
Abstract
In Arabidopsis, variant in methylation (VIM) proteins are required for the maintenance of DNA methylation in the CpG dinucleotide context. VIM1 acts as a cofactor of DNA methyltransferase 1 (MET1), although the mechanism for this co-regulation remains unclear. In this study, we used RNA-seq analysis to profile the transcriptomes of vim1, vim1 vim2 vim3, and met1 null mutants. Consistent with previous studies indicating functional redundancy between these VIM proteins, we found no transcripts that were significantly misregulated in vim1 mutants. However, we identified a large set of VIM protein regulatory targets through analysis of vim1 vim2 vim3 mutants, and we observed that this set is essentially identical to that regulated by MET1. Log2 fold changes in gene expression relative to wild type are strongly correlated between vim1 vim2 vim3 and met1 mutants. While the largest subset of these transcripts is upregulated and enriched with transposable elements, we also found small subsets of downregulated genes in each mutant, which are enriched with protein-coding genes. Together, these results expand on previous studies that profiled cytosine methylation in the vim1 vim2 vim3 mutant, and show that VIM proteins function in transcriptional regulation via their roles in the MET1 DNA methylation pathway.
Introduction
Cytosine methylation is a mechanism used by many eukaryotes to mediate transcriptional repression. In many organisms, such as mammals, cytosine methylation exists predominantly in the CG dinucleotide context.Citation1,Citation2 In plants such as Arabidopsis, methylation can additionally occur in the CHG or CHH contexts (where H represents A, C, or T), but CG dinucleotides are still the most abundant methylation sequence context.Citation3,Citation4 Cytosine methylation in Arabidopsis functions primarily to silence transposons and other repetitive elements,Citation5,Citation6 although the bodies of expressed genes can also be methylation targets.Citation7,Citation8 Methylation is established through the activity of the Domains Rearranged Methyltransferase 2 (DRM2), an ortholog of mammalian DNMT3 methyltransferases that can act on all three sequence contexts.Citation9 Maintenance of CG and CHG methylation are performed by DNA methyltransferase 1 (MET1) and chromomethylase 3 (CMT3), respectively.Citation10,Citation11 MET1 is an ortholog of mammalian DNMT1, which is present at DNA replication forks and actively methylates CG sites on the newly synthesized daughter strands of DNA.Citation12,Citation13
The maintenance of CG methylation in plants also requires the activity of additional proteins, such as the Variant In Methylation (VIM/ORTH) family.Citation14-Citation16 In Arabidopsis, this family is composed of five highly similar proteins, some of which (VIM1, 2 and 3) are expressed in vegetative tissues, while VIM5 is more highly expressed in endosperm.Citation17,Citation18 Its first identified member, VIM1, was found through a screen for trans-acting mutations that affect DNA methylation in natural Arabidopsis strains.Citation14 Later, it was found that VIM1 functions redundantly along with VIM2 and VIM3 to maintain CG methylation at centromeres and other repetitive loci.Citation17 VIM1 has been shown to bind DNA containing methylcytosine in vitro, with a particular specificity for hemimethylated cytosines.Citation19 Because of this activity, it was proposed that VIM proteins function to recruit MET1 to CG sites through the activity of its methylcytosine binding SET- and RING-associated (SRA) domain, as MET1 lacks a methylcytosine binding module.
Studies on the VIM animal homolog UHRF1 have shown that it fills a very similar biological role,Citation20 with at least two potential mechanisms having been proposed for its function. One group suggested that a direct interaction occurs between the UHRF1 SRA domain and a region of DNMT1, supporting their model with a yeast two-hybrid assay and in vitro approaches.Citation21 More recently, a separate set of results was published showing that UHRF1 ubiquitylates histone H3 in frog oocyte extracts through the activity of its RING domain.Citation22 This study found that DNMT1 was recruited to target loci by binding the ubiquitylated histone H3. In both of the proposed mechanisms, UHRF1 and DNMT1 coordinately regulate cytosine methylation at target loci.
The recent emergence of whole-genome bisulfite sequencing (BS-seq) has allowed DNA methylation profiling in Arabidopsis across a variety of conditions.Citation3,Citation4 Notably, methylomes were recently mapped in a large number of gene silencing mutants.Citation23 That study concluded that VIM1, VIM2, and VIM3 function entirely redundantly, and noted that the methylation profile of a vim1 vim2 vim3 mutant strongly resembled that of a met1 null mutant. However, these profiling results did not address the question of whether VIM proteins serve additional roles in transcriptional regulation outside of their functions in maintaining DNA methylation. Uncertainty remains over whether VIM proteins are involved in other chromatin modification pathways, raising the possibility that VIM proteins transcriptionally regulate additional loci outside the set of MET1 targets. Alternatively, VIM proteins might function exclusively as MET1 cofactors, in which case there would be a complete overlap in the sets of genes regulated by VIM proteins and MET1.
In this study, we investigate the transcriptional regulatory function of VIM proteins using RNA-seq. We sequenced whole transcriptomes of met1, vim1, and vim1 vim2 vim3 mutants to compare the sets of regulatory targets. Strikingly, the met1 and vim1 vim2 vim3 target sets are almost identical, suggesting that VIM proteins do not have additional transcriptional regulatory functions outside their role in maintaining DNA methylation. We also found no significant differentially expressed targets in the vim1 mutant, confirming studies that have shown functional redundancy between VIM1 and other VIM proteins.Citation17 We have generated an extensive list of regulated targets, including transposable elements, protein coding genes, and noncoding RNA transcripts. Comparable to previous studies in CG methylation mutants,Citation4,Citation7 we found that most of the regulated targets are transposable elements. Together, these analyses provide a comprehensive picture of the targets regulated by CG methylation in Arabidopsis.
Results
VIM protein and MET1 target gene sets overlap extensively
We extracted RNA from vim1, vim1 vim2 vim3, met1, and wild type plants, then prepared strand-specific Illumina RNA-seq libraries in biological triplicates according to a protocol developed by Wang et al.Citation24 After final library cleanup and amplification, the average fragment size of each library ranged between 330 and 370 base pairs. Sequencing data was generated on the Illumina Hi-Seq 2000 platform, and the resulting reads were mapped to the Arabidopsis TAIR10 genome build using TopHat. Because transposable elements are the predominant targets of DNA methylation in Arabidopsis, we included the TAIR10 annotated transposable elements in our transcriptome reference file. To increase confidence in our results, we included only reads that mapped uniquely to a single genomic location in our differential expression analysis by filtering the TopHat output files for a MAPQ value of 10 or greater. Reads were aligned with no more than two mismatches. Summary statistics of the data are presented in .
Table 1. Summary statistics for the RNA-seq data
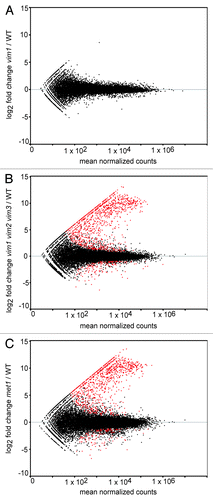
We analyzed differential expression between mutant and wild type samples using the R package DESeq.Citation25 No transcripts were significantly differentially expressed in the vim1 mutant (), reflecting the functional redundancy that our group and others have observed between VIM1, VIM2, and VIM3.Citation17,Citation23 The analysis identified 2387 transcripts differentially expressed between vim1 vim2 vim3 mutant and wild type, and 2748 transcripts differentially expressed between met1 and wild type (; Tables S1 and S2). Owing partially to advances in high-throughput sequencing technology and an additional focus on transposable elements in our analysis, the number of targets identified in the met1 mutant is substantially greater than that found by Lister et al. (2008).Citation4 As expected, the distribution of p-values for each differential expression analysis is uniform, except for the low p-values associated with differentially expressed transcripts and the high p-values associated with transcripts having low read counts (Fig. S1).
Figure 1.Identification of differentially expressed transcripts in vim1 vim2 vim3 and met1 mutants. MA-plots of log2 fold change transcript expression vs. mean normalized counts in vim1 (A), vim1 vim2 vim3 (B) and met1 (C) mutants relative to wild type. Red points represent differentially expressed transcripts meeting the adjusted P value cut-off of 0.05 or lower. Black points represent all other transcripts not called differentially expressed. Notably, no transcripts, other than VIM1, were identified as differentially expressed in the vim1 mutant, while over 2000 transcripts were differentially expressed in vim1 vim2 vim3 and met1 relative to wild type. The majority of the red points fall above the gray horizontal line at zero, indicating that most differentially expressed genes are upregulated in the vim1 vim2 vim3 and met1 mutants.
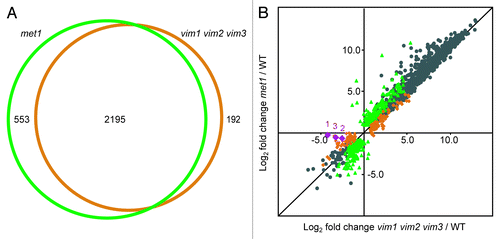
Remarkably, the sets of genes differentially expressed in vim1 vim2 vim3 and in met1 overlapped to a very large extent (). To determine whether the magnitude of up- or downregulation was well correlated between the two data sets, we generated a scatter plot based on the expression profiles of the vim1 vim2 vim3 and met1 differentially expressed genes (). Based on the paired log2 fold change data for the transcripts shared between the two data sets, the Pearson’s correlation coefficient r = 0.984, indicating a very strong correlation exists between the expression profiles. When we examined the transcripts identified as misregulated in vim1 vim2 vim3 only or in met1 only, we also found a correlation between the two mutants in log2 fold change relative to wild type (r = 0.938 and r = 0.920, respectively). Despite similar fold changes in expression for both mutants, many of these transcripts were called differentially expressed in only one mutant due to our adjusted p-value filtering.
Figure 2. VIM proteins and MET1 regulate extensively overlapping sets of transcripts. (A) Venn diagram illustrating the large overlap between the targets differentially expressed in met1 mutants (green circle) and the targets differentially expressed in vim1 vim2 vim3 mutants (orange circle). Numbers of targets specific to each mutant and shared between the two mutants are indicated. (B) Scatter plot showing high correlation between the log2 fold change expression values in met1 relative to wild type (y-axis) and vim1 vim2 vim3 relative to wild type (x-axis). Green data points represent transcripts differentially expressed only in met1; orange data points correspond to transcripts differentially expressed only in vim1 vim2 vim3; and gray data points represent transcripts differentially expressed in both mutants relative to wild type. As a confirmation of the vim1 vim2 vim3 mutant genotype, the VIM1, VIM2, and VIM3 transcripts are represented with purple diamonds labeled 1, 2, and 3, respectively. Transcripts with an expression value of zero in the mutant or wild type sample were excluded from this analysis. A black line representing y = x is shown for reference.
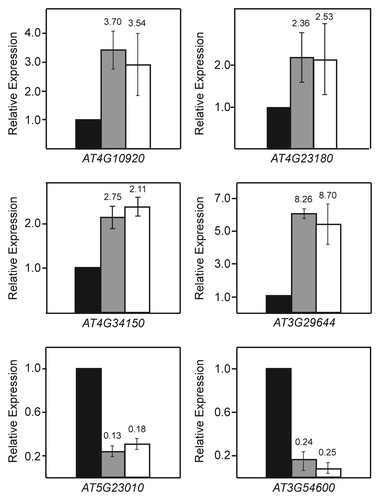
Small subsets of differentially regulated transcripts were identified as specific to the vim1 vim2 vim3 or met1 mutant plants. We chose six targets each from the vim1 vim2 vim3-specific set and the met1-specific set for follow-up expression analysis. However, none of these targets could be confirmed independently through reverse transcription quantitative PCR (RT-qPCR) (Fig. S2). On the other hand, several of the transcripts found to be differentially expressed in both vim1 vim2 vim3 mutants and met1 mutants relative to wild type were validated through further expression analysis (). One possible explanation for this discrepancy is that the identified vim1 vim2 and vim3-specific and met1-specific transcripts are primarily false-positive results. In accordance with this interpretation, many of the false discovery rate (FDR) adjusted p-values of the transcripts in these subsets are very near the cutoff of 0.05 that we established for significance, making them marginal differential expression calls. We conclude that the VIM proteins and MET1 regulate an essentially identical set of targets.
Figure 3. Validation of selected transcripts regulated by VIM proteins and MET1. RT-qPCR relative expression analysis of four upregulated targets and two downregulated targets from the set of transcripts co-regulated by VIM proteins and MET1 is shown. Similar fold changes in expression are seen in the vim1 vim2 vim3 mutant (gray bars) and met1 mutant (white bars) relative to wild type (black bars). Expression values represent the averages calculated from three independent biological replicates. Error bars represent the standard error of the mean. For comparison of RT-qPCR with the RNA-seq results, fold changes relative to wild type from the RNA-seq data set are indicated above each bar.
Transcriptional regulation of transposable elements and protein-coding genes by VIM proteins
Consistent with the established role of CG methylation in transcriptional repression, most of the transcripts identified as differentially expressed relative to wild type in the vim1 vim2 vim3 and met1 mutants were upregulated. We observed that a great majority of the upregulated transcripts were transposable elements (), with a smaller number of protein-coding genes and other transcripts. This result matches previous studies that have established CG methylation as a mechanism for transposon silencing.Citation5
Figure 4. Comparison of upregulated and downregulated targets differentially expressed in vim1 vim2 vim3 and met1 mutants. Pie charts show the proportions of transposable elements (black), protein-coding genes (white) and other transcripts such as microRNAs or unclassified RNAs (gray) among the genes upregulated (left) and downregulated (right) in both vim1 vim2 vim3 and met1 mutants. Transposable elements are highly represented in the upregulated set, while most of the downregulated genes are protein-coding. Numbers of targets in each category are indicated on the pie charts, with total numbers of targets in the upregulated and downregulated sets indicated in parentheses.
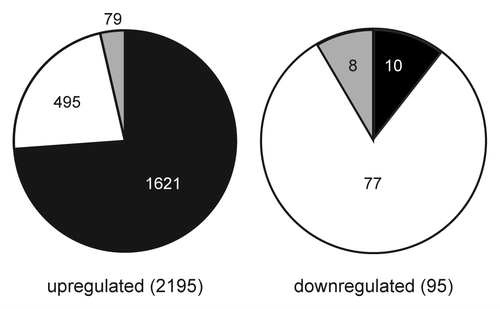
In each mutant, a smaller set of transcripts was downregulated relative to wild type, with protein-coding genes highly represented in this category (). Although protein-coding genes are enriched in the sets of downregulated genes, it is notable that the numbers of upregulated protein-coding genes is still at least double the number of downregulated protein-coding genes in each mutant. This bias may reflect that there are multiple possible modes of misregulation for protein-coding genes in the mutants. It is well established that protein-coding genes can be transcriptionally repressed when adjacent to a methylated transposable element.Citation26-Citation28 One well-known target of this regulatory mechanism is the FWA locus, which has a promoter region comprised of repeats derived from a SINE retrotransposon.Citation26 Protein-coding genes might also be up- or downregulated in vim1 vim2 vim3 and met1 mutants due to secondary regulatory effects from de-repressed transcription factors.
Discussion
The functional interface between VIM proteins and MET1 is a subject of ongoing research. Until now, it has been unclear whether VIM proteins have transcriptional regulatory functions outside their roles in the maintenance of CG methylation. Here, we show that VIM proteins and MET1 regulate very similar sets of transcriptional targets, strongly indicating that the influence of VIM proteins on gene expression is exclusively via their function as MET1 cofactors. This conclusion expands upon previous studies that showed highly similar DNA methylation profiles between vim1 vim2 vim3 mutants and met1 mutants.Citation23,Citation29
Beyond showing that the targets regulated by VIM proteins and MET1 are essentially the same, we demonstrated a high correlation between gene expression levels in vim1 vim2 vim3 and met1 mutants. This further reflects the mechanistic interplay between VIM proteins and MET1 in regulating CG methylation. Although the details of this process are unconfirmed in Arabidopsis, research on animal homologs suggests that VIM/UHRF proteins either recruit MET1 to target loci through direct interactions,Citation21 or by depositing an ubiquitylated histone mark that serves as a binding substrate for MET1.Citation22 Our results showing high correlation between the vim1 vim2 vim3 and met1 expression profiles are consistent with either model. Further research is necessary to elucidate how much similarity exists between the plant and animal systems for maintaining CG methylation.
Our previous research indicated that VIM proteins have a role in the regulation of chromatin structure aside from maintenance of DNA methylation. A comparison of centromeric structure between vim1 mutants and wild type plants showed that centromere repeats were relatively decondensed in the vim1 mutant. Additionally, the centromeric histone H3 variant HTR12 was mislocalized relative to the centromeric repeats in vim1.Citation14 This subnuclear phenotype contrasted with that of a ddm1 mutant also deficient in centromeric CG methylation, which more closely resembled the wild type centromere organization. This observation indicated that VIM1, in particular, might have a role in the maintenance of chromatin structure unconnected from its function in DNA methylation maintenance. Although the results of our transcriptional profiling analysis do not exclude this possibility, it does suggest that any additional role VIM1 might have in chromatin modulation does not impact gene expression. We propose that VIM1 has a centromere-specific function in the maintenance of chromatin structure that does not affect gene or transposon mRNA expression. VIM1 may interact with centromere-specific epigenetic marks or proteins to produce cytosine methylation-independent chromatin modifications at centromeres.
Our comparison of gene expression between vim1 mutant and wild type plants showed that no transcripts were significantly differentially expressed in the mutant. This result is consistent with previous studies that show high functional redundancy between VIM1, VIM2, and VIM3 in vegetative tissues.Citation17,Citation23 However, we demonstrated that the vim1 mutation was sufficient to produce a hypomethylation phenotype at centromeric repeats, while vim2 and vim3 mutants did not have a significant loss of methylation at centromeres.Citation14,Citation17 It is surprising that vim1 centromeres are hypomethylated, yet no effect on mRNA transcription was detected in vim1 plants. It is possible that transcriptional changes are occurring within the vim1 centromere, as other epigenetic silencing mutants derepress transcription of 180-bp centromere repeats,Citation30 but our enrichment for poly-adenylated transcripts does not allow us to effectively assess the expression of these repeats. Further research designed to detect such transcripts could be undertaken in vim1 samples to determine whether VIM1 is required to silence centromere repeat transcription.
Materials and Methods
Plant materials
The met1-3 and vim1-2 vim2-2 vim3-1 mutants used in this study have been previously described.Citation17,Citation31 Since met1-3 homozygous mutants are sterile, we obtained met1-3 homozygotes from the segregating first-generation progeny of a met1–3 heterozygote provided by Jerzy Paszkowski (University of Geneva). The homozygotes were confirmed through genotyping PCR. Plants were grown under a 16 h photoperiod at 22 °C in controlled growth chambers.
RNA-seq library preparation
Floral bud tissue was collected from wild type, vim1 vim2 vim3, and met1 plants in three independent biological replicates, and total RNA was isolated using a Qiagen RNEasy Plant Miniprep Kit. Illumina RNA-seq libraries were prepared according to a strand-specific protocol.Citation24 Briefly, poly-A mRNA was selected using oligo dT Dynabeads (Life Technologies 61002) and fragmented in SuperScript II (Life Technologies 18064-014) First Strand Synthesis Buffer at a final concentration of 5 mM MgCl2 for 10 min at 95 °C. Double-stranded cDNA was then synthesized from the mRNA, with the second strand labeled with dUTP. After end repair and adenylation, adapters were ligated to the cDNA, and libraries were size selected using a double-sided SPRI method with Ampure XP beads (Beckman Coulter A63880). The libraries were PCR amplified in 15 cycles using barcoded primers. Samples were multiplexed and sequenced on an Illumina Hi-Seq 2000 machine by the Genomics Resources Core Facility at Weill Medical College. Raw read data has been deposited in the NCBI Sequence Read Archive (SRX471361).
Sequence read alignment and differential expression analysis
Reads were aligned to the TAIR10 assembly of the Arabidopsis thaliana genome using TopHat version 2.0.4.Citation32 Since transposons are known targets of regulation by CG methylation, we included the TAIR10 transposable element annotation based on Quesneville et al.Citation33 in our GTF reference file. Since there is some redundancy between this annotation and the AGI-annotated transcriptome, we filtered out all AGI-annotated transcripts that overlapped with the transposable element annotation. The output bam files were sorted with SAMtools,Citation34 and differential expression analysis was performed using the R package DESeq.Citation25 To define differentially expressed genes, an adjusted P value cutoff of 0.05 and FDR threshold of 0.05 were used. No fold-change cutoff for expression was applied.
Reverse transcription quantitative PCR
Total RNA was extracted from inflorescence buds and used as a template for cDNA synthesis with SuperScript III reverse transcriptase (Life Technologies 18080-051). Quantitative PCR analysis was performed on a Bio-Rad iCycler machine with a home-made SYBR Green master mix. Actin2 (At3g18780) was used as a constitutively expressed transcript for normalization. The reported results are averages of at least three independent biological replicates.
Additional material
Download Zip (429.7 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge Natalie Henkhaus, Erika Hughes, and Haiyi Wang for contributing helpful insights on the results. We thank Jerzy Paszkowski (University of Geneva) for providing the met1-3 line. This research was funded by the National Institutes of Health (GM078256 to E.J.R.), the Department of Defense (BC093067 to M.S.S.), and institutional funds from the Boyce Thompson Institute.
References
- Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 1982; 10:2709 - 21; http://dx.doi.org/10.1093/nar/10.8.2709; PMID: 7079182
- Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 2005; 33:5868 - 77; http://dx.doi.org/10.1093/nar/gki901; PMID: 16224102
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008; 452:215 - 9; http://dx.doi.org/10.1038/nature06745; PMID: 18278030
- Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008; 133:523 - 36; http://dx.doi.org/10.1016/j.cell.2008.03.029; PMID: 18423832
- Kato M, Miura A, Bender J, Jacobsen SE, Kakutani T. Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr Biol 2003; 13:421 - 6; http://dx.doi.org/10.1016/S0960-9822(03)00106-4; PMID: 12620192
- Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 2001; 411:212 - 4; http://dx.doi.org/10.1038/35075612; PMID: 11346800
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW-L, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 2006; 126:1189 - 201; http://dx.doi.org/10.1016/j.cell.2006.08.003; PMID: 16949657
- Vaughn MW, Tanurdzić M, Lippman Z, Jiang H, Carrasquillo R, Rabinowicz PD, Dedhia N, McCombie WR, Agier N, Bulski A, et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol 2007; 5:e174; http://dx.doi.org/10.1371/journal.pbio.0050174; PMID: 17579518
- Cao X, Jacobsen SE. Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol 2002; 12:1138 - 44; http://dx.doi.org/10.1016/S0960-9822(02)00925-9; PMID: 12121623
- Finnegan EJ, Peacock WJ, Dennis ES. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc Natl Acad Sci U S A 1996; 93:8449 - 54; http://dx.doi.org/10.1073/pnas.93.16.8449; PMID: 8710891
- Lindroth AM, Cao X, Jackson JP, Zilberman D, McCallum CM, Henikoff S, Jacobsen SE. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001; 292:2077 - 80; http://dx.doi.org/10.1126/science.1059745; PMID: 11349138
- Soppe WJJ, Jasencakova Z, Houben A, Kakutani T, Meister A, Huang MS, Jacobsen SE, Schubert I, Fransz PF. DNA methylation controls histone H3 lysine 9 methylation and heterochromatin assembly in Arabidopsis. EMBO J 2002; 21:6549 - 59; http://dx.doi.org/10.1093/emboj/cdf657; PMID: 12456661
- Chen T, Li E. Structure and function of eukaryotic DNA methyltransferases. Curr Top Dev Biol 2004; 60:55 - 89; http://dx.doi.org/10.1016/S0070-2153(04)60003-2; PMID: 15094296
- Woo HR, Pontes O, Pikaard CS, Richards EJ. VIM1, a methylcytosine-binding protein required for centromeric heterochromatinization. Genes Dev 2007; 21:267 - 77; http://dx.doi.org/10.1101/gad.1512007; PMID: 17242155
- Kraft E, Bostick M, Jacobsen SE, Callis J. ORTH/VIM proteins that regulate DNA methylation are functional ubiquitin E3 ligases. Plant J 2008; 56:704 - 15; http://dx.doi.org/10.1111/j.1365-313X.2008.03631.x; PMID: 18643997
- Liu S, Yu Y, Ruan Y, Meyer D, Wolff M, Xu L, Wang N, Steinmetz A, Shen W-H. Plant SET- and RING-associated domain proteins in heterochromatinization. Plant J 2007; 52:914 - 26; http://dx.doi.org/10.1111/j.1365-313X.2007.03286.x; PMID: 17892444
- Woo HR, Dittmer TA, Richards EJ. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet 2008; 4:e1000156; http://dx.doi.org/10.1371/journal.pgen.1000156; PMID: 18704160
- Hsieh T-F, Shin J, Uzawa R, Silva P, Cohen S, Bauer MJ, Hashimoto M, Kirkbride RC, Harada JJ, Zilberman D, et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc Natl Acad Sci U S A 2011; 108:1755 - 62; http://dx.doi.org/10.1073/pnas.1019273108; PMID: 21257907
- Yao Q, Song C-X, He C, Kumaran D, Dunn JJ. Heterologous expression and purification of Arabidopsis thaliana VIM1 protein: in vitro evidence for its inability to recognize hydroxymethylcytosine, a rare base in Arabidopsis DNA. Protein Expr Purif 2012; 83:104 - 11; http://dx.doi.org/10.1016/j.pep.2012.03.003; PMID: 22459921
- Bostick M, Kim JK, Estève P-O, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007; 317:1760 - 4; http://dx.doi.org/10.1126/science.1147939; PMID: 17673620
- Achour M, Jacq X, Rondé P, Alhosin M, Charlot C, Chataigneau T, Jeanblanc M, Macaluso M, Giordano A, Hughes AD, et al. The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene 2008; 27:2187 - 97; http://dx.doi.org/10.1038/sj.onc.1210855; PMID: 17934516
- Nishiyama A, Yamaguchi L, Sharif J, Johmura Y, Kawamura T, Nakanishi K, Shimamura S, Arita K, Kodama T, Ishikawa F, et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 2013; 502:249 - 53; http://dx.doi.org/10.1038/nature12488; PMID: 24013172
- Stroud H, Greenberg MVC, Feng S, Bernatavichute YV, Jacobsen SE. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013; 152:352 - 64; http://dx.doi.org/10.1016/j.cell.2012.10.054; PMID: 23313553
- Wang L, Si Y, Dedow LK, Shao Y, Liu P, Brutnell TP. A low-cost library construction protocol and data analysis pipeline for Illumina-based strand-specific multiplex RNA-seq. PLoS One 2011; 6:e26426; http://dx.doi.org/10.1371/journal.pone.0026426; PMID: 22039485
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106; http://dx.doi.org/10.1186/gb-2010-11-10-r106; PMID: 20979621
- Lippman Z, Gendrel A-V, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004; 430:471 - 6; http://dx.doi.org/10.1038/nature02651; PMID: 15269773
- Whitelaw E, Martin DIK. Retrotransposons as epigenetic mediators of phenotypic variation in mammals. Nat Genet 2001; 27:361 - 5; http://dx.doi.org/10.1038/86850; PMID: 11279513
- Saze H, Kakutani T. Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. EMBO J 2007; 26:3641 - 52; http://dx.doi.org/10.1038/sj.emboj.7601788; PMID: 17627280
- Feng S, Cokus SJ, Zhang X, Chen P-Y, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A 2010; 107:8689 - 94; http://dx.doi.org/10.1073/pnas.1002720107; PMID: 20395551
- May BP, Lippman ZB, Fang Y, Spector DL, Martienssen RA. Differential regulation of strand-specific transcripts from Arabidopsis centromeric satellite repeats. PLoS Genet 2005; 1:e79; http://dx.doi.org/10.1371/journal.pgen.0010079; PMID: 16389298
- Saze H, Mittelsten Scheid O, Paszkowski J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 2003; 34:65 - 9; http://dx.doi.org/10.1038/ng1138; PMID: 12669067
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009; 25:1105 - 11; http://dx.doi.org/10.1093/bioinformatics/btp120; PMID: 19289445
- Quesneville H, Bergman CM, Andrieu O, Autard D, Nouaud D, Ashburner M, Anxolabehere D. Combined evidence annotation of transposable elements in genome sequences. PLoS Comput Biol 2005; 1:166 - 75; http://dx.doi.org/10.1371/journal.pcbi.0010022; PMID: 16110336
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078 - 9; http://dx.doi.org/10.1093/bioinformatics/btp352; PMID: 19505943