
Abstract
A formalin-fixed paraffin-embedded (FFPE) sample usually yields highly degraded DNA, which limits the use of techniques requiring high-quality DNA, such as Infinium Methylation microarrays. To overcome this restriction, we have applied an FFPE restoration procedure consisting of DNA repair and ligation processes in a set of paired fresh-frozen (FF) and FFPE samples. We validated the FFPE results in comparison with matched FF samples, enabling us to use FFPE samples on the Infinium HumanMethylation450 Methylation array.
Introduction
DNA methylation, which involves the covalent addition of a methyl group to cytosine bases, is the most extensively studied type of epigenetic modification. In mammals, it occurs mainly in the context of CG dinucleotides (CpG). These are enriched in approximately 40% of mammalian promoter genes,Citation1 and especially in housekeeping and tumor suppressor genes. Particularly in these types of genes, the repression of gene expression is known to depend on the methylation level of their promoter.Citation2,Citation3 Interest in this crosstalk between DNA methylation and gene expression explains the growing number of studies demonstrating the role of methylation in metabolic diseases,Citation4 neurodegenerative diseases,Citation5-Citation7 aging,Citation8,Citation9 cancer,Citation10,Citation11 and drug response to chemotherapy.Citation12 Thus, the importance of DNA methylation signatures as tools in many biomedical studies is becoming increasingly well established.
To interrogate the methylation status of CpGs, array-based methods have recently been used for large sets of samples, while the whole-genome bisulfite sequencing (WGBS) approach has been reserved for projects with smaller sets of samples, because of its high cost. Since the release of the Infinium HumanMethylation450 array (Illumina, Inc.)Citation13 in 2011, it has been possible to interrogate almost half a million CpG sites in the human genome at less cost than with next-generation sequencing (NGS). The Infinium Methylation assay relies on a number of serial reactions (bisulfite conversion, whole-genome amplification [WGA], fragmentation and resuspension, hybridization, single-base extension, and fluorescent antibody labeling) to accurately measure the methylation level at single-base resolution. The greatest limitation of this platform is imposed by the WGA reaction, which is known to affect the overall success of the Infinium process when the input DNA material is degraded (<1 kb),Citation14 so this platform has only been used with high-quality (non-fragmented) DNA. This restricts the potentially analyzable samples to those whose DNA has been extracted from fresh-frozen (FF) tissues, blood samples or in vitro cultured cells. Conversely, samples with previous associated clinical and follow-up data are routinely formalin-fixed and paraffin-embedded (FFPE) for histopathological diagnosis, which is known to affect DNA integrity, and to prevent these valuable samples being subjected to methods, such as Infinium, that are sensitive to DNA fragmentation.
To overcome these limitations, we have evaluated the combination of DNA repair and ligation steps with an FFPE restoration kit. Restored DNA was used as input for the well characterized Infinium Methylation assay. This enabled us to use the HumanMethylation450 array to successfully process tumor samples for which FF and matched FFPE material was available.
Results and Discussion
Samples from eight tumor types were represented on hybridized samples, breast (n = 3, tumors 1–3), colon (n = 4, tumors 4–7), kidney (n = 2, tumors 8–9), lung (n = 5, tumors 10–14), ovary (n = 4, tumors 15–18), pancreas (n = 1, tumors 19), prostate (n = 1, tumors 20), and stomach (n = 5, tumors 21–25). For each patient, two biopsies of the same tumor were taken; one was stored as fresh-frozen tissue embedded in optimal cutting temperature (OCT) compound stored at –80 °C, while the other piece underwent FFPE fixation and was stored at room temperature.
Each of the 485 512 CpG sites interrogated on the HumanMethylation450 array is present 16 times on average, allowing the significance (P value) for each single CpG site to be calculated based on the dispersion of the reads obtained when compared with background probes. The percentage of correctly detected CpG sites (P < 0.01) in conjunction with the internal controls present on the array, are the main measures used to check whether the processing of samples was performed successfully. As expected from the product specifications, data obtained from the 25 FF samples were of high quality, with an average detection rate of 99.93% (range, 99.85%–99.98%), while the average FFPE sample detection rate was slightly lower, 99.65% (range, 97.63%–99.95%). Nevertheless, they indicate that FF and FFPE results were both highly robust and, therefore, that the FFPE restoration process overcomes the limitations of non-degraded input material in the Infinium assay.
To test whether data obtained from the array were biased because of the two origins of DNA (FFPE or FF), we performed a principal component analysis in the R statistical environment, but found no difference between the 50 samples with respect to their origin (). As expected from randomizing samples at the array positions, neither array position or array itself had an effect on the data obtained ().
Figure 1. (A) Scatter plots of the first two principal components of variance, grouping samples on the basis of the DNA source (FFPE or FF), Array Position (Sentrix Position) and Array (SentrixID). (B) A representative example of the comparison of SNP values obtained from matched FF and FFPE samples, indicating correct matched FF-FFPE sample is being compared. (C) A comparison of the methylation values from paired FFPE and FF sample for tumor 17. (D) Percentage of CpG sites statistically significant among sample origin by ANOVA analysis.
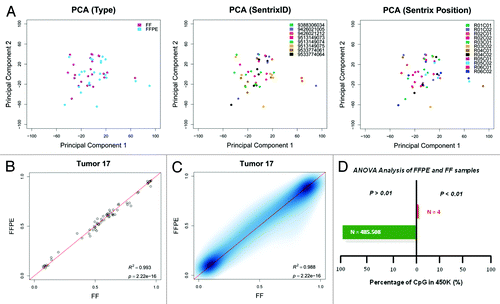
To ascertain that the matched FFPE-FF samples were correctly compared, we used the 65 SNPs included on the humanMethylation450 array for fingerprinting purposes, comparing the β-values of paired samples. In these cases, the β-values should cluster as they do on a standard genotyping theta graph, so paired samples should lie near the identity line in a scatter plot graph. As shown for tumor 17 (), our analysis revealed that all 25 paired FF/FFPE tumors were compared in correct pairs (Fig. S1).
Methylation values obtained from corresponding FF and FFPE samples were compared by considering both values for each CpG site in a linear model forced to pass by axis intersection. A representative comparison is shown on . This revealed a very strong correlation between methylation (β) values from paired FF and FFPE samples (average Pearson R2 = 0.9721; range 0.9258 – 0.9945; P = 2.2 × 10−16) (Fig. S2) with a mean methylation difference between FF vs FFPE of 2.54 × 10−3 (standard deviation = 0.0771). Thus, FFPE restoration prior to conducting the Infinium methylation assay gave results that were highly comparable to those obtained from FF samples.
Univariate analysis of variance was performed on FFPE and FF samples, resulting in 99.99% of the CpGs being not differentially methylated among both groups (FF and FFPE) of samples with Δβ > 0.50 (); moreover, when relaxing the threshold to Δβ > 0.33, the analysis resulted in 99.93% of the CpG sites not being differentially methylated (data not shown). Interestingly, the four CpGs that were statistically significant between the paired set of samples when Δβ > 0.50 was used, and 99.03% of the 311 CpG found when Δβ > 0.33 was applied, were methylated on FF samples (mean β-value Δβ0.50 = 0.78; mean β-value Δβ0.33 = 0.76), but unmethylated on FFPE samples (mean β-value Δβ0.50 = 0.27; mean β-value Δβ0.33 = 0.39). This observation could be explained by the fact that FFPE samples undergo a reparation process in which cohesive ends are repaired and then ligated one to another. If a CpG site falls within a cohesive end, the methylation status cannot be restituted and, thus, the CpG locus would be read as non-methylated.
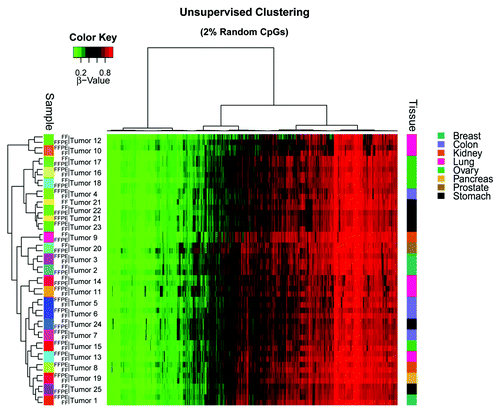
To confirm that DNA origin is not affecting the results, we performed unsupervised clustering of 2% randomly selected CpG β-values obtained from the array and found 96.0% (n = 24) of tumors for which the clustering was achieved by patient, while for the remaining 4.0% (n = 1) of the tumors, the FF and FFPE samples appeared close together (in the same tumor type [stomach cancer] but not in the same sample cluster []). Despite the same patient origin for FF and FFPE tumor samples, tumor cell heterogeneity might not be the same in different tissue sections from a given tumor piece, thereby giving rise to a degree of variation in the methylation values observed in FF samples compared with those of matched FFPE samples.
Figure 2. Unsupervised hierarchical clustering of a randomly selected 2% of CpGs from the array.
Reliable methylation profiles can be obtained from FFPE samples if DNA repair and ligation are performed after bisulfite conversion and before the whole-genome amplification step of the Infinium Methylation assay. As our results showed, methylation values of FFPE samples are comparable to those from FF samples. Therefore, applying this procedure provides a new opportunity to analyze all those samples that have been preserved on FFPE, due to the daily practice in histopathological diagnosis, benefiting from the rich clinical information that these samples usually provide. Our results validate the Infinium HumanMethylation450 Methylation array platform for the study of diseases for which it is difficult to obtain samples other than FFPE and that have not until now been suitable for comprehensive DNA methylation analysis.
Materials and Methods
DNA Extraction
Tumor samples were obtained from the Department of Pathology and Molecular Genetics, and Research Laboratory, Arnau de Vilanova Universitary Hospital, University of Lleida IRBLLEIDA, under the approval of the Institutional Review Board. Four sections of 10 µm from FF samples OCT blocks were cut, and washed in PBS to remove OCT. DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s instructions, while four 10-µm sections of FFPE blocks were processed using the E.Z.N.A. FFPE DNA kit (Omega Bio-Tek), with a xylene wash to remove paraffin. The DNA obtained was treated with RNaseA for 1 h at 45 °C.
All DNA samples were quantified by the fluorometric method (Quant-iT PicoGreen dsDNA Assay, Life Technologies), and assessed for purity by NanoDrop (Thermo Scientific) 260/280 and 260/230 ratio measurements. The integrity of FF DNAs was checked by electrophoresis in a 1.3% agarose gel.
Quality Check of FFPE DNAs
All DNAs from FFPE blocks were checked for their suitability for FFPE restoration, as indicated by the Infinium HD FFPE QC Assay (Illumina, Inc.), by performing a quantitative PCR with 2 ng of FFPE DNA. ΔCq was calculated by subtracting the average value of Cq of the interrogated sample from the Cq value of a standard provided by the manufacturer. All FFPE samples had a ΔCq < 5, which is the recommended threshold for suitability for FFPE Restoration.
Bisulfite Conversion
An amount of 300 ng of FFPE DNA, or 600 ng of FF DNA were randomly distributed on a 96-well plate, and processed using the EZ-96 DNA Methylation kit (Zymo Research Corp) following the manufacturer’s recommendations for Infinium assays. Elution step was performed with 10 µl of M-Elution buffer.
FFPE Restoration
An amount of 8 µl of bisulfite-converted DNA (bs-DNA) from FFPE samples was processed as described in the Infinium FFPE Restoration guide (Illumina, Inc.). The DNA (>100 ng) was denatured with 4 µl of NaOH 0.1N for 10 min at room temperature. A 1 h reaction at 37 °C was then performed with PPR and AMR reagents supplied by the kit manufacturer, in which DNA repair is accomplished.
DNA was cleaned with a ZR-96 DNA Clean and Concentrator-5 kit (Zymo Research) by mixing the previous reaction with 560 µl Binding buffer and dispensing the mixture into a Zymo-Spin column. Then the column was centrifuged for 2 min at 2250 × g, and eluate was discarded. A washing step with 600 µl of Zymo-Wash buffer was performed followed by a centrifugation step for 2 min at 2250 × g. Zymo-spin columns were placed on new empty tubes, and 13 µl of ERB were dispensed to each column.
After a incubation at room temperature for 5 min, columns were centrifuged for 1 min at 2250 × g, and eluate was incubated for 2 min at 95 °C. Immediately after the 95 °C incubation, samples were incubated on ice for 5 min. Maintaining the eluate on ice, the ligation reaction was performed by dispensing 10 µl of CMM reagent to each reaction, and incubating the reaction for 1 h at 37 °C. Ligation reaction material was cleaned with ZR-96 DNA Clean and Concentrtor-5 kit (Zymo Research), as previously described, and eluted in 10 µl of DiH2O. The resulting material was used as input for the hybridization on the Infinium HumanMethylation450 BeadChip.
Array hybridization
An amount of 8 µl of restored FFPE bs-DNA or 4 µl of FF bs-DNA was processed following the Illumina Infinium HumanMethylation450 protocol, as previously described.Citation15
Data Normalization
The resulting raw data (IDATs) were normalized in the R statistical environment, using the minfi package for SWAN normalization.Citation16
| Abbreviations: | ||
| bs-DNA | = | bisulfite-converted DNA |
| CpG | = | CG dinucleotides |
| FF | = | fresh-frozen |
| FFPE | = | formalin-fixed paraffin-embedded |
| NGS | = | next-generation sequencing |
| OCT | = | optimal cutting temperature |
| SNP | = | single nucleotide polymorphism |
| WGBS | = | whole-genome bisulfite sequencing |
| WGA | = | whole-genome amplification |
Additional material
Download Zip (3.1 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Carles Arribas and Diana García for their support. Supported by Cellex Foundation, Olga Torres Foundation, ERC grant agreement no. 268626 – EPINORC project, MINECO Project no. SAF2011–22803, the Institute of Health Carlos III (ISCIII) RTICC Grant RD12/0036/0039 and the Health and Science Departments of the Generalitat de Catalunya. M.E. is an ICREA Research Professor.
References
- Fatemi M, Pao MM, Jeong S, Gal-Yam EN, Egger G, Weisenberger DJ, Jones PA. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res 2005; 33:e176; http://dx.doi.org/10.1093/nar/gni180; PMID: 16314307
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003; 349:2042 - 54; http://dx.doi.org/10.1056/NEJMra023075; PMID: 14627790
- Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343:1350 - 4; http://dx.doi.org/10.1056/NEJM200011093431901; PMID: 11070098
- Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, Daunay A, Busato F, Mein CA, Manfras B, et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet 2011; 7:e1002300; http://dx.doi.org/10.1371/journal.pgen.1002300; PMID: 21980303
- Masliah E, Dumaop W, Galasko D, Desplats P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 2013; 8:1030 - 8; http://dx.doi.org/10.4161/epi.25865; PMID: 23907097
- Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ. Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci 2011; 31:16619 - 36; http://dx.doi.org/10.1523/JNEUROSCI.1639-11.2011; PMID: 22090490
- Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol 2009; 8:1056 - 72; http://dx.doi.org/10.1016/S1474-4422(09)70262-5; PMID: 19833297
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013; 49:359 - 67; http://dx.doi.org/10.1016/j.molcel.2012.10.016; PMID: 23177740
- Heyn H, Moran S, Esteller M. Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome. Epigenetics 2013; 8:28 - 33; http://dx.doi.org/10.4161/epi.23366; PMID: 23257959
- Sandoval J, Mendez-Gonzalez J, Nadal E, Chen G, Carmona FJ, Sayols S, Moran S, Heyn H, Vizoso M, Gomez A, et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol 2013; 31:4140 - 7; http://dx.doi.org/10.1200/JCO.2012.48.5516; PMID: 24081945
- Asmar F, Punj V, Christensen J, Pedersen MT, Pedersen A, Nielsen AB, Hother C, Ralfkiaer U, Brown P, Ralfkiaer E, et al. Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica 2013; 98:1912 - 20; http://dx.doi.org/10.3324/haematol.2013.088740; PMID: 23831920
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352:997 - 1003; http://dx.doi.org/10.1056/NEJMoa043331; PMID: 15758010
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288 - 95; http://dx.doi.org/10.1016/j.ygeno.2011.07.007; PMID: 21839163
- Thirlwell C, Eymard M, Feber A, Teschendorff A, Pearce K, Lechner M, Widschwendter M, Beck S. Genome-wide DNA methylation analysis of archival formalin-fixed paraffin-embedded tissue using the Illumina Infinium HumanMethylation27 BeadChip. Methods 2010; 52:248 - 54; http://dx.doi.org/10.1016/j.ymeth.2010.04.012; PMID: 20434562
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692 - 702; http://dx.doi.org/10.4161/epi.6.6.16196; PMID: 21593595
- Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol 2012; 13:R44; http://dx.doi.org/10.1186/gb-2012-13-6-r44; PMID: 22703947