplementation on reproductive success
Table 2. Effects of maternal choline supplementation in fetal and 21d weanling livers
Hepatic copper and iron concentrations in fetal and 21d pup livers
Hepatic Cu levels were markedly lower in tx-j fetal livers compared with levels in C3H mice, and maternal choline supplementation resulted in about additional 50% reduction of mean fetal Cu in the tx-j mice but had no effect on C3H Cu levels (). At 21d weaning, mean hepatic Cu concentrations in the tx-j mice were 2.5 times higher than in the C3H mice, while choline supplementation of fostering dams had no effect on these values within either genetic group. There were no differences in Fe concentrations among different genotype and maternal diet groups.
Hepatic S-adenosylmethionine and S-adenosylhomocysteine
Untreated fetal liver SAM and S-adenosylhomocysteine (SAH) concentrations were similar in the two genotypes and were unaffected by maternal choline supplementation (). However, each group of 21d tx-j mice had higher liver SAH levels than C3H mice, a finding consistent with our previous report of a reduced expression of SAH hydrolase in this strain,Citation12 and liver SAH levels were further increased by maternal choline supplementation in each genotype.
Light microscopy and histology
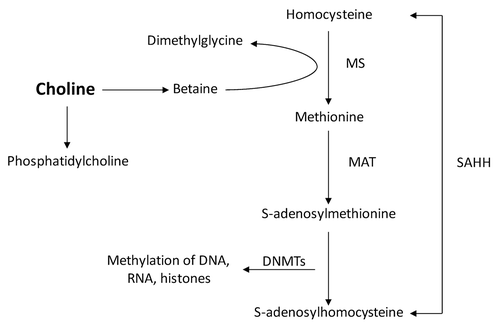
There were no histological changes in hepatic inflammation, steatosis, or fibrosis in both untreated and choline supplemented tx-j and C3H weanling mice at 21 d. Hepatocyte mitotic bodies were present in livers of untreated mice and persisted after choline supplementation. Nuclei diameters were similar before and after choline supplementation in both C3H and tx-j mice ().
Figure 2. Liver histology from control (C3H) and tx-j mice at 3 weeks of age (21d). All hematoxilin and eosin, 40x. (A) Control (C3H) mouse, untreated; (B) Tx-j mouse, untreated; (C) Control (C3H) treated with choline (no mitotic bodies were identified in this representative sample); (D) Tx-j mouse treated with choline. Tx-j mice livers did not show any significant degree of inflammation, steatosis, or fibrosis. Arrows indicate mitotic bodies. Nuclei diameter was similar in all groups (C3H vs tx-j 63.3 ± 9.4 vs 56.2 ± 8 µm; C3H choline vs tx-j choline 69.4 ± 15.5 vs 63.9 ± 2.8 µm. All P > 0.05).
Immunohistochemistry for Cyclin D1
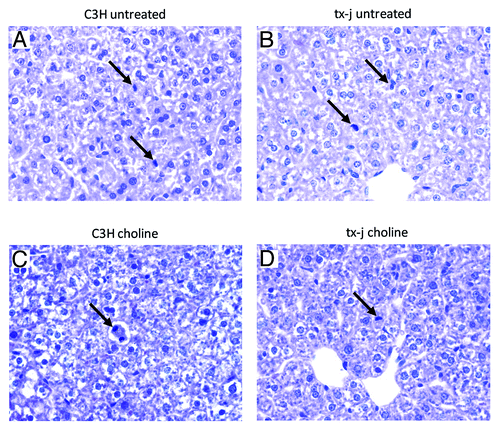
Since the regulation of Cyclin D1 expression is both transcriptional and post-transcriptional,Citation21 we performed immunohistochemistry on 21-d livers. There were no differences in percentages of Cyclin D1 positive nuclei between C3H and tx-j mice from the control diet groups but the tx-j mice in the choline supplemented group had higher percentages of Cyclin D1 positive nuclei than the C3H supplemented mice ().
Figure 3. Immunohistochemistry for Cyclin D1 in 21d pups. (A) Control (C3H) mouse, untreated; (B) Tx-j mouse, untreated; (C) Control (C3H) treated with choline; (D) Tx-j mouse treated with choline. All 40x. Arrows indicate some of the Cyclin D1 positive nuclei. (A and C). The percentages of Cyclin D1 positive nuclei were similar in C3H and tx-j mice from the control diet groups (C3H vs tx-j 66 ± 7.6% vs 66.6 ± 5.8%; P = n.s.). Hepatocyte nuclei in choline treated tx-j mice liver (D) presented a higher percentage of Cyclin D1 positivity compared with untreated mice (B) (53.7 ± 9.5% vs 72.4 ± 3.7%; P = < 0.05).
