
Abstract
Many epidemiologic studies of environmental exposures and disease susceptibility measure DNA methylation in white blood cells (WBC). Some studies are also starting to use saliva DNA as it is usually more readily available in large epidemiologic studies. However, little is known about the correlation of methylation between WBC and saliva DNA. We examined DNA methylation in three repetitive elements, Sat2, Alu, and LINE-1, and in four CpG sites, including AHRR (cg23576855, cg05575921), cg05951221 at 2q37.1, and cg11924019 at CYP1A1, in 57 girls aged 6–15 years with blood and saliva collected on the same day. We measured all DNA methylation markers by bisulfite-pyrosequencing, except for Sat2 and Alu, which were measured by the MethyLight assay. Methylation levels measured in saliva DNA were lower than those in WBC DNA, with differences ranging from 2.8% for Alu to 14.1% for cg05575921. Methylation levels for the three repetitive elements measured in saliva DNA were all positively correlated with those in WBC DNA. However, there was a wide range in the Spearman correlations, with the smallest correlation found for Alu (0.24) and the strongest correlation found for LINE-1 (0.73). Spearman correlations for cg05575921, cg05951221, and cg11924019 were 0.33, 0.42, and 0.79, respectively. If these findings are replicated in larger studies, they suggest that, for selected methylation markers (e.g., LINE-1), methylation levels may be highly correlated between blood and saliva, while for others methylation markers, the levels may be more tissue specific. Thus, in studies that differ by DNA source, each interrogated site should be separately examined in order to evaluate the correlation in DNA methylation levels across DNA sources.
Introduction
DNA methylation is an important epigenetic mechanism involved in the regulation of gene expression and genomic stability.Citation1 Data from identical twins indicates that older twins exhibit greater differences in genomic DNA methylation patterns later in life compared with younger twins, suggesting that the environment can influence DNA methylation changes throughout life.Citation2 White blood cells (WBC) are a common source of DNA to study DNA methylation changes related to disease susceptibility and the associated risk factors.Citation3-Citation8 In epidemiological studies, global DNA methylation in WBC (measured in particular repetitive elements, LINE-1 and Sat2) has been associated with increased risk of cancer, including breast cancer.Citation3,Citation5,Citation6,Citation9 In addition to examining the association between WBC methylation and disease susceptibility, epidemiologic studies have investigated the association between environmental exposures and DNA methylation (reviewed in ref. Citation3). For example, several studies measuring genome-wide DNA methylation levels using Illumina HumanMethylation 27K and 450K BeadChips in WBC DNA have identified tobacco smoking-related differential DNA methylation in specific sites in aryl hydrocarbon receptor repressor (AHRR), CYP1A1, and 2q37.Citation4,Citation7,Citation8 We previously reported that prenatal exposure to tobacco smoke can alter global DNA methylation levels in adult blood DNA, suggesting that life-long effects of in utero exposures may be mediated through alterations in DNA methylation.Citation10,Citation11
DNA methylation profiles are tissue specific.Citation12 Epidemiologic studies have generally investigated WBC DNA methylation, although saliva collection is a non-invasive approach to collect DNA for methylation analysis and has been used extensively for genotyping studies. However, little is known about the correlation of DNA methylation between DNA derived from WBC and saliva. To examine the correlation between the two most frequently used sources for DNA methylation in epidemiological studies,, we compared DNA methylation levels in 57 girls who gave both a blood and saliva sample on the same day. We measured methylation levels in three repetitive elements (Sat2, Alu, and LINE-1) and in 4 CpG sites that, in other studies, have been shown to display statistically significant different levels of DNA methylation after exposure to tobacco smoke using HumanMethylation 450K arrays.Citation7,Citation8
Results
shows the demographic characteristic of the study girls. The average age was 9.8 y (SD = 2.3). Mean BMI was 18.9 kg/M2 (SD = 3.1). Thirty-seven girls were white, and 20 girls were other races. Only one girl was exposed to prenatal smoking. presents the mean DNA methylation and standard deviation (SD) of each marker using different genomic DNA sources. Methylation levels measured in saliva DNA were lower than in WBC DNA. The mean Sat2 methylation levels were 130.2 ± 76.0% (WBC DNA) and 126.8 ± 64.0% (saliva DNA). The mean Alu methylation was 69.0 ± 25.9% in WBC DNA and 67.1 ± 24.4% in saliva DNA. The methylation for LINE-1, cg05575921, cg05951221, and cg11924019 in WBC DNA were 79.0 ± 2.6, 80.0 ± 3.4, 52.4 ± 7.1 and 27.3 ± 5.6, respectively; the corresponding methylation levels in saliva DNA were 75.2 ± 3.4, 65.9 ± 6.1, 46.6 ± 6.7, and 21.8 ± 4.4, respectively.
