
Abstract
Cornelia de Lange syndrome (CdLS) is a rare multisystem disorder characterized by facial dysmorphisms, limb anomalies, and growth and cognitive deficits. Mutations in genes encoding subunits (SMC1A, SMC3, RAD21) or regulators (NIPBL, HDAC8) of the cohesin complex account for approximately 65% of clinically diagnosed CdLS cases. The SMC1A gene (Xp11.22), responsible for 5% of CdLS cases, partially escapes X chromosome inactivation in humans and the allele on the inactive X chromosome is variably expressed. In this study, we evaluated overall and allele-specific SMC1A expression. Real-time PCR analysis conducted on 17 controls showed that SMC1A expression in females is 50% higher than in males. Immunoblotting experiments confirmed a 44% higher protein level in healthy females than in males, and showed no significant differences in SMC1A protein levels between controls and patients. Pyrosequencing was used to assess the reciprocal level of allelic expression in six female carriers of different SMC1A mutations and 15 controls who were heterozygous at a polymorphic transcribed SMC1A locus. The two alleles were expressed at a 1:1 ratio in the control group and at a 2:1 ratio in favor of the wild type allele in the test group. Since a dominant negative effect is considered the pathogenic mechanism in SMC1A-defective female patients, the level of allelic preferential expression might be one of the factors contributing to the wide phenotypic variability observed in these patients. An extension of this study to a larger cohort containing mild to borderline cases could enhance our understanding of the clinical spectrum of SMC1A-linked CdLS.
Introduction
Cornelia de Lange syndrome (CdLS; OMIM #122470, 300590, 610759, 300882, and 614701) is a genetically and clinically heterogeneous neurodevelopmental disorder with an incidence of 1:10 000 to 1:30 000 live births.Citation1-Citation4 The clinical presentation is marked by typical facial dysmorphisms including synophrys, fine arched eyebrows, long eyelashes, anteverted nares, a depressed nasal bridge, a long philtrum and a thin upper lip with down-turned commissures. It is also characterized by pre- and post-natal growth retardation, malformations of the upper limbs ranging from small hands to complete limb reduction, psychomotor delay, intellectual disability, and other specific medical complications including gastresophageal reflux, cardiac defects, hypoacusia, and genitourinary anomalies.Citation4-Citation6 Patients present with variable phenotypes, ranging from mild to severe, as assessed by the number and severity of the clinical signs.Citation7 This wide clinical expressivity has led to the need to establish a scoring system, based on a list of consensus diagnostic criteria, to rank patients according to the severity of the clinical phenotype.Citation8 The clinical variability is partially accounted for by the locus heterogeneity underlying the syndrome. CdLS can be caused by mutations in at least five different genes, encoding either regulators (NIPBL, HDAC8) or structural elements (SMC1A, SMC3, RAD21) of the cohesin complex.Citation9-Citation14 Cohesin was originally identified to mediate sister chromatid cohesion during both mitosis and meiosis and to participate in DNA double-strand break repair.Citation15 Less canonical roles, including long-range regulation of gene expression and three-dimensional organization of chromatin, have emerged more recently.Citation16,Citation17
Currently, 35 CdLS-relevant mutations in SMC1A have been identified in 52 patients.Citation11,Citation12,Citation18-Citation25 Patients carrying SMC1A mutations tend to present with a less severe phenotype than those with mutations in NIPBL, the major causative gene of CdLS. According to its X-linked dominant transmission, females represent more than 70% of SMC1A-mutated CdLS patients. Notably, female patient phenotypes encompass the entire broad spectrum of the syndrome and present with mild-to-severe phenotypes, while males have more homogeneous clinical presentations at the severe end of the phenotypic spectrum. The SMC1A mutations identified so far comprise only missense mutations or small in-frame deletions that preserve the protein reading frame.Citation11,Citation12,Citation18-Citation25 Mutations map across the entire protein, but spare the highly conserved hinge domain through which SMC1A interacts with the specular domain of SMC3 to form the core cohesin heterodimer. This suggests that mutations altering the protein reading frame or affecting the hinge domain are not compatible with cell viability.
A peculiarity of SMC1A is its localization at Xp11.22, a region that partially escapes X inactivation in humans. The allele on the inactive X chromosome is expressed at levels ranging from 15% to 30%.Citation26 Correspondingly, although data on sex-relative expression of the protein are lacking, females exhibit higher expression of the SMC1A transcript than males.Citation19,Citation27,Citation28 The altered protein is expressed in all patients carrying mutations, irrespective of whether they are male or female.Citation11,Citation19,Citation29 This suggests that the pathogenesis of SMC1A CdLS in females is not due to altered levels of the SMC1A transcript, but rather to a possible dominant negative effect exerted by the altered protein on the wild type (WT) protein.Citation19
In the present study we analyzed SMC1A expression at both the transcript and protein levels in healthy controls of both sexes and in a cohort of CdLS SMC1A-mutated females. We then applied a pyrosequencing allele-specific assay to determine whether SMC1A is differentially expressed between mutant and WT alleles in heterozygous CdLS females.Citation19
Results
SMC1A is expressed at higher levels in healthy females than in healthy males
As the SMC1A gene belongs to a class of genes that escape X inactivation to some extent, variable expression is predicted both among females and between males and females. Evidence for such variable expression has been reported by different studies in which expression levels in females were found to be 20–100% higher than those in males.Citation19,Citation27,Citation28 To expand upon these observations, we analyzed SMC1A transcript levels by real-time PCR in 17 healthy controls (7 males and 10 females). As shown in , SMC1A expression was approximately 50% higher in females than in males (***P = 0.0001; one-tailed Mann-Whitney U test).
Figure 1.SMC1A expression in healthy males and females. (A) Graphical representation of real-time PCR analysis of SMC1A transcript levels in seven males and ten females. Histograms show the average expression level of the gene in the two groups. Male and female data were compared using a one-tailed Mann-Whitney U test (***P = 0.0001). Expression data were normalized to RPLP0, and relative mRNA levels were determined using the ΔΔCt method. Data are expressed as mean ± standard deviation. (B and C) SMC1A protein expression in healthy male and females obtained through semi-quantitative immunoblotting. (B) Histograms show the relative expression of the SMC1A protein in the five female controls (F) compared with the average expression level in the four male controls (set as 1 and represented by a horizontal black line). (C) Average expression of SMC1A protein in four males and five females. Male and female data were compared using a one-tailed Mann-Whitney U test (**P = 0.0079). Expression data were normalized to GAPDH. Data are expressed as mean ± standard deviation.
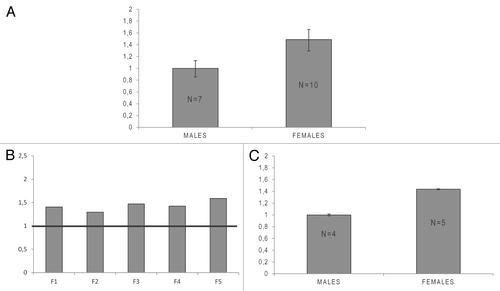
To verify if the higher expression of the SMC1A transcript observed in females was maintained at the protein level, we analyzed SMC1A protein in nine normal LCLs (five from females and four from males) by semi-quantitative immunoblotting. This analysis revealed that SMC1A expression was approximately 44% higher in females than in males (**P = 0.0079; one-tailed Mann-Whitney U test), which confirms the data obtained by RNA analysis ().
SMC1A protein is expressed at similar levels in healthy and CdLS populations
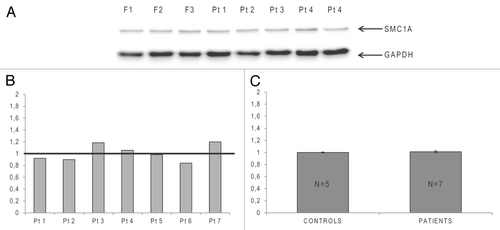
We next investigated whether missense or in-frame deletion mutations in SMC1A CdLS females affect SMC1A protein synthesis. Immunoblotting experiments were conducted on five LCLs from normal females and seven LCLs from CdLS females carrying six previously described mutations.Citation11,Citation20,Citation24 Patients 1 and 6 carry in-frame deletions D831E/Q832del and V58-R62del, respectively. The other mutations (all missense) comprised R693Q in patient 7, I784T in patients 4 and 5, N1166T in patient 3, and L1189F in patient 2.
The experiments revealed that all patients expressed the SMC1A protein at comparable levels (), independently of the type of mutation and with only small variance between samples (). Moreover, a comparison of CdLS patients with control females revealed no significant differences in protein levels between the two groups (P = 0.5; one-tailed Mann-Whitney U test), as shown in the histograms in .
Figure 2. Semi-quantitative immunoblotting analysis of SMC1A in Cornelia de Lange syndrome (CdLS) X-linked patients and female controls. (A)Representative immunoblot performed on lymphoblastoid cell lines (LCLs) of three controls (F) and five CdLS SMC1A-mutated patients. The upper band represents the ~150 kDa SMC1A protein, and the lower migrating band represents the ~36 kDa GAPDH protein, which was used for normalization. (B) Histograms showing relative expression of the SMC1A protein in the mutated CdLS female patients (Pt) compared with the average expression level in five female controls (set as 1 and represented by a horizontal black line). (C) Graphical representation of the average SMC1A protein expression level in a group of five female controls and seven CdLS patients. Data were compared using a one-tailed Mann-Whitney U test (P = 0.5).
Wild type SMC1A allelic expression is higher than mutant allele expression in CdLS females
Given the peculiar pattern of SMC1A expression, we next investigated the expression of WT and mutant alleles in affected females to determine whether these alleles differentially expressed SMC1A. We first examined the percentage of X inactivation in female patients to identify skewed or random inactivation of the X chromosome at HUMARA and DXS6673E loci. Using an X inactivation ratio ≥ 90:10 as a criterion of severe skewing, four of the seven CdLS female patients, namely patients 3, 4, 5, and 6, showed complete preferential X inactivation (see Table S1).
We subsequently evaluated the expression level of both SMC1A alleles by pyrosequencing analysis. SMC1A allele-specific primers were used to analyze samples from 15 female controls and six CdLS females (patient 6 was excluded as an effective pyrosequencing assay could not be established). Patients were analyzed using assays specific for each mutation, while the two alleles in the controls were discriminated using a heterozygous coding SNP, rs1264011. The regions analyzed in each assay are indicated on the schematic representation of the SMC1A protein in . RNA from both peripheral blood and respective LCLs from four of the six CdLS patients (3, 4, 5, and 7) was analyzed to determine if SMC1A allele expression ratios were affected by the nature of the starting material. As indicated in , no significant differences were observed between the two sample types.
Figure 3. Pyrosequencing assay. (A) Schematic representation of the SMC1A protein showing relative positions of the mutations on the functional domains. Boxes indicate the N- and C-terminal NTPase-binding cassettes (ABC) (amino acids 4–148 and 1117–1220, respectively) and the crucial hinge motif (amino acids 513–628), while the black line represents the coiled-coil domains. The position of the coding SNP rs1264011 at the N-terminus of the protein is indicated. Mutation-specific pyrosequencing assays were performed for each of the six investigated CdLS patients, and a specific assay for the coding SNP rs1264011 was used for the 15 heterozygous controls. (B) Pyrosequencing results. Expression levels of the wild type (allele 1) and mutant (allele 2) SMC1A alleles in six patients (Pt). In four patients, RNA expression analysis of both peripheral blood (PB) and LCLs are shown. (C) Histograms of pyrosequencing analysis data. Patients’ (Pt) wild type allele expression is shown in light gray and mutant allele expression is in dark gray. For comparison, the relative allele expression in controls (1:1) is indicated with a horizontal black line.
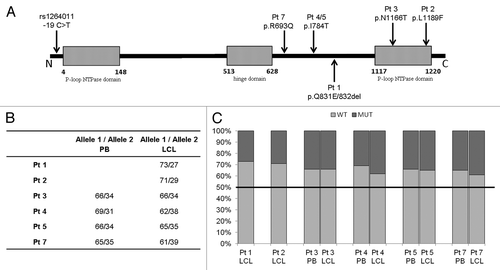
Pyrosequencing analysis showed that the expression ratio of two SNP rs1264011 alleles in female controls was 1:1 using RNA extracted from blood and LCLs. By contrast, the average expression ratio between the WT and mutant alleles in CdLS patients was 2:1 (). In all patients, >60% of the total SMC1A transcript was derived from the WT allele, and the greatest difference in expression was observed in patient 1 (73% WT allele vs. 27% mutant allele).
Discussion
Recent advances, such as the identification of the novel causative genes RAD21Citation13 and HDAC8,Citation14 and increasing knowledge about the multiple functions of cohesin,Citation17 have cast new light on the molecular mechanisms of CdLS. However, the effects of mutations in cohesin genes on the clinical phenotype have proven hard to determine, not only because of the genome-wide dysregulation of multiple genes caused by mutations in cohesin genes, but also because of the presumed differential expression of mutant and WT alleles.Citation30,Citation31 Indeed, with regards to the most investigated gene, NIPBL, insights into how the WT allele is upregulated to compensate for the lower expression of the mutant allele have been obtained in both mouse and human.Citation31 Autoregulation of the WT allele is even more relevant for the X-linked SMC1A gene, which belongs to a category of genes that escape X inactivation in humans and display variable expression of the allele localized on the inactive X chromosome.Citation26 Data on the gender-related expression of this category of X-linked genes have been provided by a few studies, all of which showed a trend toward higher expression in females. However, the percentage of genes escaping inactivation and the level of overexpression in females, as well as the precise increment in SMC1A expression, varied in the different studies according to the tissue analyzed and the X inactivation detection technique used.Citation19,Citation27,Citation28,Citation32 The rate of X inactivation escape predicts further variability in the expression of the clinical phenotype among heterozygous mutated females, making its assessment of utmost importance for the clinical evaluation of patients and genetic counseling of families.
To date, 35 unique SMC1A mutations have been described, and these are invariably missense or in-frame deletions that maintain the coding frame of the gene.Citation11,Citation12,Citation18-Citation25 A 2:1 male:female ratio has been observed in the 52 SMC1A-mutated individuals examined to date. Compared with males, affected females display a wider spectrum of clinical variability, ranging from severe to mild, enough to allow the carriers to be identified only through their affected progeny.Citation11 Expression studies in heterozygous females confirmed the presence of the mutant transcript, normal levels of the SMC1A protein,Citation11,Citation19,Citation29 and incorporation of the mutated proteins into the cohesin complex.Citation33 Nevertheless, based on the finding that SMC hinge dimers containing a mutated SMC1A protein display impaired association with DNA,Citation34 the function of SMC hinge dimers involved in the bridging of chromatin remodeling complexes to basic transcription factors might be also affected. According to the emerging role of cohesin genes as “epigenetic regulators”Citation35, we speculate that a dominant negative effect of the mutant SMC1A allele could be a driving force for the clinical phenotype of CdLS X-linked female patients, although the possibility of a loss-of-function pathomechanism should be also taken into accountCitation19,Citation29,Citation34 To gain further insights into the pathogenic mechanism, we analyzed overall and allele-specific SMC1A expression in healthy controls and X-linked CdLS females. To date, three different studies observed variable SMC1A expression levels in females that were 20–100% higher than in males,Citation19,Citation27,Citation28 but no quantitative data regarding protein levels were obtained. In line with these data, our real-time PCR analysis revealed that the level of SMC1A transcript in females is 50% higher than in males. The immunoblotting experiments showed that females express 44% more SMC1A protein than males, which agrees with the results of transcriptional analysis. When this study was extended to our cohort of CdLS female patients, it revealed that there were no significant differences in the total amount of the SMC1A protein between healthy females and patients. This consolidates the view that the pathogenic mechanism underlying the syndrome in females is not linked to haploinsufficiency.
While SMC1A total amount does not differ between healthy females and patients, a previous study by Liu et al. reported a difference in SMC1A transcription between mutant and WT alleles in a single patient. To address this issue, we used pyrosequencing analysis to systematically determine the transcriptional expression ratio between WT and mutant alleles in a clinically heterogeneous group of six SMC1A-mutated females and 15 SMC1A-heterozygous controls. Notably, three of the six analyzed patients showed complete preferential inactivation, with a ratio higher than 90:10; thus, the finding that all of the patients express the mutant allele at levels > 30% provides further evidence that SMC1A escapes X inactivation. The pyrosequencing analysis showed that all the female carriers of SMC1A mutations expressed the two alleles differentially. The WT allele was expressed at a higher level in all patients, with an average WT:mutant allele expression ratio of 2:1 that did not correlate with XCI status. These data may partially explain reports of asymptomatic, pauci-symptomatic,Citation11 and moderately affected carriers,Citation12 making the determination of the expression levels of mutant vs. WT SMC1A a prerequisite to understanding variable clinical expressivity in females.
Given the likely dominant negative effect of the altered protein, it is possible that the expression level of the mutant allele may act to modulate the CdLS phenotype. Consistent with this hypothesis, the patient with the lowest expression level of the mutated SMC1A allele (Patient 1) had the least severe phenotype.Citation11 Nevertheless, the firm establishment of a relationship between expression levels and severity of the phenotype requires extension of the analysis described in this study to a larger cohort containing mild to borderline cases who are frequently overlooked unless they are identified through the affected progeny.
Patients and Methods
Subject recruitment and generation of lymphoblastoid cell lines
Blood samples were collected from seven previously described CdLS patients enrolled in different Clinical Genetics Units. All affected individuals or their legal guardians gave written informed consent for participation in the study.
Patient lymphoblastoid cell lines (LCLs) were established by Epstein-Barr virus transformation by Galliera Genetic Bank. LCLs were cultured in RPMI 1640 medium supplemented with 20% fetal bovine serum (FBS) and 1% antibiotics at 37 °C in 5% CO2.
DNA extraction
Genomic DNA was isolated from peripheral blood and LCLs. Extraction from blood was performed with a Promega DNA Blood Kit (Promega), while the QIAamp DNA Mini Kit (Qiagen) was used for gDNA isolation from LCLs. Both extractions were performed following the manufacturer’s instructions.
X inactivation analysis
Detection of X chromosome inactivation status was performed at HUMARA and DXS6673E loci. Each sample contained 250 ng of blood or LCL gDNA, which were digested with the appropriate enzymes.
HUMARA digestion was performed with 2.5 U of HhaI and HpaII (New England Biolabs), while DXS6673E digestion was performed with 5 U of HhaI and RsaI (New England Biolabs). In both cases, gDNA was digested for 2 h at 37 °C, and the enzymes were then inactivated at 65 °C for 20 min. A male control was included in the digestion experiment to verify the completeness of enzymatic cleavage. Digested and undigested samples were subsequently polymerase chain reaction (PCR)-amplified. The HUMARA locus was amplified with 5′-FAM-TCCAGAATCT GTTCCAGAGC GTGC-3′ (forward) and 5′-GCTGTGAAGG TTGCTGTTCC TCAT-3′ (reverse) primers, and the DXS6673E locus was amplified with 5′-FAM-ATGCTAAGGA CCATCCAGGA-3′ (forward) and 5′-GGAGTTTTCC TCCCTCACCA-3′ (reverse) primers.
PCR products were separated using an ABI Prism 310 Genetic Analyzer (Applied Biosystems, Life Technologies), and GeneScan 4.0 software was used to calculate peak position and the area intensity of each allele. The level of X chromosome inactivation was calculated by dividing the ratio of the allele peak areas in the digested sample by the ratio of the allele peak areas in the undigested sample.Citation36
RNA extraction and cDNA synthesis
Total RNA was extracted from blood and LCLs. Extraction from blood was performed using the Tempus Spin RNA Isolation Reagent Kit (Applied Biosystems) according to the manufacturer’s instructions. RNA isolation from LCLs was performed with TRI reagent RNA Isolation Reagent (Sigma-Aldrich). To avoid genomic DNA contamination, all extracted RNA samples were treated with DNase I (RNase-free, New England Biolabs).
cDNA synthesis was performed on 250 ng RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and random hexamers. All samples were reverse transcribed in two independent experiments.
Real-time PCR assay
Relative SMC1A mRNA levels were determined by real-time PCR using a StepOne Real Time PCR System Instrument (Applied Biosystems). The real-time PCR reaction was performed using a TaqMan gene expression assay (assay ID: Hs01091953-m1) which probe spans exons 19–20 junction. RPLP0 (assay ID: 4333761F) was selected as an endogenous normalizing gene based on the results of previously performed efficiency and stability experiments. Relative gene expression was determined using the ΔΔCt method.Citation37
Pyrosequencing assay
To assess the reciprocal level of allelic expression, pyrosequencing analysis was performed on samples from 15 controls and from six female patients carrying different SMC1A mutations. Allelic expression was discriminated by examining for the specific heterozygous mutation in each patient and the heterozygous coding SNP rs1264011 in the controls. Approximately 50 ng of cDNA template were amplified in 50 µl reactions using GoTaq Flexi DNA Polymerase (Promega) and mutation/SNP specific primers (available upon request). The analysis was performed with the Pyro Gold Reagent Kit (Biotage AB). Briefly, the sequencing primer (at a final concentration of 2 µM) was hybridized to single biotinylated PCR strands and then transferred to the Pyro Mark ID instrument (Biotage AB) for determination of transcriptional allele frequencies.
Protein extraction and immunoblotting
LCLs were lysed using lysis buffer (150 mM NaCl, 50 mM Tris pH 7.5, 1% NP-40, and 0.25% deoxycholic acid) containing a protease inhibitor cocktail (complete, EDTA-free; Roche Diagnostic). Protein concentrations were determined using a BCA Protein Assay Kit (Pierce) and an ND-1000 Spectrophotometer (Thermo Fisher Scientific Inc., NanoDrop Products), according to the manufacturers’ protocols.
Each sample (20 µg protein) was diluted in reducing SDS Loading Buffer (Blue Loading Buffer Pack, Cell Signaling Technology Inc.) and the proteins were denatured at 99 °C for 3 min. Proteins were separated by 8% SDS polyacrylamide gels and subsequently transferred to a polyvinylidene fluoride (PVDF) membrane (Roche) by electroblotting. After electroblotting, membranes were washed twice in PBS-T (100 mM NaCl, 80 mM Na2HPO4, 20 mM NaH2PO4, and 0.3% Tween20) and nonspecific binding was blocked by incubating the membranes in 5% skimmed milk in PBS-T for 1 h at room temperature, with agitation. Membranes were cut horizontally immediately after the blocking step and incubated with agitation overnight at 4 °C with rabbit anti-SMC1A (1:2,000) (ab21583; Abcam) and mouse anti-GAPDH (1:12,500) (ab8245; Abcam) antibodies diluted in PBS-T. Membranes were then washed four times in PBS-T and incubated with agitation at RT for 1 h with HRP-conjugated secondary antibodies diluted in PBS-T. Goat anti-rabbit IgG (1:10,000) (sc-2004; Santa Cruz Biotechnology) and goat anti-mouse IgG (1:25,000) (sc-2005; Santa Cruz Biotechnology) were used respectively for SMC1A and GAPDH detection. After four washes in PBS-T and two washes in PBS, bound antibodies were detected using enhanced chemiluminescence (Westar ηC; Cyangene).
Blot images were acquired using a Gbox Chemi XT4 system (Syngene) and semi-quantitative analysis of SMC1A protein expression was performed using Gene Tools Gel Analysis software (Syngene) and the results were normalized to GAPDH protein expression.
Statistical analysis
Real-time PCR and immunoblotting experiments were performed in triplicate for each sample; values with standard deviations exceeding 0.5% or standard errors exceeding 0.3% were excluded and the experiments were repeated. The Mann-Whitney U test was used to compare gene expression data, and differences between experimental groups were considered significant when P < 0.05. All statistical analyses were performed using the GraphPad Prism 5 program.
| Abbreviations: | ||
| CdLS | = | Cornelia de Lange syndrome |
| FBS | = | fetal bovine serum |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| HDAC8 | = | histone deacetylase 8 |
| LCLs | = | lymphoblastoid cell lines |
| NIPBL | = | Nipped-B-like |
| PVDF | = | polyvinylidene fluoride |
| RAD21 | = | homolog of S. pombe Rad21 |
| SMC1A | = | structural maintenance of chromosomes 1A |
| SMC3 | = | structural maintenance of chromosomes 3 |
| WT | = | wild type |
Additional material
Download Zip (181.1 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank the study participants and their families, as well as the healthy control individuals, without whom this work would not have been possible. The authors would like to thank Paolo Verderio (Istituto Nazionale dei Tumori, Milano) for his expert assistance in the statistical analysis of the data and helpful discussion. We also thank the Galliera Genetic Bank – Network of Telethon Genetic Biobank (project GTB07001) for providing us the LCLs of both patients and controls. This study was supported by “Accordo quadro Università-Regione Lombardia no: 17292” (to L.L.) and by a Ministry of Health “Ricerca Corrente” grant to Istituto Auxologico Italiano IRCCS (08C001–2010).
References
- Opitz JM. The Brachmann-de Lange syndrome. Am J Med Genet 1985; 22:89 - 102; PMID: 3901753
- Ireland M, Donnai D, Burn J. Brachmann-de Lange syndrome. Delineation of the clinical phenotype. Am J Med Genet 1993; 47:959 - 64; PMID: 8291539
- Jackson L, Kline AD, Barr MA, Koch S. de Lange syndrome: a clinical review of 310 individuals. Am J Med Genet 1993; 47:940 - 6; http://dx.doi.org/10.1002/ajmg.1320470703; PMID: 8291537
- Kline AD, Krantz ID, Sommer A, Kliewer M, Jackson LG, FitzPatrick DR, Levin AV, Selicorni A. Cornelia de Lange syndrome: clinical review, diagnostic and scoring systems, and anticipatory guidance. Am J Med Genet A 2007; 143A:1287 - 96; PMID: 17508425
- Brachmann E. Ein Fall von symmetrischer Monodaktylie durch Ulnadefekt, mit symmetrischer Flughaurbildung in den Ellenbogen sowie anderen Abnormalitäten. Jahrbuch für Kinderheilkunde und physische Erziehung 1916; 84:225 - 235
- de Lange C. Sur un type nouveau de dégénération (typus Amstelodamensis). Arch Med Enfants 1933; 36:713 - 7193
- Van Allen MI, Filippi G, Siegel-Bartelt J, Yong SL, McGillivray B, Zuker RM, Smith CR, Magee JF, Ritchie S, Toi A, et al. Clinical variability within Brachmann-de Lange syndrome: a proposed classification system. Am J Med Genet 1993; 47:947 - 58; PMID: 8291538
- Selicorni A, Russo S, Gervasini C, Castronovo P, Milani D, Cavalleri F, Bentivegna A, Masciadri M, Domi A, Divizia MT, et al. Clinical score of 62 Italian patients with Cornelia de Lange syndrome and correlations with the presence and type of NIPBL mutation. Clin Genet 2007; 72:98 - 108; PMID: 17661813
- Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet 2004; 36:636 - 41; http://dx.doi.org/10.1038/ng1363; PMID: 15146185
- Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 2004; 36:631 - 5; http://dx.doi.org/10.1038/ng1364; PMID: 15146186
- Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, Russo S, Vezzoni P, Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet 2006; 38:528 - 30; http://dx.doi.org/10.1038/ng1779; PMID: 16604071
- Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, Pié J, Gil-Rodríguez C, Arnedo M, Loeys B, Kline AD, et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet 2007; 80:485 - 94; http://dx.doi.org/10.1086/511888; PMID: 17273969
- Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, Mönnich M, Yan Y, Xu W, Gil-Rodríguez MC, et al. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet 2012; a 90:1014 - 27; http://dx.doi.org/10.1016/j.ajhg.2012.04.019; PMID: 22633399
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012; b 489:313 - 7; http://dx.doi.org/10.1038/nature11316; PMID: 22885700
- Dorsett D, Ström L. The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr Biol 2012; 22:R240 - 50; http://dx.doi.org/10.1016/j.cub.2012.02.046; PMID: 22497943
- Dorsett D. Cohesin: genomic insights into controlling gene transcription and development. Curr Opin Genet Dev 2011; 21:199 - 206; http://dx.doi.org/10.1016/j.gde.2011.01.018; PMID: 21324671
- Mehta GD, Kumar R, Srivastava S, Ghosh SK. Cohesin: functions beyond sister chromatid cohesion. FEBS Lett 2013; 587:2299 - 312; http://dx.doi.org/10.1016/j.febslet.2013.06.035; PMID: 23831059
- Borck G, Zarhrate M, Bonnefont JP, Munnich A, Cormier-Daire V, Colleaux L. Incidence and clinical features of X-linked Cornelia de Lange syndrome due to SMC1L1 mutations. Hum Mutat 2007; 28:205 - 6; http://dx.doi.org/10.1002/humu.9478; PMID: 17221863
- Liu J, Feldman R, Zhang Z, Deardorff MA, Haverfield EV, Kaur M, Li JR, Clark D, Kline AD, Waggoner DJ, et al. SMC1A expression and mechanism of pathogenicity in probands with X-Linked Cornelia de Lange syndrome. Hum Mutat 2009; 30:1535 - 42; http://dx.doi.org/10.1002/humu.21095; PMID: 19701948
- Limongelli G, Russo S, Digilio MC, Masciadri M, Pacileo G, Fratta F, Martone F, Maddaloni V, D’Alessandro R, Calabro P, et al. Hypertrophic cardiomyopathy in a girl with Cornelia de Lange syndrome due to mutation in SMC1A. Am J Med Genet A 2010; 152A:2127 - 9; http://dx.doi.org/10.1002/ajmg.a.33486; PMID: 20635401
- Pié J, Gil-Rodríguez MC, Ciero M, López-Viñas E, Ribate MP, Arnedo M, Deardorff MA, Puisac B, Legarreta J, de Karam JC, et al. Mutations and variants in the cohesion factor genes NIPBL, SMC1A, and SMC3 in a cohort of 30 unrelated patients with Cornelia de Lange syndrome. Am J Med Genet A 2010; 152A:924 - 9; http://dx.doi.org/10.1002/ajmg.a.33348; PMID: 20358602
- Mannini L, Menga S, Musio A. The expanding universe of cohesin functions: a new genome stability caretaker involved in human disease and cancer. Hum Mutat 2010; 31:623 - 30; http://dx.doi.org/10.1002/humu.21252; PMID: 20513141
- Hoppman-Chaney N, Jang JS, Jen J, Babovic-Vuksanovic D, Hodge JC. In-frame multi-exon deletion of SMC1A in a severely affected female with Cornelia de Lange Syndrome. Am J Med Genet A 2012; 158A:193 - 8; http://dx.doi.org/10.1002/ajmg.a.34360; PMID: 22106055
- Gervasini C, Russo S, Cereda A, Parenti I, Masciadri M, Azzollini J, Melis D, Aravena T, Doray B, Ferrarini A, et al. Cornelia de Lange individuals with new and recurrent SMC1A mutations enhance delineation of mutation repertoire and phenotypic spectrum. Am J Med Genet A 2013; 161A:2909 - 19; PMID: 24124034
- http://grenada.lumc.nl/LOVD2/CDLS/home.php?select_db=SMC1A
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005; 434:400 - 4; http://dx.doi.org/10.1038/nature03479; PMID: 15772666
- Craig IW, Mill J, Craig GM, Loat C, Schalkwyk LC. Application of microarrays to the analysis of the inactivation status of human X-linked genes expressed in lymphocytes. Eur J Hum Genet 2004; 12:639 - 46; http://dx.doi.org/10.1038/sj.ejhg.5201212; PMID: 15114374
- Johnston CM, Lovell FL, Leongamornlert DA, Stranger BE, Dermitzakis ET, Ross MT. Large-scale population study of human cell lines indicates that dosage compensation is virtually complete. PLoS Genet 2008; 4:e9; http://dx.doi.org/10.1371/journal.pgen.0040009; PMID: 18208332
- Revenkova E, Focarelli ML, Susani L, Paulis M, Bassi MT, Mannini L, Frattini A, Delia D, Krantz I, Vezzoni P, et al. Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum Mol Genet 2009; 18:418 - 27; PMID: 18996922
- Liu J, Zhang Z, Bando M, Itoh T, Deardorff MA, Clark D, Kaur M, Tandy S, Kondoh T, Rappaport E, et al. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol 2009; 7:e1000119; PMID: 19468298
- Kawauchi S, Calof AL, Santos R, Lopez-Burks ME, Young CM, Hoang MP, Chua A, Lao T, Lechner MS, Daniel JA, et al. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/-) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet 2009; 5:e1000650; http://dx.doi.org/10.1371/journal.pgen.1000650; PMID: 19763162
- Castagné R, Zeller T, Rotival M, Szymczak S, Truong V, Schillert A, Trégouët DA, Münzel T, Ziegler A, Cambien F, et al. Influence of sex and genetic variability on expression of X-linked genes in human monocytes. Genomics 2011; 98:320 - 6; PMID: 21763416
- Gimigliano A, Mannini L, Bianchi L, Puglia M, Deardorff MA, Menga S, Krantz ID, Musio A, Bini L. Proteomic profile identifies dysregulated pathways in Cornelia de Lange syndrome cells with distinct mutations in SMC1A and SMC3 genes. J Proteome Res 2012; 11:6111 - 23; PMID: 23106691
- Mannini L, Cucco F, Quarantotti V, Krantz ID, Musio A. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum Mutat 2013; 34:1589 - 96; http://dx.doi.org/10.1002/humu.22430; PMID: 24038889
- Kleefstra T, Schenck A, Kramer JM, van Bokhoven H. The genetics of cognitive epigenetics. [Epub ahead of print] Neuropharmacology 2014; 80C:83 - 94; http://dx.doi.org/10.1016/j.neuropharm.2013.12.025; PMID: 24434855
- Sharp A, Robinson D, Jacobs P. Age- and tissue-specific variation of X chromosome inactivation ratios in normal women. Hum Genet 2000; 107:343 - 9; http://dx.doi.org/10.1007/s004390000382; PMID: 11129333
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods 2001; 25:402; PMID: 11846609