
Abstract
A commonly deleted region in chronic lymphocytic leukemia (CLL) is the 11q22–23 region, which encompasses the ATM gene. Evidence suggests that tumor suppressor genes other than ATM are likely to be involved in CLL with del(11q). A microRNA (miR) cluster including the miR-34b and miR-34c genes is located, among other genes, within the commonly deleted region (CDR) at 11q. Interestingly, these miRs are part of the TP53 network and have been shown to be epigenetically regulated. In this study, we investigated the expression and methylation status of these miRs in a well-characterized cohort of CLL, including cases with/without 11q-deletion. We show that the miR-34b/c promoter was aberrantly hypermethylated in a large proportion of CLL cases (48%, 25/52 cases). miR-34b/c expression correlated inversely to DNA methylation (P = 0.003), and presence of high H3K37me3 further suppressed expression regardless of methylation status. Furthermore, increased miR-34b/c methylation inversely correlated with the presence of 11q-deletion, indicating that methylation and del(11q) independently silence these miRs. Finally, 5-azacytidine and trichostatin A exposure synergistically increased the expression of miR-34b/c in CLL cells, and transfection of miR-34b or miR-34c into HG3 CLL cells significantly increased apoptosis. Altogether, our novel data suggest that miR-34b/c is a candidate tumor suppressor that is epigenetically silenced in CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is a clinically and biologically heterogeneous malignancy, where detection of certain recurrent genomic aberrations can aid prognostication and therapy selection. There are four recurrent chromosomal aberrations that are more frequent in CLL, i.e., trisomy 12, deletion of 13q, 11q, and 17p.Citation1,Citation2
Cases with deletion of 11q are often characterized by an aggressive clinical course, suggesting the existence of a disease driving gene or genes within the commonly deleted region (CDR). Previous studies have delimited this region to 11q22.3–q23.1, encompassing the ATM gene in almost all cases,Citation2-Citation4while some studies have narrowed it down to a smaller region that does not include the ATM gene.Citation5 Since ATM is only mutated in a minority of CLL samples, even in those with del(11q),Citation6,Citation7 this suggests that there might be another tumor suppressor gene within the region. Several potential candidate genes have been suggested, including BTG4 and POU2AF1.Citation8,Citation9 We previously described a CLL case with a microdeletion involving six genes in the CDR, adjacent to the implicated tumor suppressor gene ATM but separated from ATM by a small heterozygote region.Citation10
This part of the CDR at 11q also contains a miR cluster, containing miR-34b and miR-34c, which are regulated by a common promoter that also regulates the BTG4 gene. microRNAs are small, non-transcribed RNA oligonucleotides, typically 21–23 bp long, that negatively regulate protein synthesis through pairing to coding mRNA at their 3′ UTR, blocking translation.Citation11 Importantly, two other miRs, miR-15 and miR-16, located at 13q14 and deleted in more than 50% of all CLL cases, were the first miRs to be linked to pathogenesis in any malignancy.Citation12 Loss of these miRs leads to upregulation of BCL2 and thereby resistance to apoptosis.Citation12,Citation13
Considering that a deletion of a tumor suppressor on one allele can often be accompanied by epigenetic silencing on the other allele,Citation14-Citation16 and the fact that miR34b/c have been shown to be epigenetically regulated by DNA methylation in other cancer types,Citation17-Citation19 we decided to investigate the epigenetic status of miR-34b/c promoter region in CLL. For the first time, we show that the miR-34b/c promoter is aberrantly hypermethylated in CLL and that methylation at this site is more commonly observed in CLL patients without an 11q deletion. Methylation of the miR-34b/c promoter was also shown to correlate with expression, and repressive H3K27 trimethylation at the same site further decreased expression. Finally, overexpression of both miR-34b and miR-34c could induce apoptosis in the human CLL cell line HG3, which further suggests that miR-34b/c act as tumor suppressors in CLL.
