
Abstract
Similar to most cancers, genome-wide DNA methylation profiles are commonly altered in pediatric acute lymphoblastic leukemia (ALL); however, recent observations highlight that a large portion of malignancy-associated DNA methylation alterations are not accompanied by related gene expression changes. By analyzing and integrating the methylome and transcriptome profiles of pediatric B-cell ALL cases and primary tissue controls, we report 325 genes hypermethylated and downregulated and 45 genes hypomethylated and upregulated in pediatric B-cell ALL, irrespective of subtype. Repressed cation channel subunits and cAMP signaling activators and transducers are overrepresented, potentially indicating a reduced cellular potential to receive and propagate apoptotic signals. Furthermore, we report specific DNA methylation alterations with concurrent gene expression changes within individual ALL subtypes. The ETV6-RUNX1 translocation was associated with downregulation of ASNS and upregulation of the EPO-receptor, while Hyperdiploid patients (>50 chr) displayed upregulation of B-cell lymphoma (BCL) members and repression of PTPRG and FHIT. In combination, these data indicate genetically distinct B-cell ALL subtypes contain cooperative epimutations and genome-wide epigenetic deregulation is common across all B-cell ALL subtypes.
Introduction
DNA methylation is a well characterized epigenetic modification, involving the addition of a single methyl group to cytosine residues, primarily within a CpG dinucleotide context.Citation1 Approximately 60% of gene promoters within the human genome contain dense clusters of CpG dinucleotides (CpG islands), most of which remain unmethylated during development.Citation2,Citation3 The methylation of gene promoters is generally associated with heterochromatin states that restrict access of factors necessary for gene expression. Importantly, DNA methylation patterns are maintained during cell division, providing a mechanism for the inheritance of transcriptional programs important for tissue specification.Citation2
DNA methylation alterations affecting hundreds of genes have been described for several cancers. In both hematologic and solid tumor subtypes the alterations are often associated with underlying genetic mutations, indicating a cooperative role in tumorigenesis.Citation4-Citation12 Furthermore, patient prognosis and drug resistance have also been associated with methylated loci, indicating that de novo DNA methylation contributes to malignant phenotype.Citation10-Citation16 Whether such changes represent a cause or a consequence of malignant transformation is still debatable; nevertheless, experiments utilizing demethylating agents to re-express hypermethylated genes within cancer cell lines show that many such changes affect gene expression.Citation17-Citation19
We recently identified common genome-wide disruptions to the DNA methylation profiles of pediatric B-cell ALL patients with promoter hypermethylation as a prominent feature, affecting genes previously implicated in leukemia, other malignancies, and hematopoietic development.Citation20 Furthermore, differences in DNA methylation patterns between common genetic subtypes of pediatric ALL have also been reported.Citation21-Citation23 Recent observations highlight that DNA methylation alterations in malignancy are not always associated with changes in gene expression.Citation24 To understand the contribution of DNA methylation alterations toward disease it is important to delineate epigenetic events associated with cellular transformation from “passenger” epimutations that arise independently of altered gene expression.
Here we have integrated whole-genome DNA methylation and gene expression data from purified primary non-diseased lymphocyte pre B-cell populations and diagnostic bone marrow from B-cell ALL patients to identify genes/pathways with associated DNA methylation and gene expression change during malignant transformation. Further, by comparing pediatric B-cell ALL subtypes to non-leukemic controls, we were able to identify subtype-specific epigenetic deregulation potentially linked to disease pathobiology.
Results
B-cell ALL (common)
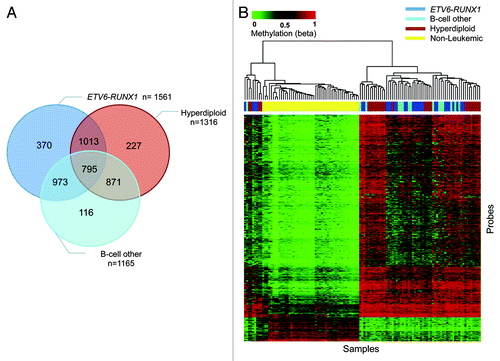
The DNA methylation status of 25 014 CpG loci was interrogated in 69 pediatric B-cell ALL patients (26 ETV6-RUNX1, 15 Hyperdiploid, and 28 “B-cell other”) and 48 non-leukemic control samples (Table S1). A common set of 795 probes (; Table S2), mapped to 602 genes, were found to be differentially methylated in all 3 subtypes relative to controls, confirming a common subtype-independent, B-cell ALL-associated DNA methylation “signature.”Citation20 As expected, unsupervised hierarchical clustering of these probes showed separation of the leukemic and non-leukemic samples (). Notably, 9 ALL cases (of mixed subtypes) clustered separately from the main leukemic group, with a profile more similar to control samples, although still distinct from the non-leukemic controls.
Figure 1. (A) Venn diagram displaying all significant probes found differentially methylated (adjusted P value < 0.05 and ∆ β > 0.3 or < –0.3), including 795 probes commonly differentially methylated between all subtypes. (B) Unsupervised hierarchical clustering of the 795 common probes showing separation of pediatric ALL samples from non-leukemic controls regardless of ALL subtype.
Gene expression data were generated for 17 of the B-cell ALL cases analyzed for DNA methylation using the Affymetrix U133Plus2 microarray (as previously describedCitation25). Gene expression data were available for 584 of the 602 differentially methylated genes identified. Of these 584 genes, 45% of their differentially methylated probes were located within the gene promoter (). Negative correlations () were observed between mean methylation and mean gene expression for probes located within the gene promoter, 5′UTR, 1st exon, and gene body, illustrating the commonly observed inverse relationship between DNA methylation and gene expression levels.