Table 1. Demographic characteristics of young girls at the New York Site of the LEGACY Girls study
Table 2. Distribution of DNA methylation in white blood cell (WBC) and saliva DNAs
Methylation levels for the three repetitive elements measured in saliva DNA were all positively correlated with those in WBC DNA. However, there was a wide range in the Spearman correlation coefficients with the smallest correlation for Alu (rs = 0.24) and the strongest correlation for LINE-1 (rs = 0.73) (). Spearman correlations between WBC and saliva DNA for cg05575921, cg05951221 and cg11924019 were 0.33, 0.42, and 0.79, respectively. The correlations were similar by race/ethnicity and age (data not shown).
Figure 1. Correlation of methylation markers between WBC and saliva DNAs. (A) Correlation on Sat2 methylation between WBC and saliva DNAs (rs = 0.32, P = 0.02). (B) Correlation of Alu methylation between WBC and saliva DNAs (rs = 0.24, P = 0.09). (C) Correlation of LINE-1 methylation between WBC and saliva DNAs (rs = 0.73, P < 0.0001). (D) Correlation of AHRR cg05575921 methylation between WBC and saliva DNAs (rs = 0.33, P = 0.01). (E) Correlation of 2q37.1 cg05951221 methylation between WBC and saliva DNAs (rs = 0.42, P = 0.001). (F) Correlation of CYP1A1 cg11924019 methylation between WBC and saliva DNAs. (rs = 0.79, P < 0.0001).
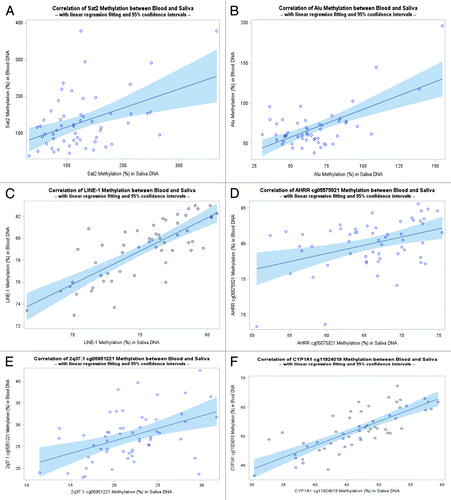
Because we previously reported that hypomethylation in Sat2 was associated with breast cancer risk,Citation5 we categorized Sat2 methylation based on the quartile values in our previous study and found a high concordance between WBC and saliva DNA (data not shown).
Discussion
In our study of 57 girls 6–15 y of age, we observed lower DNA methylation levels in saliva than in WBC DNA. For repetitive elements, the differences were about 2.77% for Alu and 3.75% for LINE-1. For loci-specific methylation, the differences ranged from 5.55% for 2q37.1 cg05951221 to 14.1% for AHRR cg05575921. We found positive correlations in methylation levels measured in saliva DNA with those in WBC DNA, with the correlation coefficient as high as 0.79 for CYP1A1 cg11924019, and as low as 0.32 for Sat2.
Studies of patients with allogeneic bone marrow transplants demonstrated that buccal swabs and mouthwash samples contain high amounts of blood DNA.Citation13,Citation14 In buccal swabs, blood cells, identified as being from the bone marrow donor, ranged from 5% to 63% of total cells present.Citation13 However, for mouthwash samples, collected as the first rinse, these values ranged from 16% to 95%.Citation14 These results suggest that saliva and WBC DNA methylation should be positively correlated.
Talens et al.Citation15 examined methylation in 8 loci in 34 individuals, and also reported different levels of DNA methylation for DNA derived from WBC and buccal cells collected by swabbing. Consistently with our results, they observed correlation coefficients ranging from as low as 0.37 to as high as 0.90.Citation15 Comparing epigenome-wide DNA methylation profiling by Illumina HumanMethylation 27K among leukocyte subtypes, Koestler et al.Citation16 reported a total of 10,370 significantly differentially methylated CpG loci. Shifts in WBC subpopulations in the buccal sample may account for the variability in correlation coefficients in different methylation markers if methylation level varies by cell type. Cell-type specificity of DNA methylation in conjunction with variability in the proportion of WBC in saliva could explain the variation in the correlation of DNA methylation levels across the various markers.
Although array-based designs have identified specific CpG loci associated with both adult and prenatal tobacco smoke exposure in DNA from WBC or buccal cells,Citation4,Citation7,Citation8,Citation17 when designing an epidemiologic study to examine the association of tobacco-related methylation markers, it is important to consider different cell types as well as the type of assay. Bisulfite-pyrosequencing has been considered as a gold standard for measuring DNA methylation. A preliminary scan of specific CpG loci suggested that eight genes were hypermethylated in exposed children, of which only two genes were validated by pyrosequencing.Citation17 The lack of validation of some array data indicates the importance of confirming results. In addition, methylation levels are tissue-specific and, thus, studies of DNA methylation in association with exposure and disease should validate which DNA source is appropriate for biomarker measurement and measure the correlation between sources if multiple sources are used across studies or even within studies.
Methylation array studies have determined that methylation levels of cg05575921, cg05951221, and cg11924019 were associated with tobacco smoking including prenatal tobacco exposure.Citation4,Citation7,Citation8 However, only one girl had exposure to prenatal smoking in our study, limiting our ability to examine the association of methylation with smoking in different sources of DNA. The strength of this study is that both WBC and saliva DNA were collected at the same day.
Our findings, if replicated, suggest that methylation of some CpG sites measured in saliva DNA are highly correlated with methylation in WBC DNA. As saliva and WBC DNA correlations are likely variable depending on the site interrogated, it is essential for studies relying on saliva DNA to perform correlational analyses in a subset of participants to understand how the correlation, or lack thereof, influences the interpretation of the overall findings.
Materials and Methods
Study participants
This pilot study includes girls ages 6–15 y participating in the LEGACY Girls Study (Lessons in Epidemiology and Genetics of Adult Cancer from Youth), a multicenter prospective study of early-life exposures, pubertal development, and other endpoints relevant to breast cancer etiology conducted across five sites in North America. As part of the study protocol, all girls are asked to provide either a blood or saliva sample at baseline and follow-up visits. At the New York site, we also asked 57 girls who provided a blood sample to also give a saliva sample on the same day of the clinic visit. We collected saliva using Oragene kits (DNA Genotek). The study was approved by the Institutional Review Board of Columbia University.
DNA extraction and bisulfite treatment
DNA was extracted from total WBC and saliva by a salting out procedure. Cells were lysed with SDS in a nuclei lysis buffer and treated with RNase A (final 133 µg/mL) and RNase T1 (final 20 units/mL) to remove RNA. Proteins were co-precipitated with NaCl (330 µL of saturated NaCl added per 1mL solution) by centrifugation. Genomic DNA was recovered from the supernatant by precipitation with 100% ethanol, washed in 70% ethanol, and dissolved in Tris-EDTA buffer.
Aliquots of DNA (500 ng) were bisulfite-treated with the EZ DNA methylation kit (Zymo Research). Modified DNA were resuspended in 20 µL of distilled water and stored at -20 °C until assayed. The laboratory investigator who performed the assays was blinded to the epidemiologic data. All methylation measurements in both WBC and saliva DNA from the same individuals were performed in the same batch.
Sat2 and Alu methylation measured by the MethyLight assay
We used the sequences of probes and forward and reverse primers designated as Sat2-M1 and Alu-M2 in Weisenberger et al.Citation18 Polymerase chain reaction (PCR) was performed in a 10µl reaction volume with 0.3 µM forward and reverse PCR primers, 0.1 µL probe, 3.5 µM MgCl2, using the following PCR program: 95 °C for 10 min, then 55 cycles of 95 °C for 15 s, followed by 60 °C for 1 min. Assays were run on an ABI Prism 7900 Sequence Detection System (LifeTechnologies).
The MethyLight data were expressed as percent of methylated reference (PMR) values and are the mean of duplicates.
PMR = 100% * 2 exp – (Delta Ct [target gene in sample - control gene in sample] - Delta Ct [100% methylated target in reference sample - control gene in reference sample]).
Pyrosequencing
The methylation status of LINE-1, AHRR (cg23576855, cg05575921), cg05951221 at 2q37.1, and cg11924019 at CYP1A1 were measured by pyrosequencing. The sequences of primers and PCR condition for LINE-1 and 2q37_p3 have been described in detail previously.Citation7,Citation19 All pyrosequencing assays for loci specific methylation including AHRR (cg23576855, cg05575921), and cg11924019 in CYP1A1 were designed to cover the same CpG sites interrogated by Illumina. The primers were AATGAGTTTT TTTTTGGTTG TAGTG (FWD), (5′biotin) ACCTAAACAA CCCCTATATC CT (REV), and AGATTTTTTA AGGTGGTTGA (sequence) for cg23576855; ATAGGGGTTG TTTAGGTTAT AGATT (FWD), (5′biotin) ACCTATCCCC TACCTCCC (REV), and ATTGTTTATT TTTGAGAGGG TA (sequence) for cg05575921; GGGTTTTTAG GAAAAAAAAA GTTGTAT (FWD), (5′biotin)-AAATACTATC AACTATATTC CCTTCTCT (REV), and AGTTTAATTT GGTTTTAGTT AATAT (sequence) for cg11924019. The biotinylated PCR products were purified and made single-stranded to act as a template in the pyrosequencing reaction as recommended by the manufacture using the Pyrosequencing Vacuum Prep Tool (Qiagen). Then, 0.3 nmol/L of pyrosequencing primer was annealed to the purified single-stranded PCR product and pyrosequencing was run on a PyroMark Q24. We used non-CpG cytosine residues as internal controls to verify efficient sodium bisulfite DNA conversion, and universal unmethylated and methylated DNA were run as controls. Methylation quantification was performed using the PyroMark Q24 1.010 software.
We excluded the results for AHRR cg23576855 because the methylation levels in both WBC and saliva DNA were over 90%. We repeated measuring 10% of both WBC and saliva DNA. The inter-assay coefficients of variation (CV) for WBC and saliva DNA were 15.6% and 22.6% for Sat2, 5.6% and 19.8% for Alu, 1.3% and 0.7% for LINE-1, 4.5% and 7.0% for cg05951221, 6.5% and 6.6% for cg05575921, and 3.4% and 1.8% for cg11924019, respectively.
Statistical methods
We used ANOVA to test for differences in methylation by source of DNA. We calculated the Spearman rank correlation coefficients (rs) to determine the correlation of each marker between WBC and saliva DNA. All analyses were performed with SAS software 9.0 (SAS Institute).
| Abbreviations: | ||
| AHRR | = | aryl hydrocarbon receptor repressor |
| LINE-1 | = | long interspersed nucleotide element-1 |
| Sat2 | = | satellite 2 |
| WBC | = | white blood cells |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to gratefully acknowledge all of the families that participate in the New York site of the LEGACY Girls Study. This work was supported by awards from the National Cancer Institute (R01CA138822 and P30 CA013696), the National Institute of Environmental Health Sciences (P30 ES009089), and the Breast Cancer Research Foundation.
References
- Thornburg KL, Shannon J, Thuillier P, Turker MS. 3 - In Utero Life and Epigenetic Predisposition for Disease. In: Zdenko H, Toshikazu U, eds. Advances in Genetics: Academic Press; 2010:57-78.
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 2005; 102:10604 - 9; http://dx.doi.org/10.1073/pnas.0500398102; PMID: 16009939
- Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics 2011; 6:828 - 37; http://dx.doi.org/10.4161/epi.6.7.16500; PMID: 21636973
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 2011; 88:450 - 7; http://dx.doi.org/10.1016/j.ajhg.2011.03.003; PMID: 21457905
- Wu HC, Delgado-Cruzata L, Flom JD, Perrin M, Liao Y, Ferris JS, Santella RM, Terry MB. Repetitive element DNA methylation levels in white blood cell DNA from sisters discordant for breast cancer from the New York site of the Breast Cancer Family Registry. Carcinogenesis 2012; 33:1946 - 52; http://dx.doi.org/10.1093/carcin/bgs201; PMID: 22678115
- Delgado-Cruzata L, Wu HC, Perrin M, Liao Y, Kappil MA, Ferris JS, Flom JD, Yazici H, Santella RM, Terry MB. Global DNA methylation levels in white blood cell DNA from sisters discordant for breast cancer from the New York site of the Breast Cancer Family Registry. Epigenetics 2012; 7:868 - 74; http://dx.doi.org/10.4161/epi.20830; PMID: 22705975
- Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet 2013; 22:843 - 51; http://dx.doi.org/10.1093/hmg/dds488; PMID: 23175441
- Joubert BR, Håberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun Ø, Cupul-Uicab LA, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 2012; 120:1425 - 31; http://dx.doi.org/10.1289/ehp.1205412; PMID: 22851337
- Wu H-C, Wang Q, Yang H-I, Tsai W-Y, Chen C-J, Santella RM. Global DNA methylation levels in white blood cells as a biomarker for hepatocellular carcinoma risk: a nested case-control study. Carcinogenesis 2012; 33:1340 - 5; http://dx.doi.org/10.1093/carcin/bgs160; PMID: 22581841
- Terry MB, Ferris JS, Pilsner R, Flom JD, Tehranifar P, Santella RM, Gamble MV, Susser E. Genomic DNA methylation among women in a multiethnic New York City birth cohort. Cancer Epidemiol Biomarkers Prev 2008; 17:2306 - 10; http://dx.doi.org/10.1158/1055-9965.EPI-08-0312; PMID: 18768498
- Flom JD, Ferris JS, Liao Y, Tehranifar P, Richards CB, Cho YH, Gonzalez K, Santella RM, Terry MB. Prenatal smoke exposure and genomic DNA methylation in a multiethnic birth cohort. Cancer Epidemiol Biomarkers Prev 2011; 20:2518 - 23; http://dx.doi.org/10.1158/1055-9965.EPI-11-0553; PMID: 21994404
- Ollikainen M, Smith KR, Joo EJ-H, Ng HK, Andronikos R, Novakovic B, Abdul Aziz NK, Carlin JB, Morley R, Saffery R, et al. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum Mol Genet 2010; 19:4176 - 88; http://dx.doi.org/10.1093/hmg/ddq336; PMID: 20699328
- Thiede C, Prange-Krex G, Freiberg-Richter J, Bornhäuser M, Ehninger G. Buccal swabs but not mouthwash samples can be used to obtain pretransplant DNA fingerprints from recipients of allogeneic bone marrow transplants. Bone Marrow Transplant 2000; 25:575 - 7; http://dx.doi.org/10.1038/sj.bmt.1702170; PMID: 10713640
- Endler G, Greinix H, Winkler K, Mitterbauer G, Mannhalter C. Genetic fingerprinting in mouthwashes of patients after allogeneic bone marrow transplantation. Bone Marrow Transplant 1999; 24:95 - 8; http://dx.doi.org/10.1038/sj.bmt.1701815; PMID: 10435742
- Talens RP, Boomsma DI, Tobi EW, Kremer D, Jukema JW, Willemsen G, Putter H, Slagboom PE, Heijmans BT. Variation, patterns, and temporal stability of DNA methylation: considerations for epigenetic epidemiology. FASEB J 2010; 24:3135 - 44; http://dx.doi.org/10.1096/fj.09-150490; PMID: 20385621
- Koestler DC, Marsit CJ, Christensen BC, Accomando W, Langevin SM, Houseman EA, Nelson HH, Karagas MR, Wiencke JK, Kelsey KT. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol Biomarkers Prev 2012; 21:1293 - 302; http://dx.doi.org/10.1158/1055-9965.EPI-12-0361; PMID: 22714737
- Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med 2009; 180:462 - 7; http://dx.doi.org/10.1164/rccm.200901-0135OC; PMID: 19498054
- Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, Ehrlich M, Laird PW. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res 2005; 33:6823 - 36; http://dx.doi.org/10.1093/nar/gki987; PMID: 16326863
- Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun H-M, Jiang J, Marinelli B, Pesatori AC, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res 2007; 67:876 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-06-2995; PMID: 17283117