
Abstract
DNA methylation is a hallmark of genomic imprinting and differentially methylated regions (DMRs) are found near and in imprinted genes. Imprinted genes are expressed only from the maternal or paternal allele and their normal balance can be disrupted by uniparental disomy (UPD), the inheritance of both chromosomes of a chromosome pair exclusively from only either the mother or the father. Maternal UPD for chromosome 7 (matUPD7) results in Silver-Russell syndrome (SRS) with typical features and growth retardation, but no gene has been conclusively implicated in SRS. In order to identify novel DMRs and putative imprinted genes on chromosome 7, we analyzed eight matUPD7 patients, a segmental matUPD7q31-qter, a rare patUPD7 case and ten controls on the Infinium HumanMethylation450K BeadChip with 30 017 CpG methylation probes for chromosome 7. Genome-scale analysis showed highly significant clustering of DMRs only on chromosome 7, including the known imprinted loci GRB10, SGCE/PEG10, and PEG/MEST. We found ten novel DMRs on chromosome 7, two DMRs for the predicted imprinted genes HOXA4 and GLI3 and one for the disputed imprinted gene PON1. Quantitative RT-PCR on blood RNA samples comparing matUPD7, patUPD7, and controls showed differential expression for three genes with novel DMRs, HOXA4, GLI3, and SVOPL. Allele specific expression analysis confirmed maternal only expression of SVOPL and imprinting of HOXA4 was supported by monoallelic expression. These results present the first comprehensive map of parent-of-origin specific DMRs on human chromosome 7, suggesting many new imprinted sites.
Introduction
Methylation of the 5′-cytosines in CpG dinucleotides is an epigenetic mark that can affect gene expression. Clusters (approximately 1 kb) of multiple CpGs form CpG islands (CGIs), which are usually unmethylated when located at gene transcription start sites (TSS), but are also found within coding regions, at 3′ ends, as well as in intra- and inter-genic regions.Citation1 Methylation near TSSs blocks transcription initiation, but methylation within gene bodies may increase expression and influence splicing.Citation1 Methylation of promoter CGIs is rare and found in genes where expression is permanently repressed, as for imprinted genes, genes on the inactive X chromosome, and genes exclusively expressed in germ cells.Citation1 Moreover, CpG methylation is a hallmark of imprinted genes, which are expressed exclusively from the maternal or paternal allele and most of which play crucial roles in growth and development.Citation2 Imprinted genes generally reside in clusters where an imprinting control center (ICR) and other additional differentially methylated regions (DMRs) control the expression of the imprinted genes.Citation3 In human, approximately 32 imprinting clusters and over 70 imprinted genes have been described (see Catalogue of Imprinted Genes and Parent-of-origin Effects in Humans and Animals, http://igc.otago.ac.nz).Citation4
The normal balance of imprinted genes can be disrupted by uniparental disomy (UPD), the inheritance of both chromosomes of a chromosome pair exclusively either from the mother (maternally, matUPD) or the father (paternally, patUPD).Citation5 UPD can lead to imprinting syndromes where loss or gain of methylation at a specific DMR/ICR leads to the syndrome phenotype, e.g., patUPD for chromosome 6 (patUPD6) in transient neonatal diabetes mellitus (TNDM [MIM 601410]), matUPD7 in Silver-Russell Syndrome (SRS [MIM 180860]), patUPD11 in Beckwith-Wiedemann syndrome (BWS [MIM 130650]), matUPD14 and patUPD14 [MIM 608149] as well as patUPD15 in Angelman syndrome (AS [MIM 105830]), and matUPD15 in Prader-Willi syndrome (PWS [MIM 176270]).Citation5-Citation10 However, aberrant methylation is not always confined to only one locus, and multilocus loss of methylation (LOM) at multiple imprinted loci has been reported for TNDM, PWS, and a matUPD7q patient with also hypomethylation at the paternally imprinted DLK1/MEG3 locus at 14q32 (MIM 176290).Citation11,Citation12
MatUPD7 is found in approximately 10% of SRS patients and hypomethylation of the ICR1 of IGF2-H19 (MIM 147470, MIM 103280) on chromosome 11p15 in 20–60%.Citation13,Citation14 SRS is characterized by severe pre- and postnatal growth restriction, macrocephaly, skeletal asymmetry, a triangular face and other variable dysmorphic features.Citation15 Conversely, patUPD7 does not affect growth and development.Citation16-Citation18 An imprinted gene on chromosome 7 has been suggested to cause SRS, but none of the known imprinted genes at three suggested domains (7p12: GRB10 [MIM 601523], 7q21.3: TFPI2 [MIM 600033], SGCE [MIM 604149], PEG10 [MIM 609810], PPP1R9A [MIM 602468], and 7q32.2: CPA4 [MIM 607635], MEST [MIM 601029], MESTIT1 [MIM 607794], COPG2IT1 [MIM 610581], KLF14 [MIM 609393]) have been conclusively linked with SRS.Citation19-Citation23 Segmental maternal duplications spanning the imprinted gene GRB10 on 7p12.2 and rare cases of segmental matUPD7q31-qter, matUPD7q, and a mosaic matUPD7q21-qter in SRS patients have however narrowed down the candidate SRS regions.Citation24-Citation28
Imprinted genes can be identified through parent-of-origin specific difference in methylation and expression.Citation29 High-throughput genome wide methylation profiling has enabled systematic identification of multiple new loci.Citation30 Whole genome methylation has already been used to identify new imprinted genes by profiling between matUPD15 and patUPD15 cases and in rare reciprocal genome-wide UPD samples.Citation31,Citation32 Global methylation arrays with 1505 and 27 500 CpG sites in SRS patients with/without ICR1 hypomethylation at IGF2-H19 did not reveal any significant common associations outside the H19 promoter.Citation33,Citation34 However, individual ICR1 hypomethylated patients showed increased methylation changes at separate loci.Citation34 No matUPD7 patients were included in these studies and to our knowledge high-throughput methylation profiling of matUPD7 has not been reported.
To identify novel DMRs on chromosome 7, we compared DNA methylation status of matUPD7 cases to controls and a rare patUPD7 case with the Illumina Infinium HumanMethylation450K BeadChip methylation assay.Citation35 We found 17 DMRs on chromosome 7, of which 14 are novel and three are the known DMR/ICRs of GRB10 and the SGCE/PEG10 and PEG/MEST clusters. Imprinted expression was suggestive for three genes with novel DMRs, HOXA4 (MIM 142953), GLI3 (MIM 165240), and SVOPL (MIM 611700) by qRT-PCR. Allele specific expression of SVOPL confirmed it as a novel imprinted gene and monoallelic expression of HOXA4 supported imprinting. These results present to our knowledge the first comprehensive map of parent-of origin specific DMRs on human chromosome 7, suggesting novel imprinted domains.
Results
Genome-wide methylation in UPD7
To identify DMRs between maternal and paternal chromosomes 7, we performed genome-wide comparisons of the methylation of individual CpG sites between nine matUPD7 cases, including one segmental matUPD7q31-qter, ten controls and one patUPD7 case. We used the Infinium HumanMethylation450K BeadChip (Illumina), which measures methylation of 485 512 individual CpG sites, of which 30 017 are located on chromosome 7. Comparison between matUPD7 and controls showed that the differentially methylated CpGs mapped exclusively to chromosome 7 (), as expected for matUPD7. None of the CpGs outside chromosome 7 exceeded the genome-wide significance level (P = 5 × 10−8), but single CpGs exceeding suggestive significance level (P = 1 × 10−5) were observed on chromosomes 2, 4, 5, 6, 14, 16, 17, and 20. None of these CpGs coincide with known imprinted gene clusters or genes previously reported to show LOM in SRS patients with H19 hypomethylation or other syndromes.Citation11,Citation33,Citation36,Citation37
Figure 1. Manhattan plot of the differentially methylated CpG sites between matUPD7 and controls. The black line indicates the genome-wide significance level of P = 5 × 10−8 and the gray line indicates the suggestive significance level of P = 1 × 10−5.
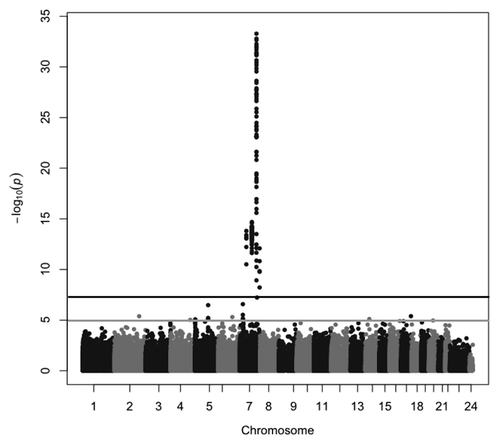
Comparing the single patUPD7 sample with controls, the strong clustering of differentially methylated CpGs was also observed on chromosome 7. Because only a single patUPD7 sample was available, the statistical analysis was disturbed by noise, and scattered CpGs above the significance threshold emerged genome-wide (data not shown). Although the information provided by the patUPD7 had limitations in statistical analyses, the sample was highly informative when analyzing imprinted loci in greater detail.