Figure 2. For the 795 (corresponding to 602 genes) methylation probes that were identified as commonly deregulated in all 3 subtypes, expression data were obtained for 584 genes (726 probes). (A) Shows the location of the 726 probes (B) Shows the correlation of mean methylation (β) vs. mean expression (Log2 ratio) for the 17 matched samples separated by probe location (C) (i) Plot of gene expression (∆Log2 Ratio) and methylation (∆ β) change between leukemic and non-leukemic control samples for 602 genes associated with the 795 differentially methylated probes. Genes displaying an inverse relationship (blue quadrants = 370 genes) were selected for ontology analysis. (ii) Shows the proportion of probes with relative gene expression differences (∆Log2 Ratio) between leukemic and non-leukemic control samples.
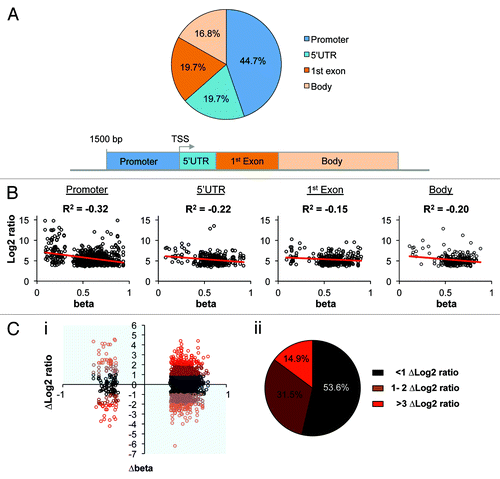
To investigate genes displaying concurrent DNA methylation and gene expression alterations in pediatric B-cell ALL as compared with non-leukemic controls, we obtained publicly available gene expression data for 6 FACS-sorted lymphoblast progenitors. Interestingly, however, 85% of gene expression probes of differentially methylated genes displayed < 2 ∆Log2 ratio between B-cell ALL samples and non-leukemic samples, indicating that, in a substantial proportion of genes, DNA methylation alterations did not translate to major changes in gene expression (; Fig. S1).
Integration of both fold-change gene expression (∆Log2 ratio) and change in DNA methylation (∆ β) data between leukemic and non-leukemic controls identified 325 genes that showed a significant increase in DNA methylation with a corresponding decrease in gene expression in B-cell ALL (bottom right shaded quadrant, , i). Conversely, we identified 45 genes that displayed a significant decrease in DNA methylation with a corresponding increase in gene expression in B-cell ALL (top left shaded quadrant, , i). These genes were therefore classified as epigenetically deregulated in B-cell ALL (Table S3). A comparison of the 370 epigenetically deregulated genes to our previously defined 115 gene signature of ETV6-RUNX1 ALL (generated from methylation data alone),Citation20 showed that less than half of the original signature was associated with corresponding gene expression change between control and diseased samples, highlighting the value of integrating both types of data (Fig. S2).
Ontology analysis of the 370 epigenetically deregulated genes revealed significant overrepresentation of molecular and cellular pathways associated with cancer/hematological malignancy, notably “cell-to-cell signaling and interaction,” “cell death/survival,” and “cellular development.” Many of the genes identified encode for plasma membrane proteins that are involved in extracellular signaling/cation transport, and are implicated in various apoptotic mechanisms (Fig. S3). Specifically, we identified hypermethylated and repressed genes encoding molecules involved in glutamate receptor signaling, G-protein signaling and cAMP mediated signaling (Fig. S3). Additionally, genes involved in cation transport were repressed, including 2 calcium channel subunits, 7 potassium channel subunits, 10 solute carriers, and 3 transient receptor potential cation channel subunits (Table S3).
Analysis revealed 4 C-X-C motif chemokines (CXCL1, CXCL3, CXCL5, and CXCL6) that are epigenetically repressed in B-cell ALL, all of which target CXC receptor 2. Two other cytokines were found to be epigenetically deregulated, including upregulation of LTA and downregulation of IL7. Notably, our analysis revealed 11 epigenetically deregulated kinases, including BLK, DDR1, and HSPB8, which were all found upregulated, while EPHA5, ERBB4, FGFR2, KDR, MAPK15, MYO3A, PAK7, and PRKD1 were downregulated. Several transcription factors were also found downregulated, notably, GATA4, HOX genes, several Zinc Fingers (ZNFs) and HLF, while TCF3 was found upregulated. Frequently reported in cancer, Wnt/β-catenin subunits were significantly overrepresented, including upregulation of WNT5b and WISP1, while pathway inhibitors SFRP1, PP2A, and SOX genes (SOX1, SOX9, SOX14, and SOX17) were all downregulated.
B-cell ALL (subtype-specific)
To investigate subtype-specific epigenetic deregulation, we performed linear modeling on the DNA methylation data between leukemic B-cell ALL subtypes. This identified 66 probes (associated with 52 genes) that delineated the ETV6-RUNX1 subtype from “B-cell other,” 172 probes (143 genes) delineating ETV6-RUNX1 from Hyperdiploid and 77 probes (67 genes) differentiating “B-cell other” from Hyperdiploid subtype (). Combining the 3 probe sets revealed 250 unique probes that clearly separated ETV6-RUNX1 and Hyperdiploid subtypes by unsupervised hierarchical clustering (Fig. S4A); however, several of the “B-cell other” samples displayed DNA methylation patterns similar to either the ETV6-RUNX1 (n = 2) or Hyperdiploid (n = 7) subtypes than to the remaining “B-cell other” group, highlighting the heterogeneity of “B-cell other” patients. Interestingly, a significant difference in the patient age of disease onset was observed between “B-cell other” patients clustering with Hyperdiploid subtype (older) than “B-cell other” patients clustering with ETV6-RUNX1 subtype (younger) (P = 0.05) and all other B-cell other patients (P = 0.03) (Fig. S4B).