Results
The BTG4/miR-34b/c shared promoter is frequently methylated in CLL but not in normal leukocytes
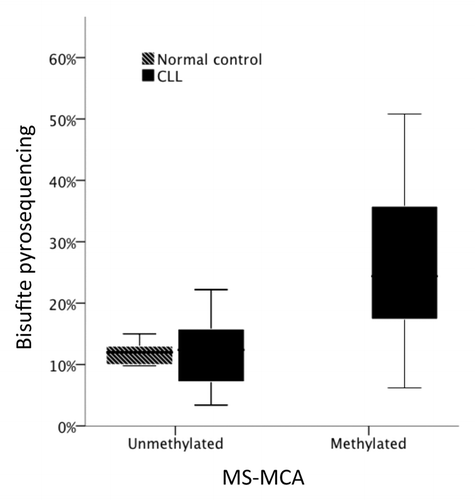
Five of the six gene promoters within the previously described microdeletion at 11q,Citation20 were selected for methylation analysis (POU2AF1, SNFLK2, PPP2R1, LAYN, BTG4/miR-34b/c). One gene, FLJ46266, did not contain a CpG island in its promoter region and was excluded from further analysis. Ten samples were initially screened for promoter methylation by MS-MCA. POU2AF1, SNFLK2, and PPP2R1 were unmethylated in all samples. LAYN was methylated in two samples and BTG4/miR-34b/c in four out of ten samples (). We extended our screening of LAYN methylation to 15 samples but no further case was found to be methylated. In contrast, when BTG4-miR-34b/c shared promoter was analyzed in a total of 52 CLL samples, a large proportion of cases (48%, 25 of 52) were deemed as methylated. Importantly, peripheral blood mononuclear cells from healthy controls (n = 2), CD19 positive B cells from healthy controls (n = 3) and commercially available mixed leukocyte DNA from multiple donors were all found to be unmethylated. To validate the MS-MCA findings, we applied bisulfite pyrosequencing to sequence 48 CLL cases and 6 control samples, including 32 samples previously analyzed by MS-MCA (28 CLL cases and CD19+ cells from three healthy donors and PBMC_s from one) (). Results were highly concurrent with the MS-MCA results (P = 0.001).
Table 1. DNA promoter methylation frequency by MS-MCA. N methylated / n tested
Figure 1. DNA methylation as measured by MS-MCA (x-axis) compared with bisulfite pyrosequencing (y-axis) in normal controls (n = 4) and CLL samples (n = 28).
Expression of miR-34b and miR34-c in relation to miR-34a
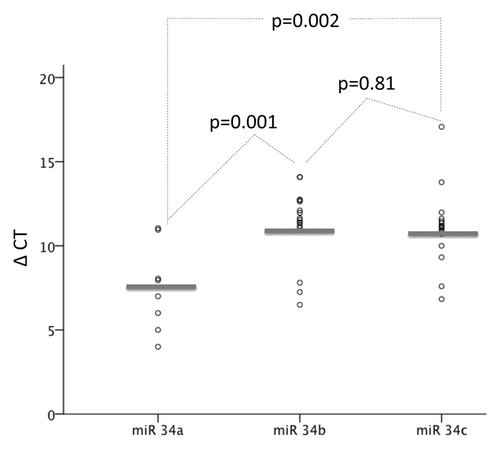
Expression levels of miR-34b and miR-34c were measured by RQ-PCR in 16 and 15 primary CLL samples, respectively, and miR-34a in 8 of those. There was no difference between miR-34b and miR-34c expression (P = 0.2), which instead was highly correlated (R2 = 0.973, P < 0.001). There was no correlation of either miR-34b or miR-34c with miR-34a (R2 = -16, P = 0.6 and R2 = 0.05, P = 0.9, respectively). Expression of miR-34a was generally higher than both miR-34b (P = 0.004) and miR-34c (P = 0.002) ().
Figure 2. Expression of miR-34a (n = 8), miR-34b (n = 16), and miR-34c (n = 15) by RQ-PCR in CLL samples. ΔΔCT for each miR compared with the reference gene RNU6 was calculated. P values indicate difference in expression between miRs.
Role of DNA methylation in silencing miR-34b/c expression
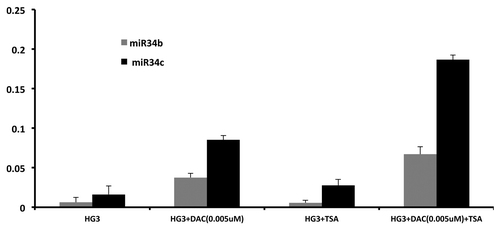
We defined a cutoff of 18.5% DNA methylation by pyrosequencing at the miR-34b/c promoter, which equaled three standard deviations above the mean DNA methylation of normal samples (n = 5 mean 11.96%, SD = 2.17%). Using this cutoff, expression levels of miR-34b and miR-34c were higher in unmethylated as compared with methylated samples (P = 0.007, ), indicating a correlation between DNA methylation and expression (Spearman’s Rho -0.39, P = 0.052). Reversibility was shown by incubating the human CLL cell line HG3, which is hypermethylated at the miR-34b/c promoter, with the demethylating agent 5-aza-2'-deoxycytidine (DAC) with and without HDAC inhibitor Trichostatin A (TSA). Interestingly, the combination of DAC and TSA showed a synergistic increase of miR-34b and miR-34c expression compared with DAC or TSA alone ().