Differentially methylated regions on chromosome 7
We screened chromosome 7 for DMRs by using a step-by-step filtering process for all 30 017 CpG sites on chromosome 7 (). The process included two separate tracks, one screening for maternally hypermethylated CpGs and the other for maternally hypomethylated CpGs. The first step of the filtering process was based on identifying a pattern where the median M-value of the different groups (matUPD7, controls, and patUPD7) would differ in the way that would be expected for an imprinted locus, i.e., methylation in controls is midway and matUPD7s and patUPD7 diverge in opposite directions. For maternally hypermethylated CpGs, we set the filter to include all CpGs where the median M-value of matUPD7s was larger than that of the controls, and the median M-value of the controls was larger than that of the patUPD7. For the maternally hypomethylated genes, we set the filter to include all CpGs where the median M-value of the patUPD7 was larger than that of the controls, and the median M-value of the controls was larger than that of the matUPD7s. Step two excluded all CpGs where the methylation difference between matUPD7s and controls did not reach nominal significance (P value < 0.05). A nominal threshold was applied in order to avoid false negative findings. The significance level of differential methylation between controls and patUPD7 was disregarded for having only a single patUPD7 sample. The third step was set to include only those CpGs where a consecutive row of three or more CpGs had passed steps one and two in the filtering process. We excluded single and only two adjacent significant CpGs because these were judged more likely to be false positives. We excluded CpGs with poor signals and chose only adjacent CpGs with a good signal across the entire locus to increase the likelihood of obtaining true positive results. Our strategy may have left some loci undetected, but should have a low false positive rate.
Figure 2. Schematic presentation of the filtering process for the DMRs.
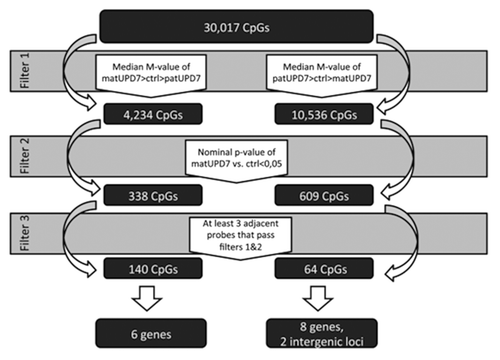
We found 338 single maternally hypermethylated CpGs scattered along chromosome 7 and 150 CpGs with at least two adjacent CpGs clustered to nine genes (MEST, SGCE, GRB10, HTR5A [MIM 601305], RPS2P32, SVOPL, IQCE, HOXA5 [MIM 142952], and HOXA6 [MIM 142951]; note that six of these genes also contained a stretch of three or more consecutive CpGs with differential methylation) and two intergenic regions (Table S1). For maternally hypomethylated CpGs, we found 609 single CpGs and 98 adjacent CpGs in 20 genes and seven intergenic regions (Tables S2 and S3).
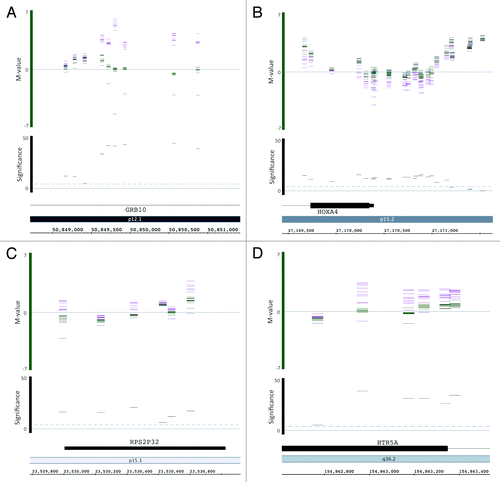
We found significant differential methylation that passed our three step filtering process for 204 CpGs at 17 DMRs/CGIs spread along chromosome 7, localizing to 14 genes and two intragenic regions at long non-coding RNAs (lncRNA) (; Tables S4 and S5; ). The majority of the DMRs were maternally hypomethylated (11/17, 65%) and six maternally hypermethylated (; Tables S4 and S5; ). As expected, the three known DMRs of the imprinted domains of GRB10 on 7p12.2, SGCE/PEG10 on 7q21.3, and MEST/MESTIT1 on 7q32 passed our filter ().Citation23,Citation38,Citation39 The well-conserved CGI2 upstream of GRB10 was entirely differentially methylated, with four CpGs encompassing the CGI2 and two CpGs in the North shore covering 113 bp (). The DMR in the promoter of MEST has been proposed to act as an ICR for the whole imprinted cluster spanning from CPA4 to KLF14.Citation22,Citation40 We identified 63 maternally hypermethylated CpGs spanning the entire MEST ICR, with 50 over the CGI and 13 CpGs located to the North shore covering 793 bp (). We found 55 maternally hypermethylated CpGs in the SGCE/PEG10 ICR with 41 located in the CGI and 14 in the South shore covering 715 bp ( and ). The SGCE/PEG10 CGI was not completely differentially methylated as 662 bp in the 5′ end lacked a DMR. CGI shores are defined as the region 0–2 kb upstream (North) or downstream (South) from the CGIs and shelves as the region 2–4 kb from the CGIs (Illumina, based on UCSC predictions).
Table 1. DMRs identified on chromosome 7
Figure 3. DMRs identified on chromosome 7. The DMRs identified in this study are shown on the left of the chromosome 7 ideogram. Known imprinted genes are highlighted in bold and predicted imprinted genes are highlighted in italics right of the chromosome 7 ideogram.
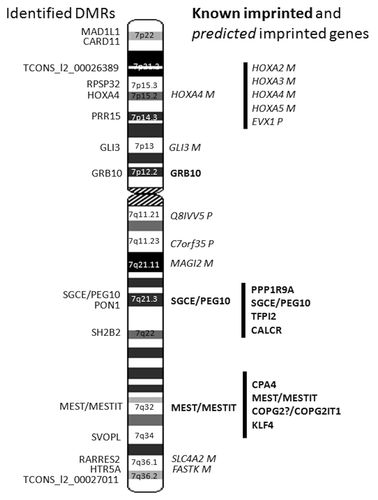
Table 2. Differential expression of genes and location of DMRs in relation to gene expression regulatory elements
Altogether ten DMRs located to genes with no prior imprinting status, two to previously predicted imprinted genes (HOXA4 and GLI3),Citation41 and one to PON1 (MIM 168820), with a disputed imprinted status (http://igc.otago.ac.nz) (). The DMR of PON1 was the only new DMR found in a known imprinting cluster (SGCE/PEG10). Previously, only one DMR, in an ICR situated between PEG10 and SGCE, has been found in this cluster out of all the CGIs ranging from CALCR (MIM 114131) to PPP1R9A (MIM 602468).Citation23,Citation42
The intergenic DMR at 7p21.1 partially overlapped the promoter and 5′ region of a possible long non-coding RNA (lncRNA), defined as a transcript of uncertain coding potential (TUCP) by the UCSC (University of California, Santa Cruz) genome browser (http://genome.ucsc.edu/) (TCONS_l2_00026389, 710 bp at 16 625 596–16 626 306 bp) and the second intergenic DMR at 7q36.2 was located 659 bp upstream of the TSS of another TUCP (TCONS_I2_00027011, 135 475 bp at 156 264 552–156 400 027 bp). Several imprinting clusters contain at least one lncRNA located in the proximity or partially overlapping a protein-coding gene, e.g., MESTIT1 overlapping MEST at the 7q32 imprinted cluster.Citation43,Citation44 Imprinted lncRNAs, e.g., Air (antisense Igf2r RNA, [MIM 604893]) and Kcnq1ot1 (Kcnq1 overlapping transcript 1, [MIM 604115]), have also been shown to play a major role in silencing multiple genes in the imprinted clusters. The ICR can lie within the lncRNA, a few kb upstream of the promoter or at the lncRNA promoter.Citation43
Interestingly, RPS2P32 at 7p15.3 is itself a 1025 bp lncRNA, with the maternally hypermethylated DMR located to the TSS and gene body (). MAD1L1 (MIM 602686) at 7p22 overlaps in its 3′ end with the antisense lncRNA AK127048 (4632 bp at 1 884 936–1 889 567 bp). HTR5A and its DMR overlap with an uncharacterized non-coding RNA LOC100128264 (4489 bp at 154 858 779–154 863 267 bp) and HOXA4 overlaps with an antisense hypothetical protein LOC285943 (6842 bp at 27 161 538–27 168 379 bp). We checked up to 500 kb up- and down-stream of each DMR for lncRNAs on the UCSC browser and found at least two up to 28 lncRNAs and TUCPs for each (not analyzed further here). The two closest protein coding genes of the intergenic DMR at 7p21.1 were LRRC72 located 5 kb upstream at 16 566 505–16 621 114 bp and ANKMY2 13 kb downstream at 16 639 401–16 685 442 bp. The two closest protein coding genes of the intergenic DMR at 7q36.2 are RNF32 (MIM 610241) 32 kb downstream at 156 433 353–156 469 820 bp and LMBR1 (MIM 605522) 72 kb also downstream at 156 473 570–156 589 186 bp. The closest protein coding genes to RPS2P32 are IGF2BP3 (MIM 608259) 20 kb upstream at 23 349 828–23 509 995 bp and TRA2A (MIM 602718) 13 kb downstream at 23 544 401–23 571 656 bp.