Figure 3. (A) The 3 heatmaps display unsupervised hierarchical clustering of significant probes delineating the 3 subtypes. A total of 66 probes were found to be differentially methylated between the ETV6-RUNX1 and “B-cell other” samples, 172 probes were found to be differentially methylated between ETV6-RUNX1 and Hyperdiploid subtypes and 77 probes were differentially methylated between Hyperdiploid and “B-cell other.” (B) Correlations between methylation and gene expression differences (∆β and ∆Log2 Ratio) between subtypes.
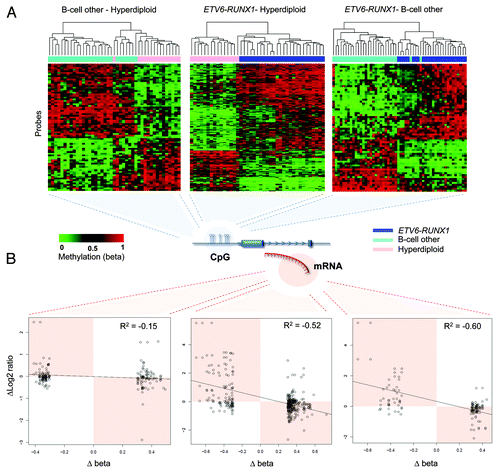
Expression data were available for 27/52 genes differentially methylated between ETV6-RUNX1 and “B-cell other,” 77/143 genes distinguishing ETV6-RUNX1 from Hyperdiploid and 39/67 genes distinguishing “B-cell other” from Hyperdiploid subtype. Gene expression levels and DNA methylation differences between each subtype were integrated to observe any relationship between methylation and expression. Although all comparisons displayed a negative correlation, a weaker correlation was observed between Hyperdiploid and “B-cell other” cases, highlighting a similarity between some “B-cell other” and Hyperdiploid patients, which is supported by the sample clustering above (; Fig. S4B).
The same approach used to identify B-cell ALL common epigenetic deregulation by comparison to non-leukemic samples identified 55, 51, and 13 epigenetically deregulated genes associated with ETV6-RUNX1, Hyperdiploid, and “B-cell other” subtypes respectively (Tables S4–6). The “B-cell other” subtype displayed upregulation of WWC1 and NELL1 compared with ETV6-RUNX1 and Hyperdiploid subtypes (Table S6). Additionally, CHST12 and LSP1 genes were upregulated while DSC3 was downregulated in “B-cell other” subtype compared with ETV6-RUNX1 (Fig. S5).
The ETV6-RUNX1 subtype displayed downregulation of LPXN and upregulation of SLC35B3, while the Hyperdiploid subtype displayed downregulation of BMPR1B, tumor suppressors FHIT and PTPRG and upregulation of LOC284837. Subtype-specific upregulation of the EPO receptor (EPOR) gene was identified in the ETV6-RUNX1 subtype, while the gene encoding the asparagine synthetase enzyme (ASNS) and CD44 were both downregulated in ETV6-RUNX1 subtype. Furthermore, the Hyperdiploid subtype was associated with upregulation of B-cell lymphoma genes (BCL2 and BCL9L) (Tables S4 and S5; Fig. S5).
Discussion
B-cell ALL epigenetic deregulation
A major challenge of identifying malignancy-associated epigenetic disruption is delineating functionally relevant epigenetic events associated with cellular transformation from those present in the original disease-free cell, or “passenger” epimutations, which arise independently of altered gene expression. In this study, we observed gross DNA methylation changes common to all subtypes of B-cell ALL relative to control, non-diseased tissue, with hypermethylation as the predominant feature (90% of probes) (Table S2). However, malignancy-associated promoter hypermethylation changes across several cancers are often associated with genes already repressed in the primary tissue.Citation24 By integrating DNA methylation and gene expression change data between leukemic and non-diseased tissue we have identified epigenetic deregulation of key genes and cellular pathways involved in pediatric B–cell ALL leukemogenesis.
Previously, we have published work on DNA methylation in pediatric B-cell ALL, which identified 115 genes displaying aberrant DNA methylation states (independent of gene expression change) that could be utilized as diagnostic biomarkers for B-cell ALL.Citation20 Recently, our work in DNA methylation has been replicated by 2 other studies that confirm many of our findings. Specifically, aberrant DNA methylation patterns in GRM7, GPR26, LPPR4, and ZNF560 were described by two genome-scale studies using similar (HM450) and distinct (HELP assay) DNA methylation analytical technologies.Citation22,Citation23 Furthermore, 447 of our 602 genes were identified by the HM450 platform as having DNA methylation alterations, highlighting the functional relevance to disease.Citation23
The significance of the epigenetically deregulated genes identified in B-cell ALL in contributing to two aspects of malignant phenotype (prevention of apoptosis and promotion of proliferation) is discussed below.
Apoptosis
The significant overrepresentation of repressed signaling pathways and plasma membrane bound subunits involved in cation transport within the list of epigenetically deregulated genes depicts functional cellular changes limiting apoptotic mechanisms and cellular interactions with the surrounding environment and explains some of the intrinsic malignant phenotypes observed in B-cell ALL.
A key apoptotic mechanism utilized by pre B-cells is the activation and propagation of cAMP mediated signaling.Citation26 The repression of genes involved in cAMP mediated signaling are protective against DNA damage-induced apoptosis in B-cell ALL.Citation27 Interestingly, we observed the epigenetic repression of a number of G-coupled receptor subunits known to initiate cAMP mediated signaling. Furthermore, we observed the epigenetic repression of adenylate cyclase and cAMP specific phosphodiesterases that catalyze the ATP-cAMP and cAMP-AMP reactions, respectively.Citation28,Citation29
Cell volume changes that are intrinsically tied to ion transportation are integral to apoptosis. We found calcium channel subunits, transient receptor potential cation channel subunits and potassium channel subunits to be hypermethylated and repressed. The deregulation of calcium channels and transient receptor potential channels has been shown to contribute to cell proliferation and apoptosis dysfunction.Citation30,Citation31 Meanwhile, sustained potassium channel opening and efflux are critical for cells to undergo apoptotic-associated cell shrinkage.Citation32
Single molecules can also exert large influences over cellular apoptotic ability. Specifically, we observed DNA methylation mediated repression of PAWR (PAR4), a potent stimulator of apoptosis in cancer cells and reported to have low levels of expression in several cancers.Citation33 Additionally, ERBB4, which has been shown to induce apoptosis in breast tumor cells,Citation34 was found to be hypermethylated and repressed.