Figure 3. Expression of miR-34b and miR-34b in unmethylated (n = 9) vs. methylated (n = 6) CLL samples by RQ-PCR. Expression levels are presented as in and p-values show the difference in expression between indicated groups.
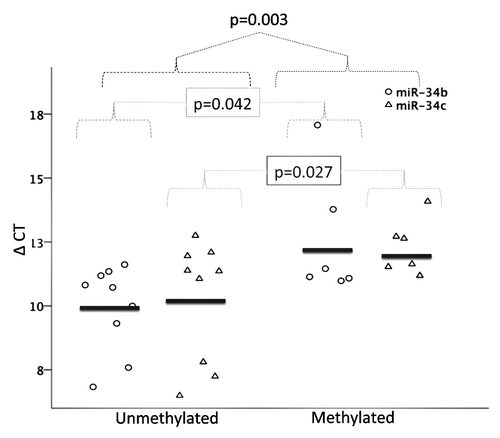
Figure 4. Effect on miR-34b and miR-34b expression by Decitabine (DAC) and trichostatin A (TSA) alone and in combination. HG3 cells were cultured either with DAC at 0,005 μM/L for 72 h,, with TSA at 500nM/L for the last 24 h or in combination with DAC for 48 h followed by TSA for 24 h. Reference gene for RQ-PCR analysis was RNU6.
Methylation in relation to clinical parameters, genomic aberrations and IGHV mutation status
Methylation of the miR-34b/c promoter region was unrelated to age or gender (P = 0.34 and P = 0.65, respectively). Since the region of interest in this study was localized on chromosome 11q23, we expected some relationship with the occurrence of deletions of that region. However, a trend toward an inverse relationship between DNA methylation and 11q deletion was found: 54% (n = 19/35) of non-11q-deleted cases were methylated compared with 31% (n = 5/16) of 11q-deleted CLL cases (P = 0.13), using a cutoff for del(11q) of 5% of the cells by FISH. There was also a trend toward increased methylation in IGHV mutated samples, 63% vs. 39% in unmutated samples (P = 0.10). Interestingly, using a more stringent cutoff for defining del(11q) cases (20% instead of 5% cells by FISH), the inverse relationship became stronger and statistically significant: 54% (n = 19/35) of non-11q-deleted cases were methylated compared with 15% (n = 2/13) of 11q-deleted CLL cases (P = 0.016). No correlations between del(13q), del(17p), trisomy 12 or the combination of 11q and 17p deletions and miR-34b/c methylation were found.
Presence of repressive chromatin mark H3K27me3 and correlation to expression
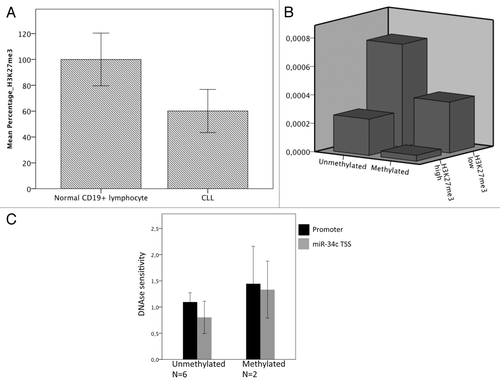
Since we found a synergistic effect of TSA and DAC on the induction of miR-34b and miR-34c and as the expression levels of miR-34b and miR-34c were highly variable even among the unmethylated DNA cases, we speculated whether other repressive epigenetic mechanisms were present. We investigated six CLL samples and three normal CD19+ cells from healthy subjects by ChIP for the repressive chromatin mark H3K27me3. H3K27me3 was found to be present in all six CLL samples and three normal controls at varying levels, with generally lower levels in the CLL samples than among the normal controls (P = 0.048) (). Out of the six samples where both promoter methylation and H3K27me3 was measured along with miR expression, the lowest expression levels of miR-34b and miR-34c were seen in samples that were both DNA methylated and marked with more than median levels of H3K23me3 (). Interestingly, the level of DNA methylation did not significantly change nucleosome occupancy measured as DNase hypersensitivity (DHSS) in eight CLL samples, neither in the promoter nor closer to the transcription start site (P = 0.4 and 0.3, respectively, ).
Figure 5. (A) Higher normalized levels of H3K27me3 at the miR-34b/c promoter in normal CD19+ lymphocytes (n = 3) compared with CLL samples (n = 6)(P = 0.048). Error bars show ± 2 SD (B) miR-34b/c expression in relation to DNA methylation (x-axis) and H3K27me3 levels (dichotomized at median) (z-axis). Sample numbers are indicated in the figure and individual measurements for each sample are given in Table S2. (C) Nucleosome occupancy measured by DHSS, normalized to GADPH. There is no difference between unmethylated and methylated CLL samples, P = 0.4 and 0.3 for promoter and TSS respectively. Error bars show ± 2 SD.
Overexpression of miR-34b and miR-34c increases apoptosis in HG3 cells
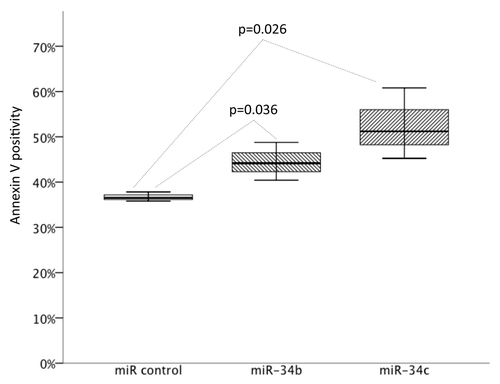
In order to study the functional role of miR-34b/c as a potential tumor suppressor, we transfected the HG3 CLL cell line either with miR-34b, miR-34c or miRNA mimic control using the Neon transfection system. The transfection efficacy was over 90%. Overexpressing either miR-34b and miR-34c each increased the number of apoptotic cells compared with mock transfected, supporting a tumor suppressor role in CLL cells (P = 0.036 and P = 0.026 for miR-34b and miR-34c, respectively)().
Figure 6. Results of miR-34b and miR-34c overexpression on apoptosis measured as Annexin V positivity by FACS in the HG3 cells incubated for 24 h. Box plots indicate the percentages of apoptosis of six individual experiments (P = 0.036 for miR-34b and P = 0.026 for miR-34c compared with control).
Discussion
It is still unclear what genes that are implicated in the leukemogenic process in CLL with 11q-deletion. The CDR at 11q encompasses the ATM gene, but ATM is only mutated, and thus biallelically lost, in a minority of the 11q-deleted cases.Citation4,Citation6 Recently, we reported a single CLL case with a microdeletion within the CDR at 11q23, which did not contain the ATM gene but instead harbored a miR cluster containing miR-34b and miR-34c among other genes.Citation10 As miR-34b/c has shown to be epigenetically silenced in other cancers,Citation17-Citation19 DNA methylation of the miR-34b/c promoter and 4 other genes within this microdeletion was analyzed. Interestingly, only miR-34b/c showed aberrant methylation in a significant proportion of CLL samples. Given the role of miR-15a/16 in the pathogenesis in CLL with deletion of 13q,Citation13 we aimed to further study miR-34b/c in CLL and its relationship to 11q deletion as well as the expression and epigenetic status of these miRs.
We here show a high prevalence (48%) of aberrant DNA methylation in the shared promoter region of BTG4 and miR-34b/c in CLL. The miR-34b/c analog miR-34a is located on chromosome 1p and has been implicated as a tumor suppressor in CLL,Citation21 making the discovery of methylation of the miR-34b/c promoter highly interesting. miR-34a and the clustered miR-34b/c are transcriptional targets of TP53 and reintroduction of miR-34s in deficient cells increases apoptosis and decreases growth.Citation22 Furthermore, miR-34b/c is silenced by hypermethylation in oral squamous cell carcinoma, malignant melanoma and colon cancer.Citation20,Citation23,Citation24
We also found an inverse correlation between 11q-deletion and promoter DNA methylation, indicating that there could be a selective pressure toward repression of this locus in CLL, either by deletion or epigenetic silencing. Using a low cutoff for defining del(11q), there was only a trend, but in samples with a higher percentage of the CLL showing a 11q deletion, the anti-correlation between DNA methylation and 11q deletion was statistically significant. This is likely explained by the fact that methylation analysis was performed on the bulk CLL cells and then, cells with del(11q) and with potentially lower methylation levels have to constitute a larger proportion of the total cell population in order to be detected.
Elevated expression levels of miR-34b/c were found among the unmethylated CLL samples, whereas in normal CD19-positive lymphocytes, levels were low while still unmethylated. We also observed a synergistic effect of DAC and TSA exposure on miR34-b/c expression levels in the HG3 CLL cell line. This prompted us to investigate the levels of the histone modifications in CLL samples. Normal cells showed the highest level of the repressive H3K27me3 mark, whereas levels in CLL cells were generally lower but variable. This indicates a switch from a more easily reversible repressive chromatin state with H3K27me3 to less easily reversible DNA methylation in CLL. We interpret this as H3K27me3 in part being exchanged to more stable DNA methylation in CLL. However, in some cases, the H3K27me3 mark remains even after induction of DNA methylation, seemingly contributing to transcriptional repression (). In prostate cancer, similar types of epigenetic switches have previous been proposed.Citation25 Somewhat surprisingly, no correlation was found between increased DNA methylation and more condensed chromatin. However, changes in epigenetic marks such as histone modifications and DNA methylation do not necessarily cause a change in nucleosome occupancy.Citation26 Still, we cannot exclude that the lack of difference between methylated and unmethylated samples regarding DNase hypersensitivity in our analysis is caused by too low resolution of the methodology. There might also be changes in nucleosome occupancy at other regions than those analyzed.Citation26
The introduction of miR-34b and miR-34c transcripts into the human CLL HG3 cell line induced a significant degree of apoptosis, further suggesting a functional role of miR-34b/c in CLL. However, since there is sequence and predicted target similarity between miR-34b/c and miR-34a, this gain of function experiment does not rule out a class effect of miR-34s and that miR-34a could substitute miR-34b/c activity. Zenz et al. found miR-34a to be an important downstream effector of TP53 in CLL and argued that miR-34b/c unlikely had a role, based on their finding of low expression levels of miR-34b/c regardless of 11q-deletion status.Citation6 On the contrary, we find that miR-34b and miR-34c have higher expression in the absence of promoter methylation and that promoter methylation is more common among CLL cases without or with a low percentage of 11q deletions. Expression levels of miR-34a were generally higher compared with miR-34b/c expression but importantly, there was no correlation between miR-34a and miR-34b/c expression. This suggests that miR-34b/c and miR-34a are differentially regulated but potentially equally important in CLL.
Since BTG4 is regulated from the same promoter as miR-34b/c, BTG4 could potentially be a tumor suppressor in CLL. However, CLL cells and normal B-cells showed equally low expression levels of BTG4 (Fig. S2) why this was not further investigated.Citation27,Citation28
We conclude that this study, for the first time, has identified a high frequency of aberrant DNA hypermethylation in the promoter region of miR-34b/c in CLL, with possible functional consequences. This aberrant methylation was more predominant in CLL cells without 11q deletion and correlated with miR-34b/c expression levels. Our findings suggest that miR-34b/c is a novel candidate tumor suppressor gene on chromosome 11q23.
No author of this paper has a conflict of interest to disclose. Supplementary information is available at the Epigenetics website.
Methods
Patient and control samples
CLL cells were collected from peripheral blood from 74 CLL patients at Karolinska University Hospital and Uppsala University Hospital after informed consent. All cases were diagnosed according to the updated iwCLL criteriaCitation29 and samples contained >70% tumor cells according to immunophenotyping. Characteristics of the patients are summarized in . Normal peripheral blood mononuclear cells (PBMCs) were collected from two healthy volunteers and from commercially available mixed leukocyte DNA from multiple donors (Roche Applied Biosystems). CD19+ normal leukocytes from three individuals were purchased from 3H Biomedicals. The study was approved by the regional ethic committees in Uppsala and Stockholm.
Table 2. Patient characteristics
FISH analysis and IGHV gene mutation status
FISH analysis for the common genomic aberrations was analyzed using standard protocols and samples with >5% of aberrant cells were considered mutated. IGHV mutational status was performed by PCR amplification and sequencing as previously described.Citation30,Citation31 FISH data for chromosome 13q, 17p, and 12 was available for 67 patients, 11q for 68 and IGHV mutational status for 60 patients.
Drug exposure
The HG3 CLL cell line was cultured in RPMI Medium 1640 with Glutamax and HEPES buffer (Invitrogen) with 10% FBS added. Cells were subsequently cultured for 72 h in supplemented RPMI media treated with the following drugs: (1) 5-aza-2-deoxycytidine (DAC) at 0.005 μM/L (Sigma- Aldrich) for 72 h (medium was changed every 24 h); (2) trichostatin A (TSA) at 500nM/L; (Sigma-Aldrich) for the last 24 h; and (3) DAC for 48 h followed by addition of TSA for 24 h. Control cells were cultured as above but with no drugs added.
DNA methylation analyses
DNA was extracted and bisulfite treated using the EZ DNA Methylation Kit.Citation32 DNA methylation analysis of extracted DNA was performed using methylation-specific PCR amplification and melting curve analysis (MS-MCA) as described previouslyCitation33 in 52 CLL samples and five controls. Validation of BTG4/miR-34b/c methylation was performed using bisulfite pyrosequencing performed on a PyroMark Q24 instrument as described previously.Citation34 Primers were designed using the Oligo1.0 software (Molecular Biology Insights) or with the PyroMark Assay Design software (Qiagen). Primer sequences are provided in Table S1.
Micro-RNA expression
Total RNA was extracted from 16 CLL primary cells and the CLL cell line HG3 using the miRNeasy kit (Qiagen). Gene expression was analyzed by real-time quantitative PCR (RQ-PCR) using the ΔΔCt method. All primers used for miR-34a/b and c were commercially available Taqman probes (ABI) using RNU6 as an internal control gene. β2-microglobulin was used as internal control for BTG4 gene expression. The RQ-PCR was performed on an ABI StepOnePlus machine (ABI). All RQ-PCR analyses were made in triplicates.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) was performed using Magnetic LowCell ChIP kit from Diagenode according to manufacturer’s instruction, using antibodies against lysine 27 of histone H3 (H3K27me3) (Abcam, ab6002) and H3 (Abcam, ab1791). Levels were assessed by RQ-PCR on an ABI StepOnePlus instrument using ChIP-primers for the miR-34b/c common promoter, GADPH and SERPINA (Table S1). Samples were normalized to histone H3 levels and in adjusted so that the mean signal of the CD19 normal controls was set to 100%. The functionality of the ChIP was validated by analyzing one active promoter (GADPH) and one repressed promoter (SERPINA) with RQ-PCR. H3K27me3/H3 was found to be higher in SERPINA than in GADPH for all samples (Fig. S1). All PCR-reactions were performed in triplicate.
miR transfection
HG3 cells were transfected with either MiRIDIAN® (Thermo Scientific) miR-34b, miR-34c or the miRNA mimic control. Transfection was performed using the Neon electroporation system (Life Technologies) according to the manufacturer’s protocol. Optimized settings were: 1450 V, 3 pulses with a band width of 10 ms. The electroporated reaction was done in a 10µl Neon tip added to the wells and incubated at 37 C° for 24 h before analysis. The BLOCK-iT™ Fluorescent Oligo control (Life Technologies) was used as an indicator of transfection efficiency of electroporation in HG3 cells, which were evaluated by fluorescent microscope.
Flow cytometry
Apoptosis was measured by flow cytometry using a FACScalibur instrument (Becton Dickinson). The Annexin V Apoptosis Detection Kit I (Becton Dickinson) was used for Annexin/PI analyses. Samples were fixed with etanol, washed and treated either with RNase A/propidium iodide or with primary antibodies. FACS data were analyzed by FCS Express 3.0 software.
DNase hypersensitivity sites
DNase hypersensitivity sites were analyzed using the EpiQ Chromatin Analysis Kit (Bio-Rad), according to manufacturer’s instructions. Briefly, chromatin was extracted and digested with DNase I. Genomic DNA was then isolated and the level of digestion was assessed using RQ-PCR with primers directed at the regions of interest and compared with reference genes—GADPH (open chromatin) and SERPINA (closed chromatin). The DNase digestion time was optimized and 45 and 60 min were used in the experiments.
Statistics
Two group comparisons were made using Student t test or Mann-Whitney U-test when appropriate and the relationship between MS-MCA methylation and genomic aberrations or IGHV status by the Chi-square test. The Pearson correlation coefficient or Spearmans Rho is used to describe correlation as indicated. PASW 18.0 (IBM) was used for statistical calculations. All p-values are two sided, P < 0.05 was regarded as significant.
| Abbreviations: | ||
| CDR | = | commonly deleted region |
| ChIP | = | chromatin immunoprecipitation |
| CLL | = | chronic lymphatic leukemia |
| DAC | = | 5-aza-2-deoxycytidine |
| DHSS | = | DNAse hypersensitivity assay |
| FISH | = | Fluorescence in situ hybridization |
| HDAC | = | Histone deacetylase |
| IGHV | = | Immunoglobulin heavy chain variable region gene |
| miR | = | microRNA |
| MS-MCA | = | methylation specific melting curve analysis |
| PBMC | = | peripheral blood mononuclear cells |
| RQ-PCR | = | real-time quantitative polymerase chain reaction |
| TSA | = | trichostatin A |
| UTR | = | untranslated region |
Additional material
Download Zip (180.1 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The work was supported by grants from Cancerfonden, Stockholms läns landsting and the Swedish Hematology Association (SFH-Roche grant).
References
- Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, Döhner K, Bentz M, Lichter P. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343:1910 - 6; http://dx.doi.org/10.1056/NEJM200012283432602; PMID: 11136261
- Gunnarsson R, Isaksson A, Mansouri M, Göransson H, Jansson M, Cahill N, Rasmussen M, Staaf J, Lundin J, Norin S, et al. Large but not small copy-number alterations correlate to high-risk genomic aberrations and survival in chronic lymphocytic leukemia: a high-resolution genomic screening of newly diagnosed patients. Leukemia 2010; 24:211 - 5; http://dx.doi.org/10.1038/leu.2009.187; PMID: 19741724
- Stilgenbauer S, Liebisch P, James MR, Schröder M, Schlegelberger B, Fischer K, Bentz M, Lichter P, Döhner H. Molecular cytogenetic delineation of a novel critical genomic region in chromosome bands 11q22.3-923.1 in lymphoproliferative disorders. Proc Natl Acad Sci U S A 1996; 93:11837 - 41; http://dx.doi.org/10.1073/pnas.93.21.11837; PMID: 8876224
- Schaffner C, Stilgenbauer S, Rappold GA, Döhner H, Lichter P. Somatic ATM mutations indicate a pathogenic role of ATM in B-cell chronic lymphocytic leukemia. Blood 1999; 94:748 - 53; PMID: 10397742
- Auer RL, Jones C, Mullenbach RA, Syndercombe-Court D, Milligan DW, Fegan CD, Cotter FE. Role for CCG-trinucleotide repeats in the pathogenesis of chronic lymphocytic leukemia. Blood 2001; 97:509 - 15; http://dx.doi.org/10.1182/blood.V97.2.509; PMID: 11154230
- Zenz T, Mertens D, Döhner H, Stilgenbauer S. Importance of genetics in chronic lymphocytic leukemia. Blood Rev 2011; 25:131 - 7; http://dx.doi.org/10.1016/j.blre.2011.02.002; PMID: 21435757
- Mansouri L, Sutton LA, Ljungstrom V, Sorqvist EF, Gunnarsson R, Smedby KE, Juliusson G, Stamatopoulos K, Nilsson M, Rosenquist R. Feasibility of targeted next-generation sequencing of the TP53 and ATM genes in chronic lymphocytic leukemia. Leukemia 2013; 28:694 6; http://dx.doi.org/10.1038/leu.2013.322; PMID: 24172824
- Kalla C, Scheuermann MO, Kube I, Schlotter M, Mertens D, Döhner H, Stilgenbauer S, Lichter P. Analysis of 11q22-q23 deletion target genes in B-cell chronic lymphocytic leukaemia: evidence for a pathogenic role of NPAT, CUL5, and PPP2R1B. Eur J Cancer 2007; 43:1328 - 35; http://dx.doi.org/10.1016/j.ejca.2007.02.005; PMID: 17449237
- Auer RL, Starczynski J, McElwaine S, Bertoni F, Newland AC, Fegan CD, Cotter FE. Identification of a potential role for POU2AF1 and BTG4 in the deletion of 11q23 in chronic lymphocytic leukemia. Genes Chromosomes Cancer 2005; 43:1 - 10; http://dx.doi.org/10.1002/gcc.20159; PMID: 15672409
- Lehmann S, Ogawa S, Raynaud SD, Sanada M, Nannya Y, Ticchioni M, Bastard C, Kawamata N, Koeffler HP. Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia. Cancer 2008; 112:1296 - 305; http://dx.doi.org/10.1002/cncr.23270; PMID: 18246537
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281 - 97; http://dx.doi.org/10.1016/S0092-8674(04)00045-5; PMID: 14744438
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 2005; 102:13944 - 9; http://dx.doi.org/10.1073/pnas.0506654102; PMID: 16166262
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2002; 99:15524 - 9; http://dx.doi.org/10.1073/pnas.242606799; PMID: 12434020
- Batova A, Diccianni MB, Yu JC, Nobori T, Link MP, Pullen J, Yu AL. Frequent and selective methylation of p15 and deletion of both p15 and p16 in T-cell acute lymphoblastic leukemia. Cancer Res 1997; 57:832 - 6; PMID: 9041181
- Sakai T, Toguchida J, Ohtani N, Yandell DW, Rapaport JM, Dryja TP. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am J Hum Genet 1991; 48:880 - 8; PMID: 1673287
- Asatiani E, Huang WX, Wang A, Rodriguez Ortner E, Cavalli LR, Haddad BR, Gelmann EP. Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res 2005; 65:1164 - 73; http://dx.doi.org/10.1158/0008-5472.CAN-04-2688; PMID: 15734999
- Muraoka T, Soh J, Toyooka S, Aoe K, Fujimoto N, Hashida S, Maki Y, Tanaka N, Shien K, Furukawa M, et al. The degree of microRNA-34b/c methylation in serum-circulating DNA is associated with malignant pleural mesothelioma. Lung Cancer 2013; 82:485 - 90; http://dx.doi.org/10.1016/j.lungcan.2013.09.017; PMID: 24168922
- Vrba L, Muñoz-Rodríguez JL, Stampfer MR, Futscher BW. miRNA gene promoters are frequent targets of aberrant DNA methylation in human breast cancer. PLoS One 2013; 8:e54398; http://dx.doi.org/10.1371/journal.pone.0054398; PMID: 23342147
- Tanaka N, Toyooka S, Soh J, Kubo T, Yamamoto H, Maki Y, Muraoka T, Shien K, Furukawa M, Ueno T, et al. Frequent methylation and oncogenic role of microRNA-34b/c in small-cell lung cancer. Lung Cancer 2012; 76:32 - 8; http://dx.doi.org/10.1016/j.lungcan.2011.10.002; PMID: 22047961
- Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, Tokino T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 2008; 68:4123 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-08-0325; PMID: 18519671
- Zenz T, Mohr J, Eldering E, Kater AP, Bühler A, Kienle D, Winkler D, Dürig J, van Oers MH, Mertens D, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood 2009; 113:3801 - 8; http://dx.doi.org/10.1182/blood-2008-08-172254; PMID: 18941118
- He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer 2007; 7:819 - 22; http://dx.doi.org/10.1038/nrc2232; PMID: 17914404
- Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res 2008; 68:2094 - 105; http://dx.doi.org/10.1158/0008-5472.CAN-07-5194; PMID: 18381414
- Lujambio A, Calin GA, Villanueva A, Ropero S, Sánchez-Céspedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso MS, Faller WJ, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A 2008; 105:13556 - 61; http://dx.doi.org/10.1073/pnas.0803055105; PMID: 18768788
- Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, Lin JC, Liang G, Jones PA, Tanay A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A 2008; 105:12979 - 84; http://dx.doi.org/10.1073/pnas.0806437105; PMID: 18753622
- Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J 2011; 30:249 - 62; http://dx.doi.org/10.1038/emboj.2010.318; PMID: 21131905
- Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Béné MC, De Vos J, Hernández JM, Hofmann WK, Mills KI, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol 2010; 28:2529 - 37; http://dx.doi.org/10.1200/JCO.2009.23.4732; PMID: 20406941
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 2004; 6:1 - 6; PMID: 15068665
- Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, et al, International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008; 111:5446 - 56; http://dx.doi.org/10.1182/blood-2007-06-093906; PMID: 18216293
- Murray F, Darzentas N, Hadzidimitriou A, Tobin G, Boudjogra M, Scielzo C, Laoutaris N, Karlsson K, Baran-Marzsak F, Tsaftaris A, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood 2008; 111:1524 - 33; http://dx.doi.org/10.1182/blood-2007-07-099564; PMID: 17959859
- Ghia P, Stamatopoulos K, Belessi C, Moreno C, Stilgenbauer S, Stevenson F, Davi F, Rosenquist R, European Research Initiative on CLL. ERIC recommendations on IGHV gene mutational status analysis in chronic lymphocytic leukemia. Leukemia 2007; 21:1 - 3; http://dx.doi.org/10.1038/sj.leu.2404457; PMID: 17167526
- Kanduri M, Cahill N, Göransson H, Enström C, Ryan F, Isaksson A, Rosenquist R. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood 2010; 115:296 - 305; http://dx.doi.org/10.1182/blood-2009-07-232868; PMID: 19897574
- Guldberg P, Worm J, Grønbaek K. Profiling DNA methylation by melting analysis. Methods 2002; 27:121 - 7; http://dx.doi.org/10.1016/S1046-2023(02)00063-4; PMID: 12095269
- Kanduri M, Marincevic M, Halldórsdóttir AM, Mansouri L, Junevik K, Ntoufa S, Kultima HG, Isaksson A, Juliusson G, Andersson PO, et al. Distinct transcriptional control in major immunogenetic subsets of chronic lymphocytic leukemia exhibiting subset-biased global DNA methylation profiles. Epigenetics 2012; 7:1435 - 42; http://dx.doi.org/10.4161/epi.22901; PMID: 23154584