The lengths of the DMRs ranged from 196 bp (GLI3) to 3164 bp (MEST/MESTIT1) (). All significant CpGs defining DMRs located primarily to CGIs and their shores (). For MAD1L1 and GLI3 all significant CpGs located only to shores and the DMR for CARD11 (MIM 607210) did not hit a predicted CGI nor the shores or shelves of a CGI. SH2B2 (MIM 605300) had two separate DMRs in two independent CGIs. Otherwise, all genes had a DMR located to a single CGI and its shores, but none extended into the shelves.
Differentially methylated regions and gene regulatory elements
The location of a DMR in regard to, e.g., promoters, enhancers, TSSs, and CTCF binding sites may affect gene expression.Citation1,Citation45 The identified DMRs were predominantly in the 5′ UTRs, TSSs, and first exons of the genes (). Only for two genes (MAD1L1, SH2B2) the DMRs located to the body of the gene or the 3′ UTR (). For HOXA4 and PON1 the DMRs were spread from the TSS to the gene body and for PRR15 from the 5′UTR to the 3′UTR. However, PRR15 is a small gene of 1717 bp (NM_175887) with only two exons. Methylation at the promoter and TSS is usually associated with decreased gene expression while gene body or 3′ UTR methylation increases expression and can affect splicing.Citation1 Altered methylation of CTCF binding sites is known for some imprinted genes, e.g., H19, where one of seven CTCF binding sites shows parent-of-origin specific methylation.Citation1 Methylation prevents the binding of CTCF that acts as an insulator, and the unblocked enhancers are able to drive the promoters to upregulate transcription of the target gene. We observed a strong signal for CTCF-binding sites in 4/6 (67%) of our maternally hypermethylated genes, including the CGI2 of GRB10, and in 3/10 of the maternally hypomethylated domains ().
Methylation of CpG sites in the recognition sequences of transcription factors can strongly influence transcription factor binding by complex mechanisms.Citation1,Citation46 Conserved transcription factor binding sites were observed for 7/17 (41%) of the DMRs ().
Validation of methylation status with pyrosequencing
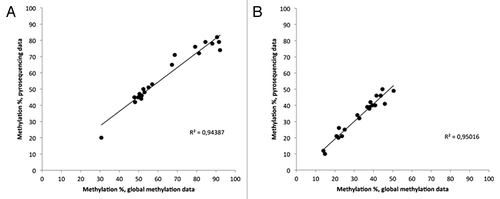
We chose three DMRs (HTR5A, RPS2P32, and HOXA4) for validation with pyrosequencing. There was a high correlation of methylation levels between the Infinium HumanMethylation450K BeadChip and pyrosequencing: 94% for the HTR5A CpG located at 154 862 969 (hg19 build) and 95% for the HOXA4 CpG located at 27 170,892 (hg19 build) (), suggesting that the Infinium microarray results were highly accurate. Pyrosequencing for RPS2P32 failed with two different primer sets.
Figure 4. Correlation of methylation levels between pyrosequencing and the Infinium HumanMethylation450K BeadChip. Dots indicate individual samples. (A) HTR5A, (B) HOXA4.
Data visualization
The methylation level of each CpG in each sample in the chromosome 7 DMRs and surrounding regions was visualized in detail with the Integrated Genome Browser (IGB, http://bioviz.org/igb/).Citation47 The full data set is available at http://publications.scilifelab.se/kere_j/imprinting7 as raw data files and IGB compatible files. As expected, GRB10, SGCE/PEG10, and MEST/MESTIT1 showed a clear parent-of-origin methylation pattern, with the methylation level of matUPD7s and patUPD7 deviating in opposite directions from the controls (). HTR5A, PON1, and RPS2P32 also showed a similar parent-of-origin specific methylation. For SVOPL, one matUPD7 case deviated from the others, suggesting individual variation (). HOXA4 showed clear hypomethylation for the matUPD7s, but the patUPD7 was indistinguishable from the controls (). For the remaining DMRs, clustering of the different groups was less apparent despite statistically significant group differences, suggesting more subtle methylation differences (data not shown).
Figure 5. Infinium HumanMethylation450K BeadChip methylation levels visualized by the Integrated Genome Browser (IGB) for (A) GRB10, (B) HOXA4, (C) RPS2P32, and (D) HTR5A. Bars show the M-values of individual samples for each CpG in the DMRs along the chromosome (horizontal axis). Bar color indicates sample type: purple, matUPD7s; green, controls; and blue patUPD7. M-value 0 indicates 50% methylation, positive values hypermethylation and negative values hypomethylation. The significance level (-log10) of matUPD7s vs. controls for each CpG is shown by the black bars. The dashed line indicates the significance threshold of P value 0.05. Figures were modified from the IGB output, and the CpG locations are approximations showing a 50 bp region calculated +/−25 bp from the Illumina annotation file coordinate corresponding to the genomic position of the C in CG dinucleotide, according to Genome build 37. The partial matUPD7 sample was excluded for genes located outside the partial matUPD7 region: GRB10, HOXA4, and RPS2P32.
Expression study of genes with DMRs
To define possible imprinted expression of the genes displaying DMRs, we performed quantitative reverse transcriptase-PCR (qRT-PCR) on freshly drawn blood cells. Differential expression between matUPD7s, controls, and patUPD7, compatible with imprinting, was seen for 7/11 (64%) of the genes with novel DMRs studied (). PEG10 was studied as a positive control for imprinting and displayed the expected paternal expression, with a significant difference between matUPD7s and controls, t test P value 0.0017 ().
Figure 6. Expression differences of differentially methylated genes. (A) HOXA4 (B) GLI3, (C) SVOPL, and (D) PEG10. Error bars indicate standard error of the mean for the matUPD7s and controls. PatUPD7 is a single sample. Regression plots for (E) HOXA4, (F) GLI3, (G) SVOPL, and (H) PEG10. Combined data from two replicate Taqman qPCRs are included for HOXA4, GLI3, and SVOPL. Only one Taqman qPCR was run for PEG10.
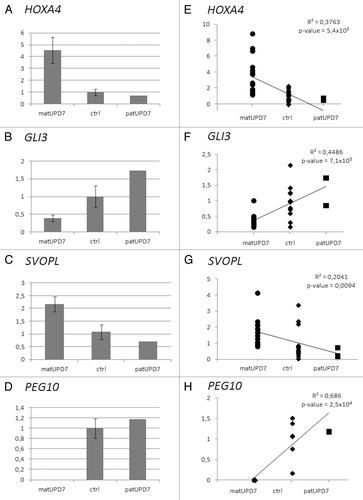
A significant difference in expression between matUPD7s and controls was found for three genes; HOXA4 at 7p15.2, GLI3 at 7p13, and SVOPL at 7q34 (; ). HOXA4 () and SVOPL () showed significantly increased expression in matUPD7s compared with controls (t test P values 0.008 and 0.017, respectively) and the patUPD7 showed markedly lower expression compared with the controls and matUPD7s (). GLI3 showed significantly decreased expression in matUPD7s compared with controls (t test P value 0.049) and the one patUPD7 showed higher expression compared with the controls and to matUPD7 (). Significant expression differences were also confirmed by performing a linear regression analysis, including the single patUPD7 case (). Maternal hypomethylation of the HOXA4 promoter region (Fig. S1A) appears to result in increased expression in matUPD7s, and hypermethylation in decreased expression in the patUPD7. However, for GLI3 maternal hypomethylation at the TSS (Fig. S1B) resulted in decreased expression in matUPD7s compared with controls and patUPD7. For SVOPL, maternal hypermethylation at the TSS and 5′UTR (Fig. S1C) results in increased maternal expression. Overall, HOXA4 and SVOPL showed maternal expression while GLI3 was paternally expressed.
In addition, CARD11, PRR15, and SH2B2 showed a clear pattern of increased maternal expression compared with controls and decreased expression in the patUPD7 suggestive of maternal expression, and RPS2P32 of decreased maternal expression indicative of paternal expression, but the difference between matUPD7s and the controls was not statistically significant for any of these genes (data not shown). For MAD1L1, and RARRES2 (MIM 601973) we did not identify differential expression indicative of imprinting and for HTR5A and PON1, expression was undetectable in lymphocytes. We did not study the lncRNAs or genes close to the two intergenic DMRs ().