The identification of repressed plasma-bound subunits involved in apoptotic transduction and cell signaling, irrespective of subtype, has implications for the development of next generation cancer therapy. Specifically, it would be interesting to examine the ability of leukemic cells to receive and propagate apoptotic signals following the use of DNA methyltransferase inhibitors.
Proliferation
Increased cellular proliferation is a hallmark of most cancers. Integral to proliferation are kinases that regulate a variety of cellular functions involved in cell homeostasis and are frequently mutated in many cancers.Citation35 Kinase activation has the potential to enhance cellular growth and proliferation; conversely, kinase repression could inhibit key developmental events potentially restricting differentiation. We identified promoter hypermethylation and concurrent downregulation of several kinases previously associated with malignancy, and promoter hypomethylation and concurrent upregulation of kinases frequently expressed in myeloid leukemia.Citation36-Citation40 Interestingly, we found promoter hypermethylation and repression of IL7 in all subtypes of B-cell ALL. Although recombinant IL7 has been reported to promote T-cell and B-cell ALL proliferation,Citation41 murine experiments have shown defective IL7R affects T-cell differentiation but not B-cell. Therefore, our results support the hypothesis that IL7 most likely contributes solely to T-cell, not B-cell, ALL progression.Citation42,Citation43 Additionally, we found important transcription factors involved in hematopoiesis (GATA4 and HOX genes) to be epigenetically repressed which could perturb differentiation.
Subtype-specific B-cell ALL epigenetic deregulation
Computational analysis of DNA methylation differences between genetic subtypes enabled us to identify subtype-specific DNA methylation patterns associated with pediatric B-cell ALL malignancy. Previous genome-scale DNA methylation interrogations have described distinct DNA methylation profiles associated with major B-cell ALL cytogenetic subtypes;Citation22,Citation44,Citation45 however, the strength of our study-design enables classification of epigenetic deregulation specific to each subtype and distinct from non-leukemic tissue. The utility of our approach is highlighted in the case of DSC3, previously reported to be hypomethylated within ETV6-RUNX1 subtype when compared with other B-cell ALL subtypes.Citation22,Citation44,Citation46 Through comparison with non-leukemic controls, we show the leukemic phenotype is in fact hypermethylation of the DSC3 promoter and concurrent downregulation within the Hyperdiploid and “B-cell other” subtypes compared with the ETV6-RUNX1 subtype and non-leukemic controls.
The ETV6-RUNX1 translocation is the most frequently occurring genetic mutation in pediatric ALL, contributing to leukemogenesis by blocking cellular differentiation and apoptosis, and is hypothesized to cooperate with growth promoting mutations for cancer onset.Citation47,Citation48 ETV6-RUNX1 binds and activates EPOR, contributing to leukemogenesis through activation of the JAK2-STAT5 pathway.Citation49 We report subtype-specific permissive promoter hypomethylation and concurrent upregulation of EPOR, in line with the results of a previous expression study.Citation50 The ETV6-RUNX1 subtype displayed hypermethylation of Asparaginase Synthetase (ASNS), integral for the synthesis of the essential amino acid, Asparagine. Although we did not observe a statistically significant difference in expression levels, the mean expression of ASNS was reduced in the ETV6-RUNX1 patients compared with other subtypes. The downregulation of ASNS reportedly increases the sensitivity of leukemic cells to L-asparaginase, an agent routinely used in ALL induction therapy.Citation51 This sensitivity is particularly pronounced in ETV6-RUNX1 patients.Citation52 Individualized dosing of L-aspariginase was recently reported to increase patient 5-y event-free survival.Citation53 It would be interesting to ascertain if individualized dosage is correlated with ASNS methylation status. If so, ASNS methylation could therefore be used as a biomarker.
The cell surface molecule, CD44, which is involved in angiogenesis and cell proliferation, differentiation, and migration,Citation54 has been shown to be downregulated in the ETV6-RUNX1 subtype.Citation55 We observed gene repression and promoter hypermethylation of CD44 within the ETV6-RUNX1 subtype as compared with other subtypes and non-leukemic controls. Increased CD44 expression has been associated with a higher risk of relapse.Citation56 As the ETV6-RUNX1 subtype has a relatively good outcome, the changes observed could represent a normo-cellular mechanism exploited by the Hyperdiploid and “B-cell other” subtypes, and could therefore contribute to malignant phenotype.
Hyperdiploid B-cell ALL has a less well-defined pathobiology than the ETV6-RUNX1 subtype. However, similar to ETV6-RUNX1, suggestions have been made that this karyotype arises prenatally.Citation57 With regard to epigenetic deregulation in Hyperdiploid B-cell ALL, we found promoter hypermethylation and reduced expression of FHIT, a tumor suppressor gene, in line with the results of a previous study.Citation58 Additionally, we observed promoter hypermethylation and repression of PTPRG, the transcriptional start site (TSS) of which runs bidirectionally to the TSS of FHIT. PTPRG is a protein tyrosine phosphatase receptor that has been previously shown to function as a tumor suppressor in myeloid leukemia.Citation59
Furthermore, we found the anti-apoptotic B-cell lymphoma gene, BCL2, and the B-cell lymphoma associated transcriptional regulator, BCL9L, displayed subtype-specific intra-genic hypomethylation with concurrent upregulation in the Hyperdiploid subtype. Expression of BCL2 contributes to many B-cell malignancies but has not previously been described within pediatric Hyperdiploid B-cell ALL.Citation60
The subtype-specific changes identified here increase biological understanding of pediatric B-cell ALL with the potential to refine current treatment regimens. For example, increasing L-Asp dosage for patients with ETV6-RUNX1 may reduce the need for other cytotoxic agents, while the use of current JAK inhibitors could target downstream effectors of EPOR activation in ETV6-RUNX1 patients. Hyperdiploid patients exhibit methylation-associated repression of a number of tumor suppressor genes and could potentially benefit from combination therapy with demethylating agents, such as azacitidine or decitabine.