Hepatic global DNA methylation
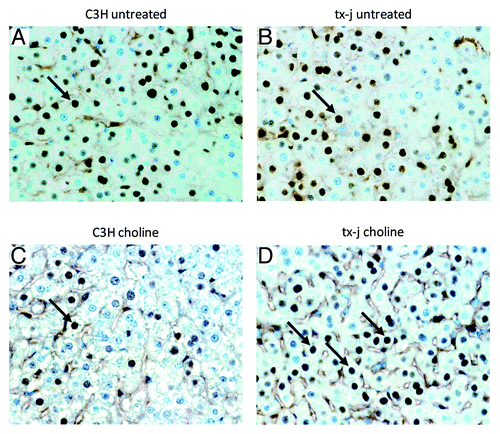
Whereas there were no significant differences in global DNA methylation levels between control C3H and tx-j fetal livers, maternal choline supplementation resulted in 17% increased hepatic global DNA methylation in tx-j fetal livers compared with untreated tx-j (). In 21d weanlings, hepatic global DNA methylation was significantly higher in tx-j mice after choline supplementation compared with C3H mice. The differences in global DNA methylation did not change after adjusting for litter size, gender, and genotype-gender interactions.
Figure 4. Global DNA methylation. Global methylation of hepatic DNA was similar in fetal livers of C3H and tx-j mice at baseline without maternal choline treatment. Tx-j mouse fetal livers from maternal choline treated mice had increased global DNA methylation compared with livers from untreated mice, whereas among choline supplemented groups at 21d, tx-j livers had increased DNA methylation compared with livers of C3H mice (*P < 0.01). Each dot for each genotype/treatment group represents an individual sample from one representative mouse.
Selected hepatic gene expression
In the non-supplemented maternal diet groups, the transcript levels of fetal liver genes related to methionine metabolism, DNA methylation, lipid synthesis, fatty acid oxidation, and Cyclin D1, representative of cell cycle, were all downregulated in the tx-j compared with C3H mice, whereas the transcript levels of each of these genes in tx-j fetal mouse livers were increased and similar to those in C3H mice in the choline supplemented groups (). The consistent pattern of decreased fetal liver transcript levels in untreated tx-j mice was preserved at 6 d of age before cross-fostering (). Conversely, there were no differences in most transcript levels in tx-j mice compared with C3H mice in 21-d tx-j mice that were fostered by C3H dams with or without dietary choline supplementation, with the exceptions of Mat1a and the DNA methyltransferases, Dnmt3a and Dnmt3b, whose deficits in 21d tx-j liver were restored by choline supplementation (). There were positive correlations between the body weight and gene transcript levels of Sterol Regulatory Element-Binding Protein (Srebp) r = 0.56; glucose-regulated protein 78 (Grp78) r = 0.38; peroxisome proliferator-activated receptor α (Ppar α) r = 0.40; DNA methyltransferase3b (Dnmt3b) r = 0.81; S-adenosylhomocysteine hydrolase (Sahh) r = 0.42; Mat2A r = 0.3 (all P < 0.05).
Table 3. Methionine metabolism, cell cycle, and lipid metabolism selected gene transcript levels
Table 4. Genes transcript levels at 6 d of age
Pyrosequencing
Out the 12 initially genes studied for transcript levels, we selected 7 genes for analysis of promoter methylation on the base of previously published data (Dnmt1,Citation22 PparαCitation23, Cpt1ACitation24) and/or relevance for methionine (Mtr, Mat2A, Sahh) and lipid metabolism pathways (Pparα, Cpt1A), and cell cycle (Cyclin D1). There were no detectable differences in methylation levels between genotypes or dietary groups, all promoters examined had expected low levels of DNA methylation ().
Table 5. Percent methylation of Mtr, Mat2A, Dnmt1, Sahh, Cpt1A, Pparα, and Cyclin D1 in samples from C3H and tx-j fetal livers
Discussion
WD is a monogenic disease that, through an established mouse model, offers the opportunity to study interactions between the genetic effects of Cu accumulation and the effects of maternal dietary supplementation with the methyl donor choline that modifies phenotype through epigenetic effects on gene expression. The present study provided several original findings on the effects of the WD genotype and maternal choline supplementation on both fetal and weanling hepatic Cu levels, global and gene-specific DNA methylation, and transcript levels. First, tx-j fetuses of dams receiving control diets presented markedly low hepatic Cu levels compared with C3H mice, which were associated with overall downregulation of transcript levels of selected genes related to methionine metabolism, DNA methylation, lipid metabolism, and liver cell regeneration. Second, reduced transcript levels of these genes in tx-j fetal livers were maintained at control levels by maternal dietary supplementation with the methyl donor choline and were correlated with the restored phenotype of body weight at 21 d. Third, maternal choline supplementation increased global DNA methylation in fetal livers of tx-j mice that was sustained in 21-d old mice. Lastly, the observation of dramatic reductions in Cyclin D1 expression in tx-j fetuses and 6-d mice that was prevented by maternal choline supplementation suggests that the uniform reductions in the transcript levels of other metabolic genes in the tx-j mice may be associated with a global reduction in cell growth and metabolism from the tx-j mutation that can be alleviated through dietary changes.
Addressing these four main observations, first, the finding of low Cu concentrations in tx-j fetal livers can be attributed to the low circulating serum ceruloplasmin in untreated tx-j dams with consequent lack of placental Cu transport.Citation25 Importantly, we observed that fetal liver Cu concentrations in the tx-j strain were even lower in the maternal choline supplemented than in the control diet group. In our previous study on adult tx-j mice, we found that betaine, a methyl group donor product of choline metabolism, was associated with reduced hepatic Cu concentration as well.Citation12 We do not have an explanation for this novel finding but it could be related to changes in methylation status and expression levels of an alternate Cu transporter. This observation is of concern given the well-recognized teratogenicity of Cu deficiency.Citation26
Regarding the second point, we previously observed a similar downregulation of Sahh, Srebp-1c, Grp78, Pparα, and Cpt1A transcripts in adult tx-j mice with hepatic Cu accumulation. However, these tx-j mice with more advanced liver disease and significant inflammation did not present any changes in gene expression after supplementation with choline derivative betaine.Citation12 By contrast, in the present study, tx-j fetal livers presented low hepatic Cu and responded to maternal choline feeding with significant changes in transcript levels. The different responses to the methyl donors betaine in the first experiment and choline in the current experiment support our previous hypothesis that inflammation, as observed in adult tx-j mice, creates a status of relative DNA hypomethylation that limits the response to methyl group supplementation.Citation12
There are several potential consequences of reduced gene expression in tx-j fetal livers. Downregulation of Dnmt1 at baseline was not associated with global DNA hypomethylation. However, increased DNA methylation after maternal choline treatment and the gene expression responses to choline treatment in tx-j mice suggest an underlying relative deficiency of methyl groups in this genotype. Also, the reduced transcript levels of Pparα and Cpt1A in fetal livers of tx-j mice may result in an increased risk for the development of fatty liver later in life. This would be in agreement with the fetal origin of adult disease hypothesis previously proposed for cardiovascular diseases potentially being primed by suboptimal nutrition during early development.Citation13 In addition, the reduction of Grp78 transcript levels may have direct effects on Cu metabolism as it has been shown that GRP78 participates in Cu homeostasis by forming complexes with Cu.Citation27
Third, the demonstration that maternal choline supplementation restored global levels of DNA methylation that were deficient in the tx-j model without a change to the methylation levels of specific dysregulated genes is consistent with genomic analyses of methylomesCitation28-Citation30 and global effects on methionine metabolism on DNA methylation.Citation31-Citation34 Although DNA hypermethylation of specific gene promoters is in general associated with gene repression, highly methylated promoters are rarely seen outside of imprinted genes, X-inactive genes, or tumor suppressor genes in cancer.Citation35 The mammalian genome is highly saturated with DNA methylation except at CpG-rich gene promoters.Citation28-Citation30,Citation36 Global methylation levels better reflect methionine metabolismCitation33,Citation34 and methylation levels outside of gene promoters and generally positively correlate with transcript levels.Citation30,Citation36-Citation38 In the present study, maternal choline supplementation of tx-j mice was associated with both increased global liver DNA methylation and increased transcript levels of genes involved in methionine and lipid metabolism in fetal livers, even though there were no significant differences in gene specific DNA methylation of any of the genes selected for pyrosequencing. Further, Cyclin D1 levels were restored to normal levels by maternal choline supplementation in the tx-j fetal livers and Cyclin D1 protein levels were significantly higher in weanlings tx-j mice livers suggesting that hepatocyte cell cycle differences are responsible for the restoration of tx-j body weights by maternal choline supplementation. The downregulation of gene expression in 6d pups, before cross-fostering, confirms the role of nutritional factors in the pre-symptomatic stages of this disease model. DNA damage and environmental stress are also associated with downregulation of Cyclin D1Citation39 and cell cycle. Of note, a previous study of the Atp7b −/− knock out mouse model of WD showed upregulation of cell cycle machinery related genes, including Cyclin G1 and G2, in adult mice with hepatic Cu accumulationCitation40 indicating a close relation between Cu levels and cell cycle regulation. In our model, maternal choline supplementation apparently provided the fetal liver the adequate conditions for the cells to respond to mitogenic signals. This is not surprising given that choline is an essential component of cell membranes, lipoproteins, and surfactants, and is crucial for liver and placenta metabolic activities.Citation41 Given that we did not observe any change in gene specific methylation, we hypothesize that choline provided an adequate environment for cell growth, with consequent increase in global DNA methylation and gene expression. Further studies are needed to investigate the potential correlations between our findings related to cell growth and DNA methylation with the well-described nuclear structural abnormalities observed both in WD patientsCitation42 and tx mice.Citation11,Citation43
The limitations of our study include the relatively short-term observations, and long-term experiments on the effect of gestational choline supplementation are needed. We explored gene specific methylation of 7 genes, which appear to be a good representation of gene regulated at the level of DNA methylation also during gestational life. Potentially, screening by methylome analyses might identify changes in methylation status of additional genes.
This is the first evidence that fetal liver metabolic transcript levels and weanling body weight is restored by maternal choline supplementation in a genetic mouse model of WD. However, our findings are in line with other reports on genetic diseases like Rett syndrome that causes mental retardation in girls and can be modified by perinatal choline supplementation.Citation44 The interaction of a genetic mutation with in utero nutritional factors and availability of methyl groups in the early phases of development demonstrate that phenotype of a genetic disease can be regulated by early nutritional and environmental factors.
Methods
Animal model and experimental protocols
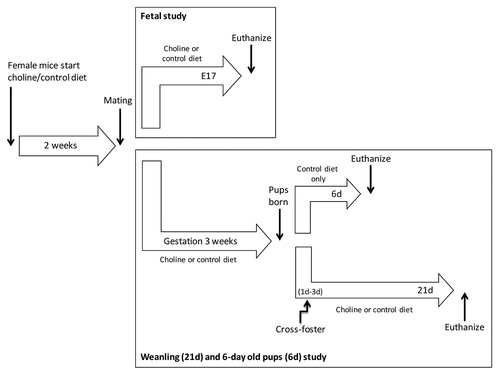
We used the C3HeB/FeJ-Atp7btx-J/J mouse (tx-j) model of WD with its background strain C3HeB/FeJ (C3H) as a control. The tx-j mouse model has a G712D missense mutation in the ATP7B Cu transporter gene which results in a phenotype similar to WD.Citation10,Citation11 Mice were obtained from our in-house colony that was developed from C3H breeder pairs and homozygous-affected tx-j breeder pairs purchased from The Jackson Laboratory (www.jax.org). The study design is diagrammed in . Control (choline 8 mmol/kg) (Dyets, Inc., #110098) or choline-supplemented (choline 36 mmol/kg) diet (Dyets, Inc., #110184) diet was fed to female C3H and tx-j mice 2 weeks before mating and continued throughout mating and pregnancy. Both diets contain the following mineral concentrations: Fe 35 mg/Kg and Cu 6 mg/Kg. In the first experiment, pregnant C3H and tx-j dams on each diet were euthanized via CO2 anesthesia followed by cervical dislocation on embryonic day 17. Their uteri were dissected and fetuses placed in cold L15 medium (Invitrogen, 11415-064). Fetal livers were collected and pooled, flash-frozen in liquid nitrogen, and stored at –80 °C for further analyses. In the second experiment, pregnant dams continued their diet treatments through birth and lactation to postpartum 21 d. Due to reduced concentrations of Cu in the tx-j dams’ milk, tx-j pups cannot be raised by their biological dams as they typically do not survive more than 7 d after birthCitation11 and must be cross-fostered to a dam with normal milk, in this case a C3H dam. All tx-j pups were cross-fostered after postpartum day 1–3 to a C3H dam who had given birth within 2 d of the tx-j pups she fostered. At 21 d, weanling livers of C3H and cross-fostered tx-j offspring were collected, flash-frozen in liquid nitrogen, and stored at –80 °C for further analyses. To study the potential effect of cross-fostering on gene expression postpartum, additional C3H and un-fostered tx-j pups were euthanized at 6 d. All mouse care and feeding protocols followed the guidelines of the American Association for Accreditation of Laboratory Animal Care and were reviewed and approved annually by the UC Davis Institutional Animal Care and Use Committee. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” (National Academy of Sciences, 1996) and published by the National Institutes of Health (NIH publication 86-23 revised 1985).
Figure 5. Diagram of study design. The study included a fetal and a 21d weanling + 6 d (6d) old pups part. C3H and tx-j dams received a control or choline supplemented diet 2 weeks before mating, and to embryonic day 17 when fetal livers were obtained for the experiments. Two additional groups of mice were euthanized at 6d, before cross-fostering, and at 21d. Tx-j mice have to be cross-fostered by C3H dams because tx-j dams milk is Cu deficient.
Hepatic copper and iron levels
Liver specimens of fetal and 21d livers were wet-ashed in 16 M nitric acid (JT Baker, 9598) and diluted in 0.1 M Ultrex nitric acid (JT Baker, 6901). The diluted digests were analyzed for Cu and Fe by inductively coupled plasma atomic emission spectroscopy (ICP-AES; Trace Scan, Thermo Electron Corp, Thermo Fisher Scientific). The ICP-AES was calibrated with multielement standards (Perkin Elmer, PE N9301721) diluted in 1 M Ultrex nitric acid. Fe was measured as there is evidence of its accumulation in animal models of WD related to hemolysis.Citation45 The Cu and Fe concentrations in bovine liver obtained from the National Institute of Standards and Technology were used as an external control on the method. Blanks were generated by the same method and their values were subtracted from the sample measurements.Citation46
Liver SAM and SAH by HPLC with fluorescence detection
50 mg of frozen liver tissue was homogenized in 0.4 ml of cold 0.5 N perchloric acid (Alfa Aesar, 33263) and then centrifuged for 10 min. at 14 000 rpm/4 °C. The supernatant was filtered through a 0.22µm syringe filter (Cole-Parmer, F2604-2) and frozen at –80 °C. HPLC analysis of SAM and SAH was performed within 4 weeks from tissue harvest to ensure sample stability.Citation47
Histology and immunohistochemistry
Liver histopathology was studied in 21-d pups (3–4 per genotype and diet) and was evaluated independently by quantitative scoring in blinded fashion using computerized software. Scoring followed published quantitative criteria for hepatocyte lipid accumulation, lobular inflammation, necrosis, fibrosis, and mitosis.Citation48 Liver sections were stained with anti-Cyclin D1 mouse monoclonal primary antibody (Abcam, ab16663) and secondary antibody conjugated with streptavidin peroxidase followed by chromogen substrate detection was used to detect binding of the primary antibody. The slides were examined using Nikon NIS Elements D morphometric software and the percentage of nuclei positive for Cyclin D1 was measured.
Global DNA methylation
DNA was isolated from fetal and 21d livers using the DNeasy Blood and Tissue Kit (QIAGEN, 69504). The concentrations and purity of extracted DNA were determined by measuring the absorbency at A260 and A280. Relative methylation dot blots were performed as previously described.Citation12,Citation49 Briefly, 50 ng of genomic DNA from each specimen was alkaline-denatured and blotted in triplicate onto a nitrocellulose membrane then bound to the membrane via UV cross-linking. Each blot was blocked with Odyssey Blocking Buffer (LI-COR, 927-40000) and incubated in Blocking Buffer + 0.1% Tween-20 with anti-5-methylcitidine (Eurogentec, BI-MECY-0100) overnight at 4 °C. After washing in 1x PBS + 0.2% Tween-20, the blot was incubated in Blocking Buffer + 0.1% Tween-20 with IRDye 680RD secondary antibody (LI-COR, 926-68072) for 1 h at room temperature. The blot was imaged using a LI-COR Odyssey Imager (LI-COR). Next, each blot was washed in 1x PBS + 0.1% Tween-20 followed by equilibration in PerfectHyb Plus Hybridization Buffer (Sigma, H&033) at 42 °C. Biotin-labeled gDNA was hybridized overnight at 42 °C. The blots were incubated with LI-COR Streptavidin IRDye 800CW secondary antibody (LI-COR, 926–32230) in Blocking Buffer + 0.1%Tween-20 for 1 h at room temperature. The blot was imaged again on the LI-COR Odyssey Imager and integrated intensities were quantified using Odyssey software with methylation signal normalized to total DNA signal.
RNA isolation, cDNA synthesis, and Quantitative Real-Time PCR
Total RNA was isolated from frozen liver specimens using the RNeasy Mini Kit (QIAGEN, 74104). The concentrations and purity of extracted RNA were determined by measuring the absorbency at A260 and A280. Lithium chloride precipitation was performed to remove any remnants of genomic DNA and total RNA integrity checked via 1% ethidium bromide agarose gel analysis. Extracted total RNA was stored at –80 °C until further use. Reverse transcription was performed using the SuperScript III First-Strand cDNA synthesis kit (Invitrogen, 18080-051). Primers for mouse cDNA sequences were designed using Primer Express 3.0 (Applied Biosystems) and blasted against the mouse genomic + transcript database using NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to check primer specificity (Table S1). The amplification efficiencies (E) of all assays were calculated from the slope of a standard curve generated in triplicate using the formula E = 10(-1/slope) -1. All assays had an efficiency of >95%. Primer specificity was verified by melt curve analysis and agarose gel electrophoresis. Reactions were performed in triplicate for all cDNA samples using SYBR Green I dye detection on the AB ViiA 7 Real-Time PCR System (Applied Biosystems). Reaction conditions were 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Cycle threshold (Ct) values were exported with a threshold of 0.15 based on the housekeeping gene GAPDH. The relative expression level of a target gene in a particular sample was calculated using the equation 2−ΔΔCt, where ΔΔCt = ΔCt(sample) − ΔCt(calibrator). Therefore, all gene transcriptions were expressed as an n-fold difference relative to the calibrator. The calibrator was the mean value of C3H fetuses or pups from dams on control diet.
DNA methylation of selected genes via pyrosequencing analysis
Pyrosequencing analyses of Mtr, Mat2A, Dnmt1, Sahh, Cpt1A, Pparα, and Cyclin D1 methylation differences were conducted on our mouse fetal liver samples. Using the EMBOSS online CpG island finder tool (http://www.ebi.ac.uk/Tools/seqstats/emboss_newcpgreport/) with minimum parameters (window = 100, length = 100, %GC = 50, Obs/Exp = 0.6), CpG islands were identified in the promoter region of each listed gene. Primers were designed using PyroMark Assay Design 2.0 software (QIAGEN); those selected for testing had a software-generated score >70. Bisulfite treatment of genomic DNA was performed using the EZ DNA Methylation-Direct Kit (Zymo Research, D5020). PCR amplifications of each target region were performed using the PyroMark PCR Kit (QIAGEN, 978703). PCR product was purified using Streptavidin Sepharose High Performance Beads (GE Healthcare, 17-5113-01) and prepared for pyrosequencing using the PyroMark Q24 Vacuum Workstation (QIAGEN). Samples were sequenced in triplicate on a PyroMark Q24 Pyrosequencer (QIAGEN) using PyroMark Gold Q24 Reagents (QIAGEN, 970802) and methylation levels were analyzed using PyroMark Q24 software.
Statistical analyses
Statistical analyses were performed using SAS statistical software, version 9.3 (SAS Institute Inc.). Descriptive statistics were calculated for each measurement by treatment group for each genotype. Prior to statistical analyses, the distributions of measurements were assessed to determine whether they were approximately normal. If necessary, natural log transformations were applied to the data in order to improve the normality of residuals and homoscedasticity of errors as appropriate, and the log-transformed values were used in the subsequent analyses. Inter-group comparisons of means were performed by analysis of covariance (ANCOVA), adjusted for each litter size and gender. Multiple comparisons were controlled by the Bonferroni correction method where appropriate. The Pearson correlation coefficient and its P value for significance of correlation were calculated to assess the magnitude and direction of an association between two given measures. Statistical significance was determined at a two-sided P value < 0.05.
| Abbreviations: | ||
| WD | = | Wilson disease |
| Cu | = | copper |
| SAM | = | S-adenosylmethionine |
| tx-j | = | toxic milk mice |
| 6d | = | 6 days old mice |
| 21d | = | 21 days old mice |
| Fe | = | iron |
| SAH | = | S-adenosylhomocysteine |
| SREBP1c | = | Sterol Regulatory Element-Binding Protein |
| PPAR alpha | = | peroxisome proliferator-activated receptor alpha |
| DNMT | = | DNA methyltransferase |
| SAHH | = | S-adenosylhomocysteine hydrolase |
| MAT | = | methionine adenosyltransferase |
Additional material
Download Zip (94.7 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This research was supported by grant number K08DK084111 from the National Institute of Diabetes and Digestive and Kidney Diseases, by Department and Divisional funds, by University of California Davis Center for Health and Nutrition Research (CHNR), by the National Center for Research Resources (NIH) through grant #UL1 RR024146 (to Medici V), by NIH 1R01ES021707 from the National Institute of Environmental Health Sciences (to LaSalle JM), by a Veterans Health Administration Biomedical Laboratory Research and Development National Merit Review grant award I01 BX001155 (to Kharbanda KK), and by NIH/NIAAA P50–011999 Morphology core (to French SW). Medici V is a full member of the University of California San Francisco Liver Center (Liver Center grant number P30 DK026743). The content is the sole responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journal/epigenetics/article/27110
Related Research Data
References
- Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet 1993; 5:327 - 37; http://dx.doi.org/10.1038/ng1293-327; PMID: 8298639
- Wilson DC, Phillips MJ, Cox DW, Roberts EA. Severe hepatic Wilson’s disease in preschool-aged children. J Pediatr 2000; 137:719 - 22; http://dx.doi.org/10.1067/mpd.2000.108569; PMID: 11060541
- Ala A, Borjigin J, Rochwarger A, Schilsky M. Wilson disease in septuagenarian siblings: Raising the bar for diagnosis. Hepatology 2005; 41:668 - 70; http://dx.doi.org/10.1002/hep.20601; PMID: 15723329
- Lee BH, Kim JH, Lee SY, Jin HY, Kim KJ, Lee JJ, Park JY, Kim GH, Choi JH, Kim KM, et al. Distinct clinical courses according to presenting phenotypes and their correlations to ATP7B mutations in a large Wilson’s disease cohort. Liver Int 2011; 31:831 - 9; http://dx.doi.org/10.1111/j.1478-3231.2011.02503.x; PMID: 21645214
- Folhoffer A, Ferenci P, Csak T, Horvath A, Hegedus D, Firneisz G, Osztovits J, Kosa JP, Willheim-Polli C, Szonyi L, et al. Novel mutations of the ATP7B gene among 109 Hungarian patients with Wilson’s disease. Eur J Gastroenterol Hepatol 2007; 19:105 - 11; http://dx.doi.org/10.1097/01.meg.0000223904.70492.0b; PMID: 17272994
- Caca K, Ferenci P, Kühn HJ, Polli C, Willgerodt H, Kunath B, Hermann W, Mössner J, Berr F. High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J Hepatol 2001; 35:575 - 81; http://dx.doi.org/10.1016/S0168-8278(01)00219-7; PMID: 11690702
- Kegley KM, Sellers MA, Ferber MJ, Johnson MW, Joelson DW, Shrestha R. Fulminant Wilson’s disease requiring liver transplantation in one monozygotic twin despite identical genetic mutation. Am J Transplant 2010; 10:1325 - 9; http://dx.doi.org/10.1111/j.1600-6143.2010.03071.x; PMID: 20346064
- Członkowska A, Gromadzka G, Chabik G. Monozygotic female twins discordant for phenotype of Wilson’s disease. Mov Disord 2009; 24:1066 - 9; http://dx.doi.org/10.1002/mds.22474; PMID: 19306278
- Senzolo M, Loreno M, Fagiuoli S, Zanus G, Canova D, Masier A, Russo FP, Sturniolo GC, Burra P. Different neurological outcome of liver transplantation for Wilson’s disease in two homozygotic twins. Clin Neurol Neurosurg 2007; 109:71 - 5; http://dx.doi.org/10.1016/j.clineuro.2006.01.008; PMID: 16545904
- Coronado V, Nanji M, Cox DW. The Jackson toxic milk mouse as a model for copper loading. Mamm Genome 2001; 12:793 - 5; http://dx.doi.org/10.1007/s00335-001-3021-y; PMID: 11668395
- Roberts EA, Robinson BH, Yang S. Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Mol Genet Metab 2008; 93:54 - 65; http://dx.doi.org/10.1016/j.ymgme.2007.08.127; PMID: 17981064
- Medici V, Shibata NM, Kharbanda KK, LaSalle JM, Woods R, Liu S, Engelberg JA, Devaraj S, Török NJ, Jiang JX, et al. Wilson’s disease: changes in methionine metabolism and inflammation affect global DNA methylation in early liver disease. Hepatology 2013; 57:555 - 65; http://dx.doi.org/10.1002/hep.26047; PMID: 22945834
- Barker DJ, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol 2002; 31:1235 - 9; http://dx.doi.org/10.1093/ije/31.6.1235; PMID: 12540728
- Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr 2007; 27:363 - 88; http://dx.doi.org/10.1146/annurev.nutr.27.061406.093705; PMID: 17465856
- Mehedint MG, Zeisel SH. Choline’s role in maintaining liver function: new evidence for epigenetic mechanisms. Curr Opin Clin Nutr Metab Care 2013; 16:339 - 45; http://dx.doi.org/10.1097/MCO.0b013e3283600d46; PMID: 23493015
- Carlin J, George R, Reyes TM. Methyl donor supplementation blocks the adverse effects of maternal high fat diet on offspring physiology. PLoS One 2013; 8:e63549; http://dx.doi.org/10.1371/journal.pone.0063549; PMID: 23658839
- Sie KK, Li J, Ly A, Sohn KJ, Croxford R, Kim YI. Effect of maternal and postweaning folic acid supplementation on global and gene-specific DNA methylation in the liver of the rat offspring. Mol Nutr Food Res 2013; 57:677 - 85; http://dx.doi.org/10.1002/mnfr.201200186; PMID: 23463647
- Haggarty P, Hoad G, Campbell DM, Horgan GW, Piyathilake C, McNeill G. Folate in pregnancy and imprinted gene and repeat element methylation in the offspring. Am J Clin Nutr 2013; 97:94 - 9; http://dx.doi.org/10.3945/ajcn.112.042572; PMID: 23151531
- Davison JM, Mellott TJ, Kovacheva VP, Blusztajn JK. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J Biol Chem 2009; 284:1982 - 9; http://dx.doi.org/10.1074/jbc.M807651200; PMID: 19001366
- Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest 2000; 105:1067 - 75; http://dx.doi.org/10.1172/JCI8814; PMID: 10772651
- Witzel II, Koh LF, Perkins ND. Regulation of cyclin D1 gene expression. Biochem Soc Trans 2010; 38:217 - 22; http://dx.doi.org/10.1042/BST0380217; PMID: 20074063
- Kovacheva VP, Mellott TJ, Davison JM, Wagner N, Lopez-Coviella I, Schnitzler AC, Blusztajn JK. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J Biol Chem 2007; 282:31777 - 88; http://dx.doi.org/10.1074/jbc.M705539200; PMID: 17724018
- Wang L, Chen L, Tan Y, Wei J, Chang Y, Jin T, Zhu H. Betaine supplement alleviates hepatic triglyceride accumulation of apolipoprotein E deficient mice via reducing methylation of peroxisomal proliferator-activated receptor alpha promoter. Lipids Health Dis 2013; 12:34; http://dx.doi.org/10.1186/1476-511X-12-34; PMID: 23497035
- Shen Y, Chow J, Wang Z, Fan G. Abnormal CpG island methylation occurs during in vitro differentiation of human embryonic stem cells. Hum Mol Genet 2006; 15:2623 - 35; http://dx.doi.org/10.1093/hmg/ddl188; PMID: 16870691
- Chu YL, Sauble EN, Cabrera A, Roth A, Ackland ML, Mercer JF, Linder MC. Lack of ceruloplasmin expression alters aspects of copper transport to the fetus and newborn, as determined in mice. Biometals 2012; 25:373 - 82; http://dx.doi.org/10.1007/s10534-011-9509-3; PMID: 22080201
- Uriu-Adams JY, Scherr RE, Lanoue L, Keen CL. Influence of copper on early development: prenatal and postnatal considerations. Biofactors 2010; 36:136 - 52; PMID: 20232410
- Qian Y, Meng B, Zhang X, Zheng Y, Taylor R, Tiffany-Castiglioni E. HSPA5 forms specific complexes with copper. Neurochem Res 2013; 38:321 - 9; http://dx.doi.org/10.1007/s11064-012-0923-x; PMID: 23161089
- Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, Bestor TH. Large-scale structure of genomic methylation patterns. Genome Res 2006; 16:157 - 63; http://dx.doi.org/10.1101/gr.4362006; PMID: 16365381
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science 2007; 315:1141 - 3; http://dx.doi.org/10.1126/science.1136352; PMID: 17322062
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315 - 22; http://dx.doi.org/10.1038/nature08514; PMID: 19829295
- Lee DH, Jacobs DR Jr., Porta M. Hypothesis: a unifying mechanism for nutrition and chemicals as lifelong modulators of DNA hypomethylation. Environ Health Perspect 2009; 117:1799 - 802; http://dx.doi.org/10.1289/ehp.0900741; PMID: 20049195
- Blusztajn JK, Mellott TJ. Choline nutrition programs brain development via DNA and histone methylation. Cent Nerv Syst Agents Med Chem 2012; 12:82 - 94; http://dx.doi.org/10.2174/187152412800792706; PMID: 22483275
- James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr 2004; 80:1611 - 7; PMID: 15585776
- James SJ, Melnyk S, Jernigan S, Hubanks A, Rose S, Gaylor DW. Abnormal Transmethylation/transsulfuration Metabolism and DNA Hypomethylation Among Parents of Children with Autism. J Autism Dev Disord 2008; 38:1976; http://dx.doi.org/10.1007/s10803-008-0614-2; PMID: 19011973
- LaSalle JM, Powell WT, Yasui DH. Epigenetic layers and players underlying neurodevelopment. Trends Neurosci 2013; 36:460 - 70; http://dx.doi.org/10.1016/j.tins.2013.05.001; PMID: 23731492
- Schroeder DI, Blair JD, Lott P, Yu HO, Hong D, Crary F, Ashwood P, Walker C, Korf I, Robinson WP, et al. The human placenta methylome. Proc Natl Acad Sci U S A 2013; 110:6037 - 42; http://dx.doi.org/10.1073/pnas.1215145110; PMID: 23530188
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013; 341:1237905; http://dx.doi.org/10.1126/science.1237905; PMID: 23828890
- Schroeder DI, Lott P, Korf I, LaSalle JM. Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome Res 2011; 21:1583 - 91; http://dx.doi.org/10.1101/gr.119131.110; PMID: 21784875
- Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer 2007; 6:24; http://dx.doi.org/10.1186/1476-4598-6-24; PMID: 17407548
- Huster D, Purnat TD, Burkhead JL, Ralle M, Fiehn O, Stuckert F, Olson NE, Teupser D, Lutsenko S. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J Biol Chem 2007; 282:8343 - 55; http://dx.doi.org/10.1074/jbc.M607496200; PMID: 17205981
- Zeisel SH. Nutrition in pregnancy: the argument for including a source of choline. Int J Womens Health 2013; 5:193 - 9; http://dx.doi.org/10.2147/IJWH.S36610; PMID: 23637565
- Sternlieb I. Mitochondrial and fatty changes in hepatocytes of patients with Wilson’s disease. Gastroenterology 1968; 55:354 - 67; PMID: 5675366
- Biempica L, Rauch H, Quintana N, Sternlieb I. Morphologic and chemical studies on a murine mutation (toxic milk mice) resulting in hepatic copper toxicosis. Lab Invest 1988; 59:500 - 8; PMID: 2845190
- Nag N, Berger-Sweeney JE. Postnatal dietary choline supplementation alters behavior in a mouse model of Rett syndrome. Neurobiol Dis 2007; 26:473 - 80; http://dx.doi.org/10.1016/j.nbd.2007.02.003; PMID: 17395475
- Kato J, Kobune M, Kohgo Y, Sugawara N, Hisai H, Nakamura T, Sakamaki S, Sawada N, Niitsu Y. Hepatic iron deprivation prevents spontaneous development of fulminant hepatitis and liver cancer in Long-Evans Cinnamon rats. J Clin Invest 1996; 98:923 - 9; http://dx.doi.org/10.1172/JCI118875; PMID: 8770863
- Clegg MS, Keen CL, Lönnerdal B, Hurley LS. Influence of ashing techniques on the concentration of trace elements in animal tissues. I: Wet ashing. Biol Trace Elem Res 1981; 3:107 - 15; http://dx.doi.org/10.1007/BF02990451
- Fu W, Dudman NP, Perry MA, Young K, Wang XL. Interrelations between plasma homocysteine and intracellular S-adenosylhomocysteine. Biochem Biophys Res Commun 2000; 271:47 - 53; http://dx.doi.org/10.1006/bbrc.2000.2587; PMID: 10777679
- French SW, Nash J, Shitabata P, Kachi K, Hara C, Chedid A, Mendenhall CL. Pathology of alcoholic liver disease. VA Cooperative Study Group 119. Semin Liver Dis 1993; 13:154 - 69; http://dx.doi.org/10.1055/s-2007-1007346; PMID: 8393214
- Woods R, Vallero RO, Golub MS, Suarez JK, Ta TA, Yasui DH, Chi LH, Kostyniak PJ, Pessah IN, Berman RF, et al. Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2308 mutation. Hum Mol Genet 2012; 21:2399 - 411; http://dx.doi.org/10.1093/hmg/dds046; PMID: 22343140