To confirm the imprinting of HOXA4, GLI3, and SVOPL, we studied parent-of-origin specific expression by sequencing selected exonic SNPs in nine parent-child trios. Four SNPs with high expected heterozygosity were chosen for HOXA4 (rs17471888, rs4722660, rs1801085, rs2158218), five for GLI3 (rs3735361, rs3823720, rs2051935, rs929387, rs846266) and three for SVOPL (rs1614641, rs3734944, rs2305816) (Table S6). Genomic DNA from the children and parents was first sequenced for all SNPs to find children heterozygous for a SNP and parents either both homozygous for different alleles or at least one parent homozygous for one allele. The child’s cDNA was then sequenced from the trios with informative SNP alleles to see whether the maternally or paternally inherited allele was expressed. For SVOPL, two trios were informative and both children showed expression of only the maternal allele for rs2305816, confirming the maternal expression as seen in the qPCR (Fig. S2A). Only one child showed two heterozygous SNPs for HOXA4, but both parents were also heterozygous. Sequencing of the child’s cDNA for rs2158218 showed monoallelic expression thus supporting imprinting (Fig. S2C). Because both parents share the same allele it is not possible to discern which parental allele is expressed. The other heterozygous SNP rs4722660 failed to give a readable cDNA sequence. For GLI3 we failed to obtain conclusive data due to high PCR failure rate. Further analyses in larger sample sets are warranted to confirm the parent-specific expression.
Discussion
We performed a genome-wide methylation study for 450 000 CpG sites to identify DMRs on chromosome 7 in matUPD7 and patUPD7 cases in comparison to biparental controls. We used a three step filtering approach to identify the most significant CpGs in terms of parent-of-origin specific methylation. We identified 17 distinct DMRs spread along chromosome 7 localizing to 14 genes and two intragenic regions at lncRNAs, including the known DMRs at the SGCE/PEG10, GRB10, and MEST/MESTIT1 imprinted domains, and also two previously predicted imprinted genes, HOXA4 on 7p15.2 and GLI3 on 7p13.Citation41 Novel DMRs were located near eight protein coding genes and two lncRNAs, none previously implicated in imprinting, and SVOPL was confirmed as imprinted by parent-specific expression.
The majority of the DMRs were maternally hypomethylated (65%) and they were predominantly located in the promoter regions of the genes. This is contradictory to the current knowledge that the majority of imprinted genes are maternally hypermethylated. Maternally methylated DMRs and ICRs of imprinted genes are typically located at the promoters.Citation45 DMRs and ICRs of imprinted genes may also be situated within the genes and gene body methylation is known to enhance expression and possibly affect splicing.Citation1 For MAD1L1 and SH2B2 the maternally hypomethylated DMRs were found only in the gene bodies. However, SHB2B showed increased maternal expression, although non-significant, and MAD1L1 biparental expression whereas gene body maternal hypomethylation would have been expected to result in decreased maternal expression in both genes. We did not study differential splicing for these genes. Paternally methylated DMRs are preferentially located in intergenic regions,Citation45 and consistently both of the DMRs we found in intergenic regions were maternally hypomethylated. The CpGs in the DMRs located to CpG islands and shores but not shelves.
CTCF-binding sites, which act as insulators between promoters and enhancers, are commonly found at imprinting clusters.Citation45,Citation48 All of the DMRs found here, except for MAD1L1, SGCE/PEG10, and MEST/MESTIT1, co-localized with CTCF-binding sites. Differential methylation at CTCF-binding sites can affect the imprinted expression of the genes regulated by the insulators, e.g., H19.Citation1 Several imprinting clusters contain also at least one antisense lncRNA located in the proximity or partially overlapping a protein coding gene.Citation43,Citation44 Imprinted lncRNAs are necessary for the imprinted expression of the genes in the cluster, but can also regulate small clusters of autosomal genes in cis.Citation44 XIST (Inactive X-specific transcript, [MIM 314670]) and TSIX (Inactive-specific transcript, antisense [MIM 300181]) are the best-known functional lncRNAs required for the epigenetic X chromosome inactivation in female mammals.Citation44 Both of the two intergenic DMRs identified here were in close proximity to possible lncRNAs defined TUCPs, suggesting that these TUCPs may have imprinted regulation by the DMRs and potentially the DMRs and lncRNAs may be part of larger imprinted domains with surrounding genes. In addition, we observed antisense lncRNAs overlapping a few genes with DMRs, namely HTR5A and MAD1L1, and an antisense hypothetical protein overlapping HOXA4. Interestingly, RPS2P32 at 7p15.3 is itself a lncRNA. More than 80% of imprinted genes reside in clusters of several imprinted genes.Citation3 It remains to be seen whether the DMRs identified here act as ICRs by regulating the imprinted expression of other genes nearby, in addition to the genes that the DMRs lie closest to and have been studied here. In depth methylation and expression analysis of the neighboring genes and lncRNAs is thus warranted, but is beyond the scope of this study.
Our results give further insight to the extent of the DMRs at the SGCE/PEG10, GRB10, and MEST/MESTIT1 imprinted domains. The entire CGIs and parts of their North shores of GRB10 and MEST/MESTIT1 proved to be DMRs, but only 61% of the SGCE/PEG10 CGI turned out to be a DMR that extended well into the South shore. We also identified a novel DMR in the CGI in the promoter and first exon of PON1 in the SGCE/PEG10 cluster. Parent-of-origin specific expression of PON1 has not been shown, although paternal expression of PON1 has been reported in mouse hybrids containing a single maternal or paternal human chromosome 7.Citation49,Citation50 We did not detect PON1 expression in lymphocytes and could thus not study its possible parent of origin specific expression. PON1 is located between PPP1R9A, imprinted in both humans and mice, and PON2 (MIM 602447) and PON3 (MIM 602720), both imprinted in mouse, but biallelically expressed in humans.Citation21,Citation23 The ICR for PPP1R9A is unknown, as a CGI in the first exon of PPP1R9A did not show differential methylation between matUPD7 and patUPD7 lymphoblastoid cell lines or fetal placenta, liver or muscle,Citation21 nor was it picked up through our screening. The PON1 DMR identified here might act as an ICR for also PPP1R9A, although it is situated over 9 kb downstream from the 3′ end of PPP1R9A. We did not observe additional DMRs for any of the other imprinted genes in the MEST/MESTIT1 cluster (CPA4, COPG2IT1, COPG2 [disputed, MIM 604355] and KLF14). No DMRs were identified for any genes near GRB10. DDC (MIM 107930) and COBL (MIM 610317) adjacent to GRB10 have been shown to be imprinted in mouse, but the DMR of GRB10 appears to control imprinting of all three genes in mouse.Citation51 However, in human DDC and COBL were biparentally expressed in multiple fetal tissues.Citation52 DLX5 (MIM 60028) at 7q21.3 was initially shown to be maternally only expressed in human lymphoblasts,Citation49 but subsequently biallelic expression has been reported.Citation23,Citation53 We did not find DMRs in or near DLX5 which would indicate imprinting.
Several predicted imprinted loci at 7q11.21, 7q11.23, 7q21.11, and 7q36.1 failed to show differential methylation, suggesting either tissue or developmental stage specific imprinting beyond the blood cells that we studied, or imprecision of the predictions.Citation41 As all CpGs are not covered by the Infinium HumanMethylation450K BeadChip, some DMRs might have been missed by the study. We set our filtering process to exclude single and two adjacent CpGs, assuming that these CpGs were more likely to be false positives, and therefore some DMRs might have been ignored. We excluded the possibility of blood lineage-specific alternative methylation as a potential source of bias by verifying all loci for the absence of such differences.Citation46 Among the detected DMRs only GRB10, MEST, and PON1 have been reported to show age-dependent methylation level changes using the same microarray method.Citation54 Thus, the methylation results for the remaining DMRs should not be affected by the age of the subjects studied.
Only loci on chromosome 7 showed genome-wide significant differences in methylation. Consistent with our findings, there have been no reports of multilocus LOM associated with UPDs for other chromosomes. Thus, our data support the finding that matUPD7 is not associated with multilocus LOM.Citation55 However, up to 73% of SRS patients with H19 hypomethylation have been reported to show multilocus LOM, suggesting a generalized defect in establishing imprints in these patients.Citation34,Citation48 We found suggestive loci on seven other chromosomes, but these loci did not hit known imprinted domains. A small proportion of them might be cross-reactive probes that have co-hybridized to both chromosome 7 and other chromosomes. We have not explored these single CpG sites further.
Our data supported the imprinted status of HOXA4 at 7p15.2, GLI3 at 7p13, and SVOPL at 7q34 by showing differential expression between matUPD7s and patUPD7 and a significant expression difference between matUPD7s and controls. Concordant with the qRT-PCR results, allele-specific expression studies revealed maternal only expression of SVOPL, thus confirming it as an imprinted gene. HOXA4 showed monoallelic expression supporting imprinting, but because the child and both parents were heterozygous for the SNP we could not discern which parental allele was expressed. We failed to confirm imprinting of GLI3 because of high PCR failure rate. Further studies are needed to confirm imprinting. Hypomethylation of the promoter of HOXA4 in matUPD7s associated with increased expression of HOXA4 compared with controls, while hypermethylation in patUPD7 resulted in decreased expression. HOXA4 belongs to the cluster of HOXA genes on 7p15.2, which is a family of homeodomain containing transcription factors key during embryonic development.Citation56 Several other genes in the HOXA cluster are also predicted to be imprinted ().Citation41 The HOXA cluster has two lncRNAs known to regulate HOXA gene expression: HOTAIRM1 at the 3′ end regulating HOXA1 (MIM 142955), HOXA4, and HOXA5 at least in myeloid cell development and HOTTIP (MIM 614060) at the 5′ end regulating HOXA9 (MIM 142956), HOXA10 (MIM 142957), HOXA11 (MIM 142958), and HOXA13 (MIM 142959).Citation57,Citation58 Also an antisense lncRNA, HOXA11_AS (27 225 027–27 228 912, 3886 bp) (MIM 607530), lies 192 bp from the 5′ end of HOXA11. LncRNAs are found in many imprinted gene clusters and have been found to be crucial for the imprinted expression of genes in the imprinted clusters.Citation44 Further studies on the other HOXA genes and lncRNAs in the HOXA cluster are needed to clarify if this is a new imprinting cluster and also if the DMR at the promoter of HOXA4 would also act as an ICR in this region.
For GLI3, maternal hypomethylation of the DMR at the TSS resulted in decreased maternal expression. GLI3 encodes a zinc finger transcription factor that functions as a transcriptional activator and a repressor of the sonic hedgehog signaling pathway, and plays a role in early development.Citation59 Defects in GLI3 are found in disorders affecting limb development, resulting in pre- and postaxial polydactyly and syndactyly of fingers and toes, and variable other malformations of other organs.
For SVOPL, maternal hypermethylation at the TSS and 5′ UTR resulted in increased maternal expression. Several imprinted genes have methylated DMRs on the active allele, where it is proposed that the methylation inactivates silencing factors.Citation2 SVOPL was found through a sequence similarity search for SLC22 anion transporters, and it shows sequence similarity with the synaptic vesicle protein SVOP, but otherwise little is known of SVOPL.Citation60
CARD11, PRR15, and SH2B2 showed increased maternal expression and RPS2P32 of decreased maternal expression, implicating imprinted expression, but we did not see a statistically significant difference between matUPD7s and the controls for these genes. Lack of statistical significance can be due to the small sample sets available for this study and also interindividual variation seen within the sample sets. CARD11 acts as a critical signal transducer for NF-kappaB activation in both B and T lymphocytes and plays a crucial role in the antigen-specific immune response in human. The function of PRR15 is poorly understood in human, but in the mouse, the expression pattern of Prr15 closely resembles that of a number of important negative cell cycle regulators and it is suggested that Prr15 could be involved in controlling cellular proliferation and/or differentiation.Citation61SH2B2 encodes an adaptor protein SH2B2, also known as APS, which belongs to the SH2B protein family, that regulates several signaling pathways and participates in various physiological responses and developmental processes.Citation62 SH2B2 interacts with insulin receptor substrate 1 (IRS1), IRS2, or Janus kinase 2 (JAK2) to regulate insulin, leptin, and growth hormone signaling. MAD1L1 and RARRES2 appeared to be biparentally expressed, as expression was similar in matUPD7s, controls and patUPD7. Imprinting is not completely excluded for these genes, as other tissues, specific isoforms or developmental time points might still reveal parent-of-origin specific differences.Citation45 For PON1 and HTR5A we did not detect any expression in the blood samples available and therefore could not make any conclusion on the effects of the DMRs on their expression.
In conclusion, we have identified 14 novel parent-of origin specific DMRs on human chromosome 7 using a genome wide methylation microarray. Expression study identified a novel imprinted gene SVOPL that was confirmed by parent-specific expression and supported the previously suggested genes HOXA4 and GLI3 as being imprinted. Imprinted genes on human chromosome 7 are implicated in the etiology of the matUPD7 phenotype of SRS. Interestingly, HOXA4 and GLI3 and many of genes close to the novel DMRs have known functions in cellular growth and development, which make them appealing as putative SRS genes in future studies.
Materials and Methods
Patients
We obtained fresh blood samples from eight SRS patients with matUPD7, of whom six have been reported before.Citation14,Citation63-Citation65 and two are first reported in this study as well as a SRS patient with segmental matUPD7q31-qterCitation26 and one individual with patUPD7.Citation16 SRS patients were recruited from the Hospital for Children and Adolescents, Helsinki University Central Hospital, Finland. Additionally three patients were referred from the Oulu University Central Hospital and one from the Päijät-Häme Central Hospital, Finland. All patients were seen by a pediatric endocrinologist and diagnosis of SRS was based on clinical features and matUPD7 was verified by microsatellite markers as described before.Citation63,Citation64 Control samples from ten unrelated normal height adults were obtained. For parent-of-origin allele-specific expression analysis we obtained fresh blood samples from nine parent-child trios. All children were form our growth retardation study cohort and had previously been excluded from having matUPD7, as described before.Citation64 Seven children had H19 hypomethylation and SRS,Citation14 one was a normally growing sister of a matUPD7 patient and one had postnatal growth retardation with no dysmorphic features. This study was approved by the Ethical Review Board of the Hospital for Children and Adolescents, Helsinki University Central Hospital, Helsinki, Finland. Written informed consent was obtained from all study participants. All clinical investigations have been conducted according to the principles expressed in the Declaration of Helsinki.
Genome-wide methylation analysis with the Infinium HumanMethylation450K BeadChip
Genomic DNA was extracted from fresh EDTA-blood samples with the FlexiGene DNA Kit (Qiagen) according to the manufacturer’s instructions. 500 ng of DNA from each subject was bisulfite converted with the EZ-96 DNA Methylation Kit (Zymo research Corporation) according to the manufacturer’s instructions. Array-based specific DNA methylation analysis was performed with the Infinium HumanMethylation450K BeadChip technology (Illumina). All samples were analyzed for more than 450 000 CpG sites at single nucleotide resolution with 99% coverage of RefSeq genes and 96% coverage of CGIs. The CpGs were distributed in CGI shelves, CGI shores, CGIs, promoter regions 5′UTRs, first exon, gene body, and 3′UTRs. Bisulfite-treated genomic DNA was whole-genome amplified and hybridized to the HumanMethylation450 BeadChips (Illumina) and scanned using the Illumina iScan at the Bioinformatic and Expression Analysis (BEA) Core Facility of the Karolinska Institutet. The intensity of the images was extracted with the GenomeStudio Methylation Software Module (v 1.9.0, Illumina).
Quality Control analysis and data validation
Quality control was conducted in GenomeStudio software (v2011.1) using the methylation module (v1.9.0) according to the manufacturer’s recommendations (Illumina). Briefly, the controls included assessment of DNP and Biotin staining, hybridization, target removal, extension, bisulfite conversion, G/T mismatch, and negative and non-polymorphic controls. The various controls indicated overall good quality of DNA preparations and chip performance.
Bioinformatics analysis of Infinium HumanMethylation450K BeadChip data
The analysis of the HumanMethylation450 BeadChips was performed as previously described.Citation46 Briefly, the raw data were analyzed with the BioConductor package lumi in R v. 2.13.Citation66 The data was adjusted for color channel imbalance and background noise, and normalized according to the quantile method. Probe-wise differential methylation was assayed by linear model followed by pair-wise comparisons by empirical Bayes t test on the normalized M values.Citation67 The M-value is calculated as the log2 ratio of the intensities of methylated probe vs. unmethylated probe and describes a measurement of how much more a probe is methylated compared with unmethylated.Citation68 A value close to 0 indicates a similar intensity between the methylated and unmethylated probes, which means the CpG site is about half-methylated.Citation68 Positive M-values mean that more molecules are methylated than unmethylated, while negative M-values mean that more molecules are unmethylated. The M values give higher resolution than the β values for extreme hyper- and hypo-methylation levels, whereas at middle-range methylation levels they are collinear.
Process of filtering for differentially methylated regions
All 30 017 CpGs on chromosome 7 were filtered according to a three-step process. For maternally hypermethylated CpGs, step-1 of the filtering process was passed if the median M-value of all eight matUPD7s was larger than that of the ten controls and median M-value of controls was larger than the M-value of the patUPD7 sample. No specific threshold was set and the filter was passed even with a minimal difference between the groups. For maternally hypomethylated CpGs, step-1 of the filtering process was passed if M-value of the patUPD7 sample was larger than the median M-value of the controls, and median M-value of the controls was larger than that of the matUPD7 samples. Step-2 of the filtering process excluded all CpGs, where the differential methylation between matUPD7s and controls did not reach the empirical Bayes significance (nominal P value < 0.05). The step-3 in the filtering process was set to include only those adjacent CpGs which were included in a consecutive row of at least three CpGs which also had passed steps one and two of the filtering process. Adjacent CpGs were considered as CpGs consecutively mapped in a given chromosomal region after one another (i.e., with no other probes in between them). A genomic distance was not used to define adjacency.
CpGs in regard to CGIs, genes, TSS, Promotors, CTCF binding sites
The loci of the CpGs in regard to CGIs, TSSs, promoters, and CTCF binding sites were obtained from the Infinium Human Methylation450K BeadChip annotation files and from the UCSC genome browser. Localization of DMRs to lncRNAs was determined according to UCSC genome browser.
Data visualization
Genome-wide visualization of the global methylation data was performed with the IGB version 6.5.1/6.5.1_5.Citation47
Pyrosequencing
Two micrograms of DNA was bisulfite-converted with EpiTect Bisulfite Kit (Qiagen) from all matUPD7, patUPD7, and controls. 10–20 ng of bisulfite modified DNA was used for PCR amplification. Primers were designed by PyroMark Assay Design SW 2.0 to encompass the DMR regions identified by the Infinium 450K Bead chip for HTR5A, RPS2P32, and HOXA4. Specific primer sequences will be given upon request. PCR was performed in standard conditions with 10 uM primers and successful PCR was verified by 1% agarose gel electrophoresis. Pyrosequencing was performed on the PyroMark Q24 system (Qiagen), according to the manufacturer’s instructions. Methylation level of the CpGs was analyzed with the PyroMark Q24 software (Qiagen).
Correlation between the pyrosequencing and the Infinium HumanMethylation450K Bead chip results was done by converting the M-values of the representative CpG sites obtained from the Infinium 450K Bead chip to Beta-values, using an approximation method.Citation68 Beta-values of the CpG sites were then compared with the methylation percentage derived from pyrosequencing experiments.
Quantitative PCR
Fresh whole blood samples were collected from all matUPD7, patUPD7, and controls into PAXgene Blood RNA tubes (PreAnalytiX, GmbH). RNA was extracted with the PAXgene Blood miRNA Kit (PreAnalytiX, GmbH), according to the manufacturer’s instructions. RNA quality was checked with Bioanalyzer (Agilent Technologies) and all samples had a RIN (RNA integrity number) value above eight.
Reverse transcription (RT) was performed using TaqMan RT reagents according to standard protocols (Applied Biosystems). qPCR reactions were run in triplicate 10 µl reactions, using 10 ng of reverse transcribed RNA and the standard reaction conditions for TaqMan Fast Advanced Master Mix and the TaqMan Gene Expression Assays: MAD1L1 (Hs002069119_m1), CARD11 (Hs00269119_m1), RPS2P32 (Hs01010823_g1), HOXA4 (Hs01573270_m1), PRR15 (Hs00828414_m1), GLI3 (Hs00609233_n1), PEG10 (Hs01122880_m1), PON1 (Hs00166557_m1), SH2B2 (Hs00184134_m1), SVOPL (Hs00736451_m1), HTR5A (Hs00225153_m1), and RARRES2 (Hs00161209_g1) were used (Applied Biosystems). The PCR runs were performed with the 7900HT Sequence Detection System (Applied Biosystems) and the results were calculated with the comparative CT method with standard protocols.
The Student’s T-test was used to calculate significance between matUPD7s and controls. ANOVA was used to calculate significance for regression plots. The partial matUPD7q31-qter was included in the matUPD7 group only for genes in the 7q31-qter region, SVOPL1 and RARRES2, otherwise the sample was excluded from the analyses.
Parent-of-origin allele-specific expression analyses
The HapMart data mining tool was used to select exonic SNPs within HOXA4, GLI3, and SVOPL, that had a minor allele frequency greater than zero reported for the CEPH (CEU) population. Primers were designed by PrimerZ and Primer-BLAST. Specific primer sequences are available upon request.
Genomic DNA from nine parent-child trios was extracted from fresh EDTA-blood samples with the FlexiGene DNA Kit (Qiagen), according to the manufacturer’s instructions. Fresh whole blood samples from the children were obtained in PAXgene Blood RNA tubes (PreAnalytiX, GmbH). RNA was extracted with the PAXgene Blood miRNA Kit (PreAnalytiX, GmbH), according to the manufacturer’s instructions. Reverse transcription was performed by High Capacity Reverse Transcription Kit (Applied Biosciences), according to the manufacturer’s protocol. Genomic DNA and cDNA were amplified by PCR using Phusion High Fidelity DNA polymerase (Thermo Scientific) by standard protocols. Products were cleaned with EXO-SAP IT® (USB) and sequenced. Genotypes were viewed using FinchTV (1.5.0, Geospiza).
| Abbreviations: | ||
| DMRs | = | differentially methylated regions |
| UPD | = | uniparental disomy |
| matUPD7 | = | maternal uniparental disomy |
| patUPD7 | = | paternal uniparental disomy |
| SRS | = | Silver-Russell syndrome |
| CGIs | = | CpG islands |
| TSS | = | transcription start sites |
| ICR | = | imprinting control center |
| TNDM | = | transient neonatal diabetes mellitus |
| BWS | = | Beckwith-Wiedemann syndrome |
| AS | = | Angelman syndrome |
| PWS | = | Prader-Willi syndrome |
| LOM | = | loss of methylation |
| lncRNA | = | long non-conding RNA |
| TUCP | = | transcript of uncertain coding potential |
| IGB | = | Integrated Genome Browser |
| qRT-PCR | = | quantitative reverse transcriptase-PCR |
| MIM | = | Mendelian Inheritance in Man |
| UCSC | = | University of California, Santa Cruz |
Additional material
Download Zip (697.9 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank all patients and families for participating in this study and Riitta Lehtinen and Riitta Känkänen for technical assistance in the laboratory. This work was supported by Academy of Finland (132559 to Hannula-Jouppi K), Päivikki and Sakari Sohlberg Foundation, Helsinki Biomedical Graduate Program, The Finnish Medical Foundation, Sigrid Jusélius Foundation, Swedish Foundation for Strategic Research (SSF, project Epigene), and Swedish Research Council.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/27160
References
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484 - 92; http://dx.doi.org/10.1038/nrg3230; PMID: 22641018
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet 2001; 2:21 - 32; http://dx.doi.org/10.1038/35047554; PMID: 11253064
- Wood AJ, Oakey RJ. Genomic imprinting in mammals: emerging themes and established theories. PLoS Genet 2006; 2:e147; http://dx.doi.org/10.1371/journal.pgen.0020147; PMID: 17121465
- Fang F, Hodges E, Molaro A, Dean M, Hannon GJ, Smith AD. Genomic landscape of human allele-specific DNA methylation. Proc Natl Acad Sci U S A 2012; 109:7332 - 7; http://dx.doi.org/10.1073/pnas.1201310109; PMID: 22523239
- Engel E. A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet 1980; 6:137 - 43; http://dx.doi.org/10.1002/ajmg.1320060207; PMID: 7192492
- Temple IK, Shield JP. 6q24 transient neonatal diabetes. Rev Endocr Metab Disord 2010; 11:199 - 204; http://dx.doi.org/10.1007/s11154-010-9150-4; PMID: 20922569
- Wakeling EL. Silver-Russell syndrome. Arch Dis Child 2011; 96:1156 - 61; http://dx.doi.org/10.1136/adc.2010.190165; PMID: 21349887
- Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 2010; 154C:343 - 54; http://dx.doi.org/10.1002/ajmg.c.30267; PMID: 20803657
- Ogata T, Kagami M, Ferguson-Smith AC. Molecular mechanisms regulating phenotypic outcome in paternal and maternal uniparental disomy for chromosome 14. Epigenetics 2008; 3:181 - 7; http://dx.doi.org/10.4161/epi.3.4.6550; PMID: 18698157
- Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet 2010; 154C:365 - 76; http://dx.doi.org/10.1002/ajmg.c.30273; PMID: 20803659
- Azzi S, Rossignol S, Le Bouc Y, Netchine I. Lessons from imprinted multilocus loss of methylation in human syndromes: A step toward understanding the mechanisms underlying these complex diseases. Epigenetics 2010; 5:373 - 7; http://dx.doi.org/10.4161/epi.5.5.11851; PMID: 20495355
- Begemann M, Spengler S, Kordass U, Schröder C, Eggermann T. Segmental maternal uniparental disomy 7q associated with DLK1/GTL2 (14q32) hypomethylation. Am J Med Genet A 2012; 158A:423 - 8; http://dx.doi.org/10.1002/ajmg.a.34412; PMID: 22246686
- Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, Danton F, Thibaud N, Le Merrer M, Burglen L, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet 2005; 37:1003 - 7; http://dx.doi.org/10.1038/ng1629; PMID: 16086014
- Bruce S, Hannula-Jouppi K, Peltonen J, Kere J, Lipsanen-Nyman M. Clinically distinct epigenetic subgroups in Silver-Russell syndrome: the degree of H19 hypomethylation associates with phenotype severity and genital and skeletal anomalies. J Clin Endocrinol Metab 2009; 94:579 - 87; http://dx.doi.org/10.1210/jc.2008-1805; PMID: 19017756
- Price SM, Stanhope R, Garrett C, Preece MA, Trembath RC. The spectrum of Silver-Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria. J Med Genet 1999; 36:837 - 42; PMID: 10544228
- Höglund P, Holmberg C, de la Chapelle A, Kere J. Paternal isodisomy for chromosome 7 is compatible with normal growth and development in a patient with congenital chloride diarrhea. Am J Hum Genet 1994; 55:747 - 52; PMID: 7942853
- Pan Y, McCaskill CD, Thompson KH, Hicks J, Casey B, Shaffer LG, Craigen WJ. Paternal isodisomy of chromosome 7 associated with complete situs inversus and immotile cilia. Am J Hum Genet 1998; 62:1551 - 5; http://dx.doi.org/10.1086/301857; PMID: 9585585
- Le Caignec C, Isidor B, de Pontbriand U, David V, Audrezet MP, Ferec C, David A. Third case of paternal isodisomy for chromosome 7 with cystic fibrosis: a new patient presenting with normal growth. Am J Med Genet A 2007; 143A:2696 - 9; http://dx.doi.org/10.1002/ajmg.a.31999; PMID: 17935233
- Hitchins MP, Monk D, Bell GM, Ali Z, Preece MA, Stanier P, Moore GE. Maternal repression of the human GRB10 gene in the developing central nervous system; evaluation of the role for GRB10 in Silver-Russell syndrome. Eur J Hum Genet 2001; 9:82 - 90; http://dx.doi.org/10.1038/sj.ejhg.5200583; PMID: 11313740
- Bentley L, Nakabayashi K, Monk D, Beechey C, Peters J, Birjandi Z, Khayat FE, Patel M, Preece MA, Stanier P, et al. The imprinted region on human chromosome 7q32 extends to the carboxypeptidase A gene cluster: an imprinted candidate for Silver-Russell syndrome. J Med Genet 2003; 40:249 - 56; http://dx.doi.org/10.1136/jmg.40.4.249; PMID: 12676894
- Nakabayashi K, Makino S, Minagawa S, Smith AC, Bamforth JS, Stanier P, Preece M, Parker-Katiraee L, Paton T, Oshimura M, et al. Genomic imprinting of PPP1R9A encoding neurabin I in skeletal muscle and extra-embryonic tissues. J Med Genet 2004; 41:601 - 8; http://dx.doi.org/10.1136/jmg.2003.014142; PMID: 15286155
- Parker-Katiraee L, Carson AR, Yamada T, Arnaud P, Feil R, Abu-Amero SN, Moore GE, Kaneda M, Perry GH, Stone AC, et al. Identification of the imprinted KLF14 transcription factor undergoing human-specific accelerated evolution. PLoS Genet 2007; 3:e65; http://dx.doi.org/10.1371/journal.pgen.0030065; PMID: 17480121
- Monk D, Wagschal A, Arnaud P, Müller PS, Parker-Katiraee L, Bourc’his D, Scherer SW, Feil R, Stanier P, Moore GE. Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing. Genome Res 2008; 18:1270 - 81; http://dx.doi.org/10.1101/gr.077115.108; PMID: 18480470
- Joyce CA, Sharp A, Walker JM, Bullman H, Temple IK. Duplication of 7p12.1-p13, including GRB10 and IGFBP1, in a mother and daughter with features of Silver-Russell syndrome. Hum Genet 1999; 105:273 - 80; http://dx.doi.org/10.1007/s004390051101; PMID: 10987657
- Monk D, Wakeling EL, Proud V, Hitchins M, Abu-Amero SN, Stanier P, Preece MA, Moore GE. Duplication of 7p11.2-p13, including GRB10, in Silver-Russell syndrome. Am J Hum Genet 2000; 66:36 - 46; http://dx.doi.org/10.1086/302717; PMID: 10631135
- Hannula K, Lipsanen-Nyman M, Kontiokari T, Kere J. A narrow segment of maternal uniparental disomy of chromosome 7q31-qter in Silver-Russell syndrome delimits a candidate gene region. Am J Hum Genet 2001; 68:247 - 53; http://dx.doi.org/10.1086/316937; PMID: 11112662
- Eggermann T, Schönherr N, Jäger S, Spaich C, Ranke MB, Wollmann HA, Binder G. Segmental maternal UPD(7q) in Silver-Russell syndrome. Clin Genet 2008; 74:486 - 9; http://dx.doi.org/10.1111/j.1399-0004.2008.01057.x; PMID: 18700897
- Reboul MP, Tandonnet O, Biteau N, Belet-de Putter C, Rebouissoux L, Moradkhani K, Vu PY, Saura R, Arveiler B, Lacombe D, et al. Mosaic maternal uniparental isodisomy for chromosome 7q21-qter. Clin Genet 2006; 70:207 - 13; http://dx.doi.org/10.1111/j.1399-0004.2006.00664.x; PMID: 16922723
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature 1993; 366:362 - 5; http://dx.doi.org/10.1038/366362a0; PMID: 8247133
- Ku CS, Naidoo N, Wu M, Soong R. Studying the epigenome using next generation sequencing. J Med Genet 2011; 48:721 - 30; http://dx.doi.org/10.1136/jmedgenet-2011-100242; PMID: 21825079
- Sharp AJ, Migliavacca E, Dupre Y, Stathaki E, Sailani MR, Baumer A, Schinzel A, Mackay DJ, Robinson DO, Cobellis G, et al. Methylation profiling in individuals with uniparental disomy identifies novel differentially methylated regions on chromosome 15. Genome Res 2010; 20:1271 - 8; http://dx.doi.org/10.1101/gr.108597.110; PMID: 20631049
- Nakabayashi K, Trujillo AM, Tayama C, Camprubi C, Yoshida W, Lapunzina P, Sanchez A, Soejima H, Aburatani H, Nagae G, et al. Methylation screening of reciprocal genome-wide UPDs identifies novel human-specific imprinted genes. Hum Mol Genet 2011; 20:3188 - 97; http://dx.doi.org/10.1093/hmg/ddr224; PMID: 21593219
- Peñaherrera MS, Weindler S, Van Allen MI, Yong SL, Metzger DL, McGillivray B, Boerkoel C, Langlois S, Robinson WP. Methylation profiling in individuals with Russell-Silver syndrome. Am J Med Genet A 2010; 152A:347 - 55; http://dx.doi.org/10.1002/ajmg.a.33204; PMID: 20082469
- Kannenberg K, Urban C, Binder G. Increased incidence of aberrant DNA methylation within diverse imprinted gene loci outside of IGF2/H19 in Silver-Russell syndrome. Clin Genet 2012; 81:366 - 77; http://dx.doi.org/10.1111/j.1399-0004.2012.01844.x; PMID: 22248018
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692 - 702; http://dx.doi.org/10.4161/epi.6.6.16196; PMID: 21593595
- Azzi S, Rossignol S, Steunou V, Sas T, Thibaud N, Danton F, Le Jule M, Heinrichs C, Cabrol S, Gicquel C, et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum Mol Genet 2009; 18:4724 - 33; http://dx.doi.org/10.1093/hmg/ddp435; PMID: 19755383
- Turner CL, Mackay DM, Callaway JL, Docherty LE, Poole RL, Bullman H, Lever M, Castle BM, Kivuva EC, Turnpenny PD, et al. Methylation analysis of 79 patients with growth restriction reveals novel patterns of methylation change at imprinted loci. Eur J Hum Genet 2010; 18:648 - 55; http://dx.doi.org/10.1038/ejhg.2009.246; PMID: 20104244
- Arnaud P, Monk D, Hitchins M, Gordon E, Dean W, Beechey CV, Peters J, Craigen W, Preece M, Stanier P, et al. Conserved methylation imprints in the human and mouse GRB10 genes with divergent allelic expression suggests differential reading of the same mark. Hum Mol Genet 2003; 12:1005 - 19; http://dx.doi.org/10.1093/hmg/ddg110; PMID: 12700169
- Riesewijk AM, Hu L, Schulz U, Tariverdian G, Höglund P, Kere J, Ropers HH, Kalscheuer VM. Monoallelic expression of human PEG1/MEST is paralleled by parent-specific methylation in fetuses. Genomics 1997; 42:236 - 44; http://dx.doi.org/10.1006/geno.1997.4731; PMID: 9192843
- Kobayashi S, Kohda T, Miyoshi N, Kuroiwa Y, Aisaka K, Tsutsumi O, Kaneko-Ishino T, Ishino F. Human PEG1/MEST, an imprinted gene on chromosome 7. Hum Mol Genet 1997; 6:781 - 6; http://dx.doi.org/10.1093/hmg/6.5.781; PMID: 9158153
- Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ. Computational and experimental identification of novel human imprinted genes. Genome Res 2007; 17:1723 - 30; http://dx.doi.org/10.1101/gr.6584707; PMID: 18055845
- Ono R, Shiura H, Aburatani H, Kohda T, Kaneko-Ishino T, Ishino F. Identification of a large novel imprinted gene cluster on mouse proximal chromosome 6. Genome Res 2003; 13:1696 - 705; http://dx.doi.org/10.1101/gr.906803; PMID: 12840045
- Pauler FM, Koerner MV, Barlow DP. Silencing by imprinted noncoding RNAs: is transcription the answer?. Trends Genet 2007; 23:284 - 92; http://dx.doi.org/10.1016/j.tig.2007.03.018; PMID: 17445943
- Koerner MV, Pauler FM, Huang R, Barlow DP. The function of non-coding RNAs in genomic imprinting. Development 2009; 136:1771 - 83; http://dx.doi.org/10.1242/dev.030403; PMID: 19429783
- Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol 2011; 3:a002592; http://dx.doi.org/10.1101/cshperspect.a002592; PMID: 21576252
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, Söderhäll C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 2012; 7:e41361; http://dx.doi.org/10.1371/journal.pone.0041361; PMID: 22848472
- Nicol JW, Helt GA, Blanchard SG Jr., Raja A, Loraine AE. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 2009; 25:2730 - 1; http://dx.doi.org/10.1093/bioinformatics/btp472; PMID: 19654113
- Ishida M, Moore GE. The role of imprinted genes in humans. Mol Aspects Med 2013; 34:826 - 40; http://dx.doi.org/10.1016/j.mam.2012.06.009; PMID: 22771538
- Okita C, Meguro M, Hoshiya H, Haruta M, Sakamoto YK, Oshimura M. A new imprinted cluster on the human chromosome 7q21-q31, identified by human-mouse monochromosomal hybrids. Genomics 2003; 81:556 - 9; http://dx.doi.org/10.1016/S0888-7543(03)00052-1; PMID: 12782124
- Parker-Katiraee L, Bousiaki E, Monk D, Moore GE, Nakabayashi K, Scherer SW. Dynamic variation in allele-specific gene expression of Paraoxonase-1 in murine and human tissues. Hum Mol Genet 2008; 17:3263 - 70; http://dx.doi.org/10.1093/hmg/ddn222; PMID: 18678600
- Shiura H, Nakamura K, Hikichi T, Hino T, Oda K, Suzuki-Migishima R, Kohda T, Kaneko-ishino T, Ishino F. Paternal deletion of Meg1/Grb10 DMR causes maternalization of the Meg1/Grb10 cluster in mouse proximal Chromosome 11 leading to severe pre- and postnatal growth retardation. Hum Mol Genet 2009; 18:1424 - 38; http://dx.doi.org/10.1093/hmg/ddp049; PMID: 19174477
- Hitchins MP, Bentley L, Monk D, Beechey C, Peters J, Kelsey G, Ishino F, Preece MA, Stanier P, Moore GE. DDC and COBL, flanking the imprinted GRB10 gene on 7p12, are biallelically expressed. Mamm Genome 2002; 13:686 - 91; http://dx.doi.org/10.1007/s00335-002-3028-z; PMID: 12514746
- Schüle B, Li HH, Fisch-Kohl C, Purmann C, Francke U. DLX5 and DLX6 expression is biallelic and not modulated by MeCP2 deficiency. Am J Hum Genet 2007; 81:492 - 506; http://dx.doi.org/10.1086/520063; PMID: 17701895
- Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST. Age-associated DNA methylation in pediatric populations. Genome Res 2012; 22:623 - 32; http://dx.doi.org/10.1101/gr.125187.111; PMID: 22300631
- Schönherr N, Meyer E, Binder G, Wollmann HA, Eggermann T. No evidence for additional imprinting defects in Silver-Russell syndrome patients with maternal uniparental disomy 7 or 11p15 epimutation. J Pediatr Endocrinol Metab 2007; 20:1329 - 31; http://dx.doi.org/10.1515/JPEM.2007.20.12.1329; PMID: 18341093
- Cillo C, Cantile M, Faiella A, Boncinelli E. Homeobox genes in normal and malignant cells. J Cell Physiol 2001; 188:161 - 9; http://dx.doi.org/10.1002/jcp.1115; PMID: 11424082
- Zhang X, Lian Z, Padden C, Gerstein MB, Rozowsky J, Snyder M, Gingeras TR, Kapranov P, Weissman SM, Newburger PE. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood 2009; 113:2526 - 34; http://dx.doi.org/10.1182/blood-2008-06-162164; PMID: 19144990
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011; 472:120 - 4; http://dx.doi.org/10.1038/nature09819; PMID: 21423168
- Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-patched-gli pathway in human development and disease. Am J Hum Genet 2000; 67:1047 - 54; PMID: 11001584
- Jacobsson JA, Haitina T, Lindblom J, Fredriksson R. Identification of six putative human transporters with structural similarity to the drug transporter SLC22 family. Genomics 2007; 90:595 - 609; http://dx.doi.org/10.1016/j.ygeno.2007.03.017; PMID: 17714910
- Meunier D, Patra K, Smits R, Hägebarth A, Lüttges A, Jaussi R, Wieduwilt MJ, Quintanilla-Fend L, Himmelbauer H, Fodde R, et al. Expression analysis of proline rich 15 (Prr15) in mouse and human gastrointestinal tumors. Mol Carcinog 2011; 50:8 - 15; http://dx.doi.org/10.1002/mc.20692; PMID: 21061267
- Wang TC, Chiu H, Chang YJ, Hsu TY, Chiu IM, Chen L. The adaptor protein SH2B3 (Lnk) negatively regulates neurite outgrowth of PC12 cells and cortical neurons. PLoS One 2011; 6:e26433; http://dx.doi.org/10.1371/journal.pone.0026433; PMID: 22028877
- Hannula K, Kere J, Pirinen S, Holmberg C, Lipsanen-Nyman M. Do patients with maternal uniparental disomy for chromosome 7 have a distinct mild Silver-Russell phenotype?. J Med Genet 2001; 38:273 - 8; http://dx.doi.org/10.1136/jmg.38.4.273; PMID: 11370636
- Hannula K, Lipsanen-Nyman M, Kristo P, Kaitila I, Simola KO, Lenko HL, Tapanainen P, Holmberg C, Kere J. Genetic screening for maternal uniparental disomy of chromosome 7 in prenatal and postnatal growth retardation of unknown cause. Pediatrics 2002; 109:441 - 8; http://dx.doi.org/10.1542/peds.109.3.441; PMID: 11875139
- Bruce S, Leinonen R, Lindgren CM, Kivinen K, Dahlman-Wright K, Lipsanen-Nyman M, Hannula-Jouppi K, Kere J. Global analysis of uniparental disomy using high density genotyping arrays. J Med Genet 2005; 42:847 - 51; http://dx.doi.org/10.1136/jmg.2005.032367; PMID: 15879501
- Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24:1547 - 8; http://dx.doi.org/10.1093/bioinformatics/btn224; PMID: 18467348
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, ed. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York, Springer. 2005:397–420.
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010; 11:587; http://dx.doi.org/10.1186/1471-2105-11-587; PMID: 21118553
- Blagitko N, Mergenthaler S, Schulz U, Wollmann HA, Craigen W, Eggermann T, Ropers HH, Kalscheuer VM. Human GRB10 is imprinted and expressed from the paternal and maternal allele in a highly tissue- and isoform-specific fashion. Hum Mol Genet 2000; 9:1587 - 95; http://dx.doi.org/10.1093/hmg/9.11.1587; PMID: 10861285
- Yoshihashi H, Maeyama K, Kosaki R, Ogata T, Tsukahara M, Goto Y, Hata J, Matsuo N, Smith RJ, Kosaki K. Imprinting of human GRB10 and its mutations in two patients with Russell-Silver syndrome. Am J Hum Genet 2000; 67:476 - 82; http://dx.doi.org/10.1086/302997; PMID: 10856193
- Zimprich A, Grabowski M, Asmus F, Naumann M, Berg D, Bertram M, Scheidtmann K, Kern P, Winkelmann J, Müller-Myhsok B, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet 2001; 29:66 - 9; http://dx.doi.org/10.1038/ng709; PMID: 11528394
- Ono R, Kobayashi S, Wagatsuma H, Aisaka K, Kohda T, Kaneko-Ishino T, Ishino F. A retrotransposon-derived gene, PEG10, is a novel imprinted gene located on human chromosome 7q21. Genomics 2001; 73:232 - 7; http://dx.doi.org/10.1006/geno.2001.6494; PMID: 11318613