Conclusions
Integrating epigenetic and transcription data are a powerful tool in identifying common and intra-disease epigenetic remodeling in a variety of disease contexts. The identification of common and subtype-specific epigenetically remodeled genes during malignant transformation defines robust biomarkers of the disease state and contributes important knowledge to disease pathogenesis. The findings have great potential to improve disease tracking and refine current therapeutics to reduce the cytotoxic burden of therapy and improve patient outcome.
Methods
Samples
This study was reviewed and approved by the Royal Children’s Hospital Human Research Ethics Committee (HREC27138). Samples for methylation analysis (Table S1) consisted of 42 archived B-cell ALL diagnostic bone marrow aspirates that were extracted from archived slides (>80% lymphoblast). DNA from a further 27 primary B-cell ALL diagnostic bone marrow aspirates were also isolated. Non-leukemic (control) samples for DNA methylation analysis consisted of 43 non-leukemic (<5% lymphoblast) bone marrow aspirates taken at patient remission, 4 Fluorescence Activated Cell Sorted (FACS) isolated hematopoietic progenitor cell populations (2 × CD19+ and 2 × CD34+) from the bone marrow of 2 unrelated and unaffected pediatric individuals and a single bone marrow sample (unsorted) from a healthy pediatric donor.
Samples for gene expression analysis consisted of 17 (of the previous 27) primary B-cell ALL diagnostic bone marrow aspirates. Our non-leukemic (control) expression data set consisted of 6 FACS sorted B-cell progenitor populations obtained from Gene Expression Omnibus (GEO) (GSE19599) which consisted of 2 immature B-cell populations (CD34-/CD19+/CD10low/CD20low), 2 pre B-cell populations (CD34-/CD19+/CD10+/CD20low) and 2 pro B-cell populations (CD34+/CD19+/CD10+).Citation61
Leukemic subtypes were determined by FISH and RT-PCR analysis of bone marrow at diagnosis. All ETV6-RUNX1 samples contained the ETV6-RUNX1 translocation, t(12;21). All Hyperdiploid samples contained >50 chromosomes. Patients lacking ETV6-RUNX1 t(12;21), E2A-PBX1 t(1;19), BCR-ABL, t(9;22) and translocated MLL t(4;11) were denoted “B-cell other.”
DNA methylation analysis
DNA extraction methods and bisulphite conversion of the archived and primary samples are as reported previously.Citation21,Citation48 Bisulphite converted DNA from samples were hybridized by the Australian Genome Research Facility (AGRF) and analyzed using the Illumina Infinium HumanMethylation27 (HM27) and HumanMethylation450 (HM450) BeadChip arrays according to manufacturer’s protocol. Illumina Genome Studio software was used to extract the raw M-values and probe intensities for downstream processing.
Data processing
Samples were retained that had an average detection value for all probes of P < 0.05.Citation62 Samples of each array were quantile normalized using lumi libraryCitation62 and batch corrected using the LIMMA program of the R statistical software package.Citation63 To eliminate sex bias, probes hybridizing to sex chromosomes were removed, leaving 25 014 usable probes common between the HM27 and HM450 bead arrays. Probe location was determined using the Illumina Infinium array annotations package (IlluminaHumanMethylation450k.db), promoter probes were defined as sitting within 1500 bp from a gene’s TSS.Citation64 Methylation M-values were used for feature selection modeling (described below) then converted to β values for correlation with expression data sets and reporting. A difference in methylation values (Δ β) for each probe was calculated for each subtype comparison using the formula:
Δβ = mean methylation β value (Subtype 1) – mean methylation β value (Subtype 2)
Gene expression analysis
Seventeen leukemic samples of mixed subtype (4 ETV6-RUNX1, 8 “B-cell other” and 5 Hyperdiploid) (Table S1) were analyzed using the Affymetrix U133Plus2 microarrays, as previously described.Citation25 Non-leukemic gene expression samples consisted of expression data obtained from GEO for 6 FACS sorted B-cell progenitors generated by Affymetrix U133 microarray (Lund University, GSE19599). Individual arrays were normalized using LIMMA R software. A difference in expression (Δ Log2 Ratio) values was calculated for each subtype comparison using the formula:
ΔLog2 ratio = mean Log2 ratio(Subtype 1) – mean Log2 ratio(Subtype2)
Feature selection
Linear modeling was applied to the normalized and batch corrected M-values, using LIMMA. B-cell ALL (common); LIMMA was applied between leukemic subtype samples and non-leukemic controls. Probes displaying an adjusted P value of < 0.05 and ∆ β of > 0.3 or < –0.3 were retained and filtered for probes common between subtypes. All correlations (R2) described in text are Pearson correlations. B-cell ALL (subtype-specific); LIMMA was applied between each subtype. Probes displaying an adjusted P value of < 0.05 were retained and then filtered for genes that displayed a negative correlation between DNA methylation and gene expression for each subtype analysis. Finally, the gene list was filtered for genes that displayed a ∆ β of > 0.3 or < –0.3 between leukemic subtype and non-leukemic controls.
Pathway analysis
Ontology analysis of the B-cell ALL (common) gene list was performed using Ingenuity Pathway Analysis (IPA) software (Ingenuity systems) on the probe Δ β values.
Additional material
Download Zip (1.5 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The Leukaemia Foundation of Australia supported this work through a Philip Desbrow Postdoctoral Fellowship (Wong N) and a Grant-in-Aid (Ashley DM, Wong N, Saffery R, and Craig JM). This work was funded by a National Health and Medical Research Project grant (Wong N, Craig JM, and Ashley DM) a Victorian Cancer Agency Grant. Wong N was also supported by My Room and the Children’s Cancer Centre Foundation. Support was also obtained from the National Institutes of Health (R01 CA140729; Carroll WL) and New York University Cancer Centre (support grant 5 P30 CA16087). MCRI was supported by the Victorian Government’s Operational Infrastructure Support Program. Morenos L is also supported by the NHMRC Dora Lush Postgraduate Research Scholarship and Saffery R by a NHMRC Senior Research Fellowship.
References
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature 1986; 321:209 - 13; http://dx.doi.org/10.1038/321209a0; PMID: 2423876
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21; http://dx.doi.org/10.1101/gad.947102; PMID: 11782440
- Mathers JC. Session 2: Personalised nutrition. Epigenomics: a basis for understanding individual differences?. Proc Nutr Soc 2008; 67:390 - 4; http://dx.doi.org/10.1017/S0029665108008744; PMID: 18847515
- Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med 2011; 3:75ra25; http://dx.doi.org/10.1126/scitranslmed.3001875; PMID: 21430268
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al, Cancer Genome Atlas Research Network. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010; 17:510 - 22; http://dx.doi.org/10.1016/j.ccr.2010.03.017; PMID: 20399149
- Teodoridis JM, Hardie C, Brown R. CpG island methylator phenotype (CIMP) in cancer: causes and implications. Cancer Lett 2008; 268:177 - 86; http://dx.doi.org/10.1016/j.canlet.2008.03.022; PMID: 18471961
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999; 96:8681 - 6; http://dx.doi.org/10.1073/pnas.96.15.8681; PMID: 10411935
- Leone G, Teofili L, Voso MT, Lübbert M. DNA methylation and demethylating drugs in myelodysplastic syndromes and secondary leukemias. Haematologica 2002; 87:1324 - 41; PMID: 12495905
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003; 349:2042 - 54; http://dx.doi.org/10.1056/NEJMra023075; PMID: 14627790
- Cancer Genome Atlas N, Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490:61 - 70; http://dx.doi.org/10.1038/nature11412; PMID: 23000897
- Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, Malik S, Pan F, Noushmehr H, van Dijk CM, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012; 22:271 - 82; http://dx.doi.org/10.1101/gr.117523.110; PMID: 21659424
- Deneberg S, Guardiola P, Lennartsson A, Qu Y, Gaidzik V, Blanchet O, Karimi M, Bengtzén S, Nahi H, Uggla B, et al. Prognostic DNA methylation patterns in cytogenetically normal acute myeloid leukemia are predefined by stem cell chromatin marks. Blood 2011; 118:5573 - 82; http://dx.doi.org/10.1182/blood-2011-01-332353; PMID: 21960591
- Baylin SB. Resistance, epigenetics and the cancer ecosystem. Nat Med 2011; 17:288 - 9; http://dx.doi.org/10.1038/nm0311-288; PMID: 21383739
- Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343:1350 - 4; http://dx.doi.org/10.1056/NEJM200011093431901; PMID: 11070098
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352:997 - 1003; http://dx.doi.org/10.1056/NEJMoa043331; PMID: 15758010
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18:553 - 67; http://dx.doi.org/10.1016/j.ccr.2010.11.015; PMID: 21130701
- Schuebel KE, Chen W, Cope L, Glöckner SC, Suzuki H, Yi JM, Chan TA, Van Neste L, Van Criekinge W, van den Bosch S, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet 2007; 3:1709 - 23; http://dx.doi.org/10.1371/journal.pgen.0030157; PMID: 17892325
- Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 2002; 21:5483 - 95; http://dx.doi.org/10.1038/sj.onc.1205699; PMID: 12154409
- Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980; 20:85 - 93; http://dx.doi.org/10.1016/0092-8674(80)90237-8; PMID: 6156004
- Wong NC, Ashley D, Chatterton Z, Parkinson-Bates M, Ng HK, Halemba MS, Kowalczyk A, Bedo J, Wang Q, Bell K, et al. A distinct DNA methylation signature defines pediatric pre-B cell acute lymphoblastic leukemia. Epigenetics 2012; 7:535 - 41; http://dx.doi.org/10.4161/epi.20193; PMID: 22531296
- Davidsson J, Lilljebjörn H, Andersson A, Veerla S, Heldrup J, Behrendtz M, Fioretos T, Johansson B. The DNA methylome of pediatric acute lymphoblastic leukemia. Hum Mol Genet 2009; 18:4054 - 65; http://dx.doi.org/10.1093/hmg/ddp354; PMID: 19679565
- Figueroa ME, Chen SC, Andersson AK, Phillips LA, Li Y, Sotzen J, Kundu M, Downing JR, Melnick A, Mullighan CG. Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. J Clin Invest 2013; 123:3099 - 111; http://dx.doi.org/10.1172/JCI66203; PMID: 23921123
- Nordlund J, Bäcklin CL, Wahlberg P, Busche S, Berglund EC, Eloranta ML, Flaegstad T, Forestier E, Frost BM, Harila-Saari A, et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol 2013; 14:r105; http://dx.doi.org/10.1186/gb-2013-14-9-r105; PMID: 24063430
- Sproul D, Kitchen RR, Nestor CE, Dixon JM, Sims AH, Harrison DJ, Ramsahoye BH, Meehan RR. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol 2012; 13:R84; http://dx.doi.org/10.1186/gb-2012-13-10-r84; PMID: 23034185
- Hogan LE, Meyer JA, Yang J, Wang J, Wong N, Yang W, Condos G, Hunger SP, Raetz E, Saffery R, et al. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 2011; 118:5218 - 26; http://dx.doi.org/10.1182/blood-2011-04-345595; PMID: 21921043
- Myklebust JH, Josefsen D, Blomhoff HK, Levy FO, Naderi S, Reed JC, Smeland EB. Activation of the cAMP signaling pathway increases apoptosis in human B-precursor cells and is associated with downregulation of Mcl-1 expression. J Cell Physiol 1999; 180:71 - 80; http://dx.doi.org/10.1002/(SICI)1097-4652(199907)180:1<71::AID-JCP8>3.0.CO;2-N; PMID: 10362019
- Naderi EH, Ugland HK, Diep PP, Josefsen D, Ruud E, Naderi S, Blomhoff HK. Selective inhibition of cell death in malignant vs normal B-cell precursors: implications for cAMP in development and treatment of BCP-ALL. Blood 2013; 121:1805 - 13; http://dx.doi.org/10.1182/blood-2012-08-452698; PMID: 23299313
- Neves SR, Ram PT, Iyengar R. G protein pathways. Science 2002; 296:1636 - 9; http://dx.doi.org/10.1126/science.1071550; PMID: 12040175
- Renna M, Jimenez-Sanchez M, Sarkar S, Rubinsztein DC. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem 2010; 285:11061 - 7; http://dx.doi.org/10.1074/jbc.R109.072181; PMID: 20147746
- Pedersen SF, Stock C. Ion channels and transporters in cancer: pathophysiology, regulation, and clinical potential. Cancer Res 2013; 73:1658 - 61; http://dx.doi.org/10.1158/0008-5472.CAN-12-4188; PMID: 23302229
- Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol 2003; 5:1041 - 3; http://dx.doi.org/10.1038/ncb1203-1041; PMID: 14647298
- Bortner CD, Cidlowski JA. Cell shrinkage and monovalent cation fluxes: role in apoptosis. Arch Biochem Biophys 2007; 462:176 - 88; http://dx.doi.org/10.1016/j.abb.2007.01.020; PMID: 17321483
- García-Cao I, Duran A, Collado M, Carrascosa MJ, Martín-Caballero J, Flores JM, Diaz-Meco MT, Moscat J, Serrano M. Tumour-suppression activity of the proapoptotic regulator Par4. EMBO Rep 2005; 6:577 - 83; http://dx.doi.org/10.1038/sj.embor.7400421; PMID: 15877079
- Das PM, Thor AD, Edgerton SM, Barry SK, Chen DF, Jones FE. Reactivation of epigenetically silenced HER4/ERBB4 results in apoptosis of breast tumor cells. Oncogene 2010; 29:5214 - 9; http://dx.doi.org/10.1038/onc.2010.271; PMID: 20603612
- Tsatsanis C, Spandidos DA. The role of oncogenic kinases in human cancer (Review). [Review] Int J Mol Med 2000; 5:583 - 90; PMID: 10812005
- Quentmeier H, Eberth S, Romani J, Weich HA, Zaborski M, Drexler HG. DNA methylation regulates expression of VEGF-R2 (KDR) and VEGF-R3 (FLT4). BMC Cancer 2012; 12:19; http://dx.doi.org/10.1186/1471-2407-12-19; PMID: 22251800
- Kober P, Bujko M, Olędzki J, Tysarowski A, Siedlecki JA. Methyl-CpG binding column-based identification of nine genes hypermethylated in colorectal cancer. Mol Carcinog 2011; 50:846 - 56; http://dx.doi.org/10.1002/mc.20763; PMID: 21438024
- Fu DY, Wang ZM, Wang BL, Chen L, Yang WT, Shen ZZ, Huang W, Shao ZM. Frequent epigenetic inactivation of the receptor tyrosine kinase EphA5 by promoter methylation in human breast cancer. Hum Pathol 2010; 41:48 - 58; http://dx.doi.org/10.1016/j.humpath.2009.06.007; PMID: 19733895
- Zhang H, Peng C, Hu Y, Li H, Sheng Z, Chen Y, Sullivan C, Cerny J, Hutchinson L, Higgins A, et al. The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat Genet 2012; 44:861 - 71; http://dx.doi.org/10.1038/ng.2350; PMID: 22797726
- Favreau AJ, Cross EL, Sathyanarayana P. miR-199b-5p directly targets PODXL and DDR1 and decreased levels of miR-199b-5p correlate with elevated expressions of PODXL and DDR1 in acute myeloid leukemia. Am J Hematol 2012; 87:442 - 6; http://dx.doi.org/10.1002/ajh.23129; PMID: 22374871
- Touw I, Pouwels K, van Agthoven T, van Gurp R, Budel L, Hoogerbrugge H, Delwel R, Goodwin R, Namen A, Löwenberg B. Interleukin-7 is a growth factor of precursor B and T acute lymphoblastic leukemia. Blood 1990; 75:2097 - 101; PMID: 2189505
- Silva A, Laranjeira AB, Martins LR, Cardoso BA, Demengeot J, Yunes JA, Seddon B, Barata JT. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res 2011; 71:4780 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-10-3606; PMID: 21593192
- Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat Genet 1998; 20:394 - 7; http://dx.doi.org/10.1038/3877; PMID: 9843216
- Busche S, Ge B, Vidal R, Spinella JF, Saillour V, Richer C, Healy J, Chen SH, Droit A, Sinnett D, et al. Integration of high-resolution methylome and transcriptome analyses to dissect epigenomic changes in childhood acute lymphoblastic leukemia. Cancer Res 2013; 73:4323 - 36; http://dx.doi.org/10.1158/0008-5472.CAN-12-4367; PMID: 23722552
- Milani L, Lundmark A, Kiialainen A, Nordlund J, Flaegstad T, Forestier E, Heyman M, Jonmundsson G, Kanerva J, Schmiegelow K, et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood 2010; 115:1214 - 25; http://dx.doi.org/10.1182/blood-2009-04-214668; PMID: 19965625
- Nordlund J, Kiialainen A, Karlberg O, Berglund EC, Goransson-Kultima H, Sonderkaer M, Nielsen KL, Gustafsson MG, Behrendtz M, Forestier E, et al. Digital gene expression profiling of primary acute lymphoblastic leukemia cells. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2012; 26:1218 - 27; http://dx.doi.org/10.1038/leu.2011.358
- Zelent A, Greaves M, Enver T. Role of the TEL-AML1 fusion gene in the molecular pathogenesis of childhood acute lymphoblastic leukaemia. Oncogene 2004; 23:4275 - 83; http://dx.doi.org/10.1038/sj.onc.1207672; PMID: 15156184
- Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 2011; 29:551 - 65; http://dx.doi.org/10.1200/JCO.2010.30.7405; PMID: 21220611
- Torrano V, Procter J, Cardus P, Greaves M, Ford AM. ETV6-RUNX1 promotes survival of early B lineage progenitor cells via a dysregulated erythropoietin receptor. Blood 2011; 118:4910 - 8; http://dx.doi.org/10.1182/blood-2011-05-354266; PMID: 21900195
- Fine BM, Stanulla M, Schrappe M, Ho M, Viehmann S, Harbott J, Boxer LM. Gene expression patterns associated with recurrent chromosomal translocations in acute lymphoblastic leukemia. Blood 2004; 103:1043 - 9; http://dx.doi.org/10.1182/blood-2003-05-1518; PMID: 14525776
- Li BS, Gu LJ, Luo CY, Li WS, Jiang LM, Shen SH, Jiang H, Shen SH, Zhang B, Chen J, et al. The downregulation of asparagine synthetase expression can increase the sensitivity of cells resistant to l-asparaginase. Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK 2006; 20:2199 - 201; http://dx.doi.org/10.1038/sj.leu.2404423
- Ramakers-van Woerden NL, Pieters R, Loonen AH, Hubeek I, van Drunen E, Beverloo HB, Slater RM, Harbott J, Seyfarth J, van Wering ER, et al. TEL/AML1 gene fusion is related to in vitro drug sensitivity for L-asparaginase in childhood acute lymphoblastic leukemia. Blood 2000; 96:1094 - 9; PMID: 10910927
- Vrooman LM, Stevenson KE, Supko JG, O’Brien J, Dahlberg SE, Asselin BL, Athale UH, Clavell LA, Kelly KM, Kutok JL, et al. Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 2013; 31:1202 - 10; http://dx.doi.org/10.1200/JCO.2012.43.2070; PMID: 23358966
- Naor D, Nedvetzki S, Golan I, Melnik L, Faitelson Y. CD44 in cancer. Crit Rev Clin Lab Sci 2002; 39:527 - 79; http://dx.doi.org/10.1080/10408360290795574; PMID: 12484499
- Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, Behm FG, Raimondi SC, Relling MV, Patel A, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002; 1:133 - 43; http://dx.doi.org/10.1016/S1535-6108(02)00032-6; PMID: 12086872
- Liu J, Jiang G. CD44 and hematologic malignancies. Cell Mol Immunol 2006; 3:359 - 65; PMID: 17092433
- Paulsson K, Johansson B. High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 2009; 48:637 - 60; http://dx.doi.org/10.1002/gcc.20671; PMID: 19415723
- Zheng S, Ma X, Zhang L, Gunn L, Smith MT, Wiemels JL, Leung K, Buffler PA, Wiencke JK. Hypermethylation of the 5′ CpG island of the FHIT gene is associated with hyperdiploid and translocation-negative subtypes of pediatric leukemia. Cancer Res 2004; 64:2000 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-03-2387; PMID: 15026336
- Della Peruta M, Martinelli G, Moratti E, Pintani D, Vezzalini M, Mafficini A, Grafone T, Iacobucci I, Soverini S, Murineddu M, et al. Protein tyrosine phosphatase receptor type gamma is a functional tumor suppressor gene specifically downregulated in chronic myeloid leukemia. Cancer Res 2010; 70:8896 - 906; http://dx.doi.org/10.1158/0008-5472.CAN-10-0258; PMID: 20959494
- Dutta C, Day T, Kopp N, van Bodegom D, Davids MS, Ryan J, Bird L, Kommajosyula N, Weigert O, Yoda A, et al. BCL2 suppresses PARP1 function and nonapoptotic cell death. Cancer Res 2012; 72:4193 - 203; http://dx.doi.org/10.1158/0008-5472.CAN-11-4204; PMID: 22689920
- Andersson A, Edén P, Olofsson T, Fioretos T. Gene expression signatures in childhood acute leukemias are largely unique and distinct from those of normal tissues and other malignancies. BMC Med Genomics 2010; 3:6; http://dx.doi.org/10.1186/1755-8794-3-6; PMID: 20211010
- Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24:1547 - 8; http://dx.doi.org/10.1093/bioinformatics/btn224; PMID: 18467348
- Smyth GK. Limma: linear models for microarray data. New York: Springer, 2005.
- Triche T Jr. IlluminaHumanMethylation450k.db: Illumina Human Methylation 450k annotation data. R package version 1.4.7. Bioconductor; 2012.
- Tong WH, Pieters R, Hop WC, Lanvers-Kaminsky C, Boos J, van der Sluis IM. No evidence of increased asparagine levels in the bone marrow of patients with acute lymphoblastic leukemia during asparaginase therapy. Pediatr Blood Cancer 2013; 60:258 - 61; http://dx.doi.org/10.1002/pbc.24292; PMID: 22961784