
Abstract
Though the mechanisms governing nuclear organization are not well understood, it is apparent that epigenetic modifications coordinately modulate chromatin organization as well as transcription. In maize, MEDIATOR OF PARAMUTATION1 (MOP1) is required for 24 nt siRNA-mediated epigenetic regulation and transcriptional gene silencing via a putative Pol IV- RdDM pathway. To elucidate the mechanisms of nuclear chromatin organization, we investigated the relationship between chromatin structure and transcription in response to loss of MOP1 function. We used a microarray based micrococcal nuclease sensitivity assay to identify genome-wide changes in chromatin structure in mop1-1 immature ears and observed an increase in chromatin accessibility at chromosome arms associated with loss of MOP1 function. Within the many genes misregulated in mop1 mutants, we identified one subset likely to be direct targets of epigenetic transcriptional silencing via Pol-IV RdDM. We found that target specificity for MOP1-mediated RdDM activity is governed by multiple signals that include accumulation of 24 nt siRNAs and the presence of specific classes of gene-proximal transposons, but neither of these attributes alone is sufficient to predict transcriptional misregulation in mop1-1 homozygous mutants. Our results suggest a role for MOP1 in regulation of higher-order chromatin organization where loss of MOP1 activity at a subset of loci triggers a broader cascade of transcriptional consequences and genome-wide changes in chromatin structure.
Introduction
Transcriptional regulation of gene expression is important for plant development and genome defense against transposable elements and exogenous DNA, such as viruses and transgenes. Since their discovery in the late 1990s (reviewed in ref. Citation1), small RNAs (sRNAs) have been demonstrated to play a critical role in transcriptional gene silencing (TGS) pathways including the RNA-directed DNA methylation (RdDM) pathway.
In plants, methylation of cytosines is observed at symmetric (CG and CHG) and asymmetric (CHH) sequence contexts and requires the activity of specific DNA methyltransferases. While symmetric methylation is maintained during DNA replication, CHH methylation is established de novo by CHROMOMETHYLASE3 (CMT3) and DOMAINS REARRANGED METHYLTRANSFERASE1 (DRM1) and DRM2 in association with RdDM, and is often considered an indication of RdDM activity (reviewed in ref. Citation2).
Relative to total cytosines, low levels of genome-wide asymmetric methyl-cytosines are observed in plants. In Arabidopsis, 1.5% of cytosines are CHH methyl-cytosines (compared with 22.3% CG and 5.9% CHG), whereas 2.2% of cytosines in rice are CHH methyl-cytosines (compared with 59.4% CG and 20.7% CHG methylation).Citation3 Although higher levels of overall methylation have been reported in maize, the levels of asymmetric methylation are significantly lower than symmetric methylation (approximately 5% CHH compared with 86% CG and 74% CHG methylation).Citation4 While the genome-wide levels are relatively low, in maize CHH methylation is enriched at intergenic loci of euchromatic chromosomes arms. Certain transposon families preferentially insert within 1 kb of maize genes and are hypothesized to drive enrichment of CHH. Near-gene CHH methylation is guided by 24 nt siRNAs and correlates with TE insertion events.Citation4,Citation5
Recent evidence in Arabidopsis thaliana suggests the existence of more than one siRNA-dependent DNA methylation TGS pathway in plants that function to establish/maintain silent chromatin states. One such pathway requires a plant-specific DNA-dependent RNA polymerase, RNA polymerase IV (Pol IV), for siRNA biogenesis and is therefore termed Pol IV-RdDM.Citation6 Pol IV transcripts are used as templates by the Pol IV-associated RNA DEPENDENT RNA POLYMERASE2 (RDR2), and processed into double-stranded RNA (dsRNA). A Dicer-like protein, DICER-LIKE3 (DCL3) cleaves RDR2-generated dsRNA into 24 nt siRNAs that are incorporated into an ARGONAUTE4 (AGO4) or AGO6 complex. This AGO4/6-siRNA complex directs transcripts produced by Pol V, an additional plant-specific DNA-dependent RNA polymerase, to target loci, ultimately acting as a guide for recruitment of chromatin remodelers and DNA methyltransferases (reviewed in ref. Citation7).
The Pol IV-RdDM pathway was initially described in Arabidopsis thaliana and several maize orthologs have been identified through genetic screens using loss of epigenetic silencing as phenotypes. The b1 and pl1 paramutation systems were used to identify the mediator of paramutation (mop) and required to maintain repression (rmr) genes, respectivelyCitation8,Citation9 and several MOP and RMR proteins are required for both paramutation and TGS (reviewed in ref. Citation10). rmr6 appears to encode the largest subunit of Pol IVCitation11 and rmr1 encodes a Snf2-like ATPase similar to DEFECTIVE IN RNA-DIRECTED DNA METHYLATION1 (DRD1) and CLASSY1 (CSLY1),Citation12 which are two Arabidopsis proteins implicated in RdDM.Citation13,Citation14 mop1 encodes a protein that is orthologous to RDR2,Citation15 whereas mop2/rmr7Citation16,Citation17 are most similar to Arabidopsis NRPD2/E2, which encodes the second largest subunit of both Pol IV and Pol V.
A separate forward genetics screen identified the transgene reactivated (tgr) mutants based upon transcriptional reactivation of the silent B1 genomic transgene (BTG-s),Citation18 which is also reactivated in mop1-1, rmr1-1, rmr2-1,Citation19 and mop3-1 (McGinnis and Sloan, unpublished data). Reactivation of BTG-s in mop1, rmr1, rmr2, tgr1, and tgr2 mutants is associated with hypomethylation of the 35SCaMV promoter sequence,Citation18,Citation19 and indicates that at specific regulatory loci, TGR1 and TGR2 likely function in the same epigenetic DNA methylation and transcriptional gene silencing pathway as RMR1, RMR2 and MOP1.
Consistent with its putative function in a Pol IV-RdDM pathway, loss of MOP1 results in a dramatic reduction of 24 nt siRNAs.Citation20,Citation21 mop1-1 mutants also display pleiotropic developmental defects and phenotypes, which may result from direct and indirect misregulation of genes and siRNAs.Citation8,Citation20,Citation22-Citation24 These developmental phenotypes are often variably penetrant and expressive, and seem to be influenced by environmental factors,Citation8,Citation23 suggesting an increased potential for variable gene expression in this mutant.
Consistently, many genes and transposable elements were differentially expressed in the shoot apical meristems (SAMs) of mop1-1 homozygous plants.Citation24 Together, these findings provide evidence that MOP1 not only plays a crucial role in progression of the Pol IV-RdDM pathway but also in maintenance of genome-wide stable epigenetic states that influence gene expression. At least one MOP1-regulated locus also exhibits changes in chromatin structure and nucleosome positioning in association with changes in transcriptional level,Citation25,Citation26 suggesting that RdDM may also influence chromatin structure.
Eukaryotic chromosomes are structurally and functionally organized in the form of chromatin. Nucleosomes, the basic unit of eukaryotic chromatin, are comprised of approximately 147 bp of DNA wrapped 1.65 times around a histone protein octamer (reviewed in ref. Citation27). In the nucleus, chromatin undergoes several levels of folding, compaction, and organization. The distribution of nucleosomes along DNA, as “beads-on-a-string,” forms the primary structure of chromatin. Nucleosomes interact with each other and give rise to secondary structures, which has been modeled as a 30 nm chromatin fiber. These secondary structures are further condensed to form tertiary (or higher-order) structures of highly condensed chromatin, as seen during the metaphase of mitotic nuclei (reviewed in refs. Citation27 and Citation28). The different levels of structural chromatin organization allow for packaging of a significant amount of charged molecules into the confined space of the nucleus while creating a structure that is permissive for essential cellular functions such as replication and transcription.
The mechanisms of higher order chromatin organization and compaction into the nucleus are not well characterized, although several models of this folding have been proposed (reviewed in ref. Citation27). Epigenetic modifications play a role in chromatin organization. For example, histone acetylation and tri-methylation of H3K4 are often associated with euchromatin, whereas DNA methylation and H3K9 di-methylation are associated with heterochromatin (reviewed in ref. Citation29). These modifications to DNA and histones are distributed non-randomly along chromosomes to modulate chromatin compaction and regulate transcription by allowing or inhibiting access of DNA to regulatory proteins.
Given the role of epigenetic modifications on higher order chromatin structure and accessibility of DNA to regulatory proteins, it is plausible that genetic mutants of epigenetic factors, such as mop1-1, could also exhibit defects in local and global chromatin organization as an element of a loss-of-function phenotype. Accordingly, mop1-1 homozygous plants were assayed for local nucleosome occupancy at transcription start sites (TSSs) of approximately 400 genes in maize, resulting in the identification of small number of distinct examples where nucleosome occupancy in mutants differed from WT.Citation30 This suggests that at some loci, MOP1-mediated pathways influence primary chromatin structure.
To date, little is known about how epigenetic mechanisms regulate higher-order chromatin structure. Because chromatin structure is modeled to be hierarchical, disruptions in primary structure may influence changes in higher order chromatin structure. Thus, it is possible that RNA-dependent regulatory mechanisms might also regulate chromatin organization. Herein, we investigated the relationship between higher-order chromatin structure and transcription in response to loss of the epigenetic regulator MOP1. We observed that, on a genome-wide scale, loss of MOP1 resulted in an increase in chromatin accessibility/sensitivity at gene-rich chromosome arms; whereas pericentromeric regions were less accessible/more resistant. We assayed expression, and observed both up and downregulated genes, physically distributed across the genome, indicating more extensive changes in chromatin organization and transcription than would be explained by specific MOP1 epigenetic silencing in localized regions. We identified a subset of differentially expressed genes (DEGs) likely to be direct targets of epigenetic transcriptional silencing. Our results suggest a possible role for MOP1-mediated pathways in regulation of higher-order chromatin organization that results in direct and indirect consequences on gene expression in maize.
Results and Discussion
The mop1-1 mutation results in global changes in nuclease sensitivity
To determine a role for MOP1 on genome-wide chromatin structure, we used a custom maize NimbleGen microarray based platform to assay higher-order chromatin structure similar to that described in humans.Citation31,Citation32 Chromatin structure was determined as a measure of sensitivity of formaldehyde-fixed nuclei to enzymatic cleavage by micrococcal nuclease (MNase). MNase cleaves naked/linker DNA (sensitive DNA) more readily than DNA bound in nucleosomes (resistant DNA) (reviewed in ref. Citation33), and can be used as a probe to assay chromatin structure (reviewed in ref. Citation34). Nuclei isolated from mop1-1 homozygous mutants and WT immature ears were digested with limiting amounts of MNase and separated by agarose gel-electrophoresis into MNase-resistant and MNase-sensitive. MNase-resistant and MNase-sensitive DNA fractions were differentially labeled and hybridized to a custom NimbleGen microarray containing ~150 000 unique, tiled and isothermal oligonucleotide probes that span the maize genome at ~14 kb spacing. We measured the signal ratio of MNase-resistant DNA/MNase-sensitive DNA for WT and mutant samples at each probe, and then calculated the deviation of the mutant response from WT ().
Figure 1. Chromatin accessibility (nuclease sensitivity) in mop1-1 and WT immature ears on three representative chromosomes. We measured the signal ratio (log2 [HMW MNase-resistant DNA/MNase-sensitive DNA]) for WT and mutant samples at each probe, then calculated the deviation of the mutant response from WT (Mutant – WT). This deviation was plotted on a genome-browser (genomaize.org) for chromosome 1 (A), chromosome 3 (B) and chromosome 9 (C). WT and mutant are the same at zero. Each plot represents the average of 3 biological replicates with smoothing at 5 pixels. The gene density plot (maizesequence.org) and relative position of the centromere (Jiang/Presting) are also illustrated for each chromosome. All ten chromosomes are included in Figures S1 and S2.
![Figure 1. Chromatin accessibility (nuclease sensitivity) in mop1-1 and WT immature ears on three representative chromosomes. We measured the signal ratio (log2 [HMW MNase-resistant DNA/MNase-sensitive DNA]) for WT and mutant samples at each probe, then calculated the deviation of the mutant response from WT (Mutant – WT). This deviation was plotted on a genome-browser (genomaize.org) for chromosome 1 (A), chromosome 3 (B) and chromosome 9 (C). WT and mutant are the same at zero. Each plot represents the average of 3 biological replicates with smoothing at 5 pixels. The gene density plot (maizesequence.org) and relative position of the centromere (Jiang/Presting) are also illustrated for each chromosome. All ten chromosomes are included in Figures S1 and S2.](/cms/asset/26175e90-98cb-40ad-98e2-8b87c93f9163/kepi_a_10929022_f0001.gif)
A consistent trend in chromatin response as a result of loss of MOP1 function was observed at all ten chromosomes of maize (; Figs. S1 and S2). The assay provides a low resolution illustration of chromatin structure in which chromatin was more accessible to cleavage by MNase at chromosome arms, whereas pericentromeric regions of chromosomes were more resistant ().
This pattern coincided with gene density distribution (maizesequence.org) across maize chromosomes (). In maize, 24 nt siRNAs are also typically enriched at chromosome arms and correspond with CHH methylation in class I TEs at a high frequency.Citation5,Citation35 Relative to other transposon families, most families of DNA elements (except the CATCA superfamily) are found more frequently at chromosome arms within and/or adjacent to genes.Citation36 It is well documented that the transcriptional silencing and DNA methylation of most Mutator (Mu) elementsCitation37,Citation38 and transcriptional silencing of most differentially expressed DNA TEs (with the exception of the CATCA superfamily) in SAMsCitation24 are reversed in mop1-1 homozygous plants.
Together, this provides evidence that MOP1-mediated activity occurs predominantly at chromosome arms, and less frequently at pericentromeres in a pattern that overlaps with specific types of TEs (e.g., Mu TEs) or their proximity to genes. An increase in chromatin accessibility at chromosome arms as a result of loss of MOP1 is consistent with loss of chromatin repression, conceivably resulting in loss of transcriptional repression at affected loci.
Misregulation of gene expression in mop1–1 immature ears
MOP1 is highly expressed in maize immature earsCitation21,Citation39 and developmental and morphological defects are observed in mop1-1 mutants in this tissue, which include late flowering, and ears that are smaller in size and/or narrower in diameter compared with WT.Citation8,Citation40 The developmental phenotypes and changes in chromatin organization we observed in mop1-1 plants could be related through changes in gene expression. While gene expression profiles for mop1-1 SAMs are available, to date, no published genome-wide expression profile exists for mop1-1 immature ears. To identify immature ear-specific DEGs in mop1–1 mutants, we used a NimbleGen expression array platform, as described by Sekhon et al.,Citation41 hybridized with cDNA prepared from the same samples used to assay global chromatin sensitivity to MNase. Because a diagnostic phenotype of mop1-1 homozygous individuals is caused by the release of epigenetic silencing of the b1 gene,Citation8 we anticipated that one hallmark of MOP1 activity would be upregulation of expression in mop1-1 ears compared with wild type.
We defined differential expression as genes with a 2-fold or greater change in expression in mop1 mutants relative to WT. Using this criterion, we identified ~762 DEGs in mop1-1 mutants in maize immature ears (two biological replicates; 2-fold change cut-off (FC ≥ 2); P < 0.05, one-tail t test). We compared our data with the published data set for expression in mop1-1 SAMs and observed that some genes were differentially expressed in both tissues in mutants compared with wild type (Table S3).
Similar to the variations observed in MNase accessibility, this differential effect in expression in immature ears spanned both arms of all chromosomes. Of these, 349 (~46% of DEGs) genes were upregupregulated in mop1-1 and 413 (~54% of DEGs) genes were downregulated in mop1-1 (Tables S1 and S2, respectively). Upregulation of expression in mop1-1 is consistent with reduced RdDM-mediated transcriptional repression. Downregulation of some loci has been previously reported for mop1-1 homozygous individuals, where approximately two-thirds of DEGs were downregulated in mop1 SAMs,Citation24 although downregulation is not an anticipated direct consequence of the loss of epigenetic silencing.
Upregulation of transposable elements apparent in mop1-1 ear shoots
Approximately 23% (79/349) of the upregulated loci are highly similar to sequences annotated as transposons in Arabidopsis and/or rice (Table S1). This was much fewer than findings by Jia et al. (2009), where ~175 transposon families were differentially expressed in mop1-1 SAMs. It is likely that our technique might be limited in this regard. Repetitive, conserved sequences, such as DNA and retro-transposons, often do not meet NimbleGen microarray probe design criteria, therefore our experiments had significantly fewer of these types of elements represented. Although the majority of these TEs identified herein were class II elements (DNA transposons), some class I TEs (retrotransposons) were also upregulated in plants deficient in MOP1 activity. MOP1 regulation of Mutator, an abundant group of class II elements, has been reported previously.Citation37,Citation38
Many genes with apparent regulatory function are upregulated in mop1-1
In addition to TEs, many genes predicted to be protein coding were differentially expressed in mop1-1 homozygous plants. Of the 117 upregulated genes with an identifiable Biological Process Gene Ontology term, 19 are predicted to encode proteins involved in regulation of transcription (). The loss of silencing observed at numerous transcription factors would be expected to have secondary consequences in mop1-1 homozygous individuals. We found that several predicted MADS-box transcription factors and related proteins were misexpressed in mop1-1 (). MADS-box genes regulate reproductive organ identity during floral development and function through interactions with chromatin-associated proteins and other transcriptional regulatorsCitation42, reviewed by.Citation43 The upregulated genes included those orthologous to the Arabidopsis SUPERMAN (SUP), SEEDSTICK (STK), AGAMOUS-LIKE 8 (AGL8), and AGL26 proteins. Additionally, expression of a maize gene similar to GIANT KILLER (GIK), a direct target of AGAMOUS (Ng KH, Yu et al. 2009), was also upregulated in mop1–1 compared with wild type. GIK is an AT-hook DNA-binding chromatin modifier and regulates patterning and differentiation of reproductive organs in Arabidopsis.Citation42 Three genes with orthology to ETTIN (ETT)/AUXIN RESPONSE FACTOR3 (ARF3), a predicted target of GIK, were also misexpressed in maize mop1-1 mutants (). In Arabidopsis, GIK-mediated repression of ETT is associated with chromatin changes (H3K9me2).Citation42 Upregulation of maize genes with similarity to both GIK and its target ETT in mop1-1 homozygous plants supports a model where loss of MOP1 triggers primary and secondary changes in gene expression.
Figure 2. Biological process of genes upregulated in mop1-1. Gene ontology was predicted and scored for all mop1-1 differentially expressed genes using BLAST2GO (Conesa, Gotz et al. 2005). The most highly scored categories are listed on the y axis, the bars represent the score for each category.
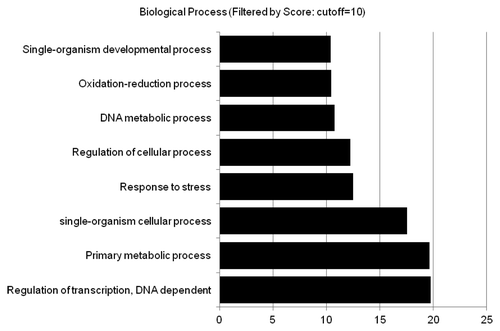
Table 1. Floral Homeotic and Chromatin-associated Genes Upregulated in mop1–1.
As previously reported,Citation24 several other chromatin associated genes were upregulated in mop1-1 (). The chromatin remodeling40 (chr40) gene encodes a SNF2 type; DRD1 class protein (www.chromdb.org) similar to REQUIRED TO MAINTAIN REPRESSION1 (RMR1), and was upregulated in mop1-1. Expression of a gene related to chromatin-associated factor REGULATOR OF CHROMOSOME CONDENSATION1 (RCC1) was also upregulated in mop1 mutants. Additionally, we found that expression of a gene highly similar to SUVR4, a SET-domain histone lysine methyltransferase, also increased in mop1-1. Proteins encoded by these genes may, in theory, function to maintain a repressed chromatin structure at specific loci in the absence of MOP1-mediated pathways, inducing downregulation at some loci.
Genes downregulated in mop1-1 may not be direct targets of MOP1-mediated regulation
In mop1 immature ears we identified 3 of the 413 (<1%) downregulated loci to encode TEs. As mentioned previously, our platform is limited in its ability to assay repetitive sequences, and so we were unable to conclude if immature ears exhibit a similar pattern of downregulation of retrotransposons as observed in shoot apical meristems.Citation24 Most downregulated loci observed in our analysis were predicted to be protein coding.
One of the genes downregulated in mop1-1 immature ears is homologous to REPRESSOR OF SILENCING 1 (ROS1)/DEMETER-LIKE1 (DML1) (FC = 2.0485; P = 0.0001; Table S2), an Arabidopsis DNA glycosylase protein that functions in active demethylation.Citation44-Citation46 ROS1 is downregulated in several Arabidopsis RdDM mutants which include pol IVa, IVb, rdr2, and drd1.Citation47,Citation48 Hypermethylation and reduced expression can occur when ROS1 function is lost in ros1 mutants.Citation49 It is possible that reduced ROS1 function leads to the downregulation of a subset of genes in mop1-1 homozygous individuals. Consistent with this, a recent report suggests that RdDM based silencing of some loci might be enhanced in plants deficient for ROS1 activity.Citation50
Another possible explanation for downregulated genes in mop1-1 homozygous individuals would be the expanded activity of an alternative silencing pathway in plants. A small RNA-dependent silencing pathway that operates independently of RDR2 activity was recently described in Arabidopsis, and is mediated by 21–22 nt siRNAs.Citation6,Citation51-Citation54 In mop1-1 homozygous individuals, while 24 nt siRNAs are substantially reduced, 22 nt siRNAs are abundant.Citation20 In the absence of the 24 nt siRNA regulatory pathway, perhaps the persisting 22 nt siRNAs downregulate a subset of genes. However, we were unable to detect an immediate correlation between an increase in homologous 22 nt siRNAs and decreased transcript abundance in mop1-1 individuals (data not shown).
Identification of putative targets of epigenetic transcriptional gene silencing
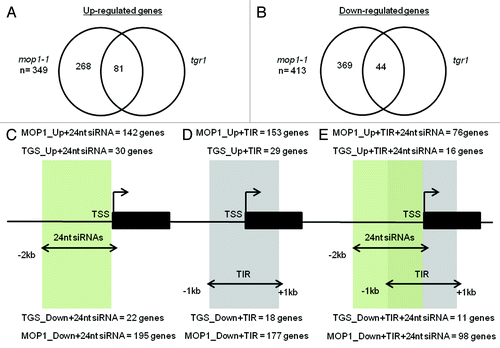
Based upon our observations, we reasoned that loss of MOP1 activity has both primary and secondary consequences, meaning that mop1-1 DEGs would include direct targets of an epigenetic transcriptional gene silencing (TGS) pathway and secondary consequences resulting from genome-wide changes triggered indirectly by the loss of MOP1. To identify genes that are likely direct targets of epigenetic TGS, we used the transgene reactivated1 (tgr1) mutant which releases silencing of some of the same loci as mop1-1.Citation18,Citation19 We identified a subset of the 349 genes upregulated in mop1-1 that were also upregulated through loss of TGR1 activity (Table S4) (log2FC ≥ 0.8) using next-generation sequencing of transcripts (RNA-seq) from immature ears of tgr1 plants. Similarly, to identify genes repressed due to loss of epigenetic TGS activity, of the 413 genes downregulated in mop1-1, we identified genes also repressed in tgr1-1 homozygous mutants (Table S5). We found 81 genes that were upregulated (; Table S4) and 44 genes that were downregulated (; Table S5) in both genotypes, and considered these to be genes most likely directly regulated by a MOP1-TGR1 epigenetic TGS pathway.
Figure 3. Identification of unifying features in mop1-1 DEG subsets. A subset of genes that were upregulated in mop1-1 were also misregulated in tgr1-1 (A). A different subset of genes were downregulated in both mop1-1 and tgr1 (B). We further categorized each subset to identify those with 24 nt siRNAs within 2 kb upstream of the transcription start site (TSS) (C), a class II transposable element within 1 kb up or downstream of the TSS (D), or both 24 nt siRNAs within 2 kb upstream of the TSS and a class II transposable element within 1 kb up or downstream of the TSS (E).
Upregulation of TGS targets associated with near-gene transposons and 24 nt siRNAs
Analysis of promoter-proximal 24 nt siRNAs at DEGs
In maize, the majority of 24 nt siRNAs are depleted in mop1-1 homozygous mutants,Citation20 indicating that they are produced in a MOP1-dependent manner. Based on the current model for RdDM, 24 nt siRNAs guide de novo methylation to target loci (reviewed in ref. Citation7). At maize genes, 24 nt siRNAs peak within 1 kb of transcription start sitesCitation4 and are depleted in mop1-1 mutants (Fig. S3).We predicted that the regulatory regions, including the promoter-proximal regions, of genes directly regulated by MOP1 activity might be expected to share homology with 24 nt siRNAs produced in a MOP1-dependent manner.
Using publically available mop1 siRNA data sets generated in the same tissue as our data,Citation20 we identified mop1 DEGs with a minimum of 5 mapped 23–24 nt siRNAs detected within 2kb upstream of transcription start sites (TSSs) in either wild type or mutant genotypes; then measured the siRNA fold change between WT and mutant. 142/349 mop1-1 upregulated genes and 195/413 downregulated genes had a ≥2-fold change in 23–24 nt siRNAs within –2kb of the TSS ( and ; Tables S6 and S7). 30/81 upregulated and 22/44 genes repressed in mop1-1 and tgr1 had a > 2-fold change in 24 nt siRNAs within –2kb of the TSS ( and ; Tables S8 and S9). At genes differentially expressed in mop1-1 immature ears, our analysis indicated that MOP1-dependent siRNAs were consistently more abundant in WT at almost all mop1-1 up- and down-regulated genes (). The average expression fold change of the subset of MOP1 upregulated genes with 24 nt siRNAs was 4.07 and the P value (0.012) was lower than the P value (0.015) for the average expression fold change of 2.99 observed for MOP1 downregulated genes with 24 nt siRNAs (). This suggests that on average, this subset of genes was more strongly affected by loss of MOP1 activity.
Figure 4. Relative expression and siRNA abundance in -2 kb region of mop1-1 DEGs. Relative expression is expressed as log2 fold change in mop1-1 mutant/wild type (black bars). Log2 fold change of 23–24nt siRNAs (gray bars) -2kb upstream of predicted transcript start site in mutant/wild type for genes differentially expressed in mop1-1 mutants (A) and both mop1-1 and tgr1-1 mutants (B).
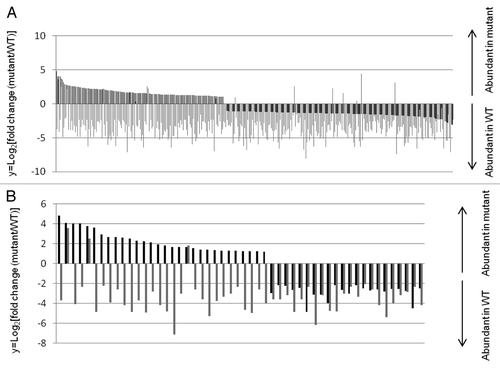
Figure 5. Average fold change of expression in mop1–1 DEG subsets. We measured the magnitude of average expression fold change for mop1-1 DEGs within different subsets. One subset included gene differentially expressed in mop1-1 alone and the other included genes differentially expressed in mop1-1 and tgr1. Within each subset, we measured the average expression of upregulated (Up) and downregulated (Down) genes; those Up and Down genes with 24 nt siRNAs within -2 kb of TSSs (+siRNA) and those that have a class II element within + or -1 kb of the TSS (+TIR) (A). The distribution of p-values for each of the genes falling within a particular subset (B), and the averages of these p-values for upregulated (black line) and downregulated (gray line) genes were calculated (C).
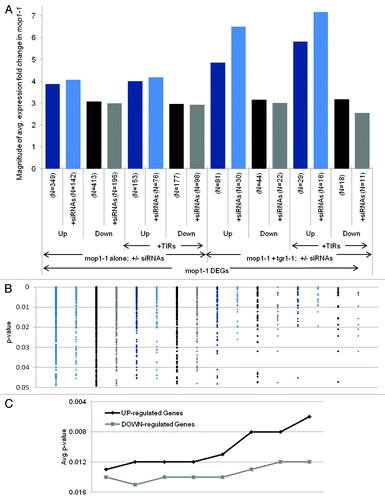
We predicted that direct targets of a MOP1-TGR1 epigenetic silencing pathway might be more likely than indirect targets to exhibit a correlation between accumulation of homologous promoter-proximal 24 nt siRNAs and gene expression. However, as observed at genes differentially expressed in mop1-1 alone, the presence or absence of siRNAs at epigenetic (MOP1 and TGR1) targets showed no direct correlation with the direction of transcriptional activity (). These results are also consistent with observations made at genes upregulated in Arabidopsis RdDM mutants (rdr2-1 and ddc); 24 nt siRNAs were detected and absent upstream of RdDM targets.Citation55 If we confined our analysis to specific subset of genes using multiple defining features, we did detect that the average expression fold change of genes upregulated in both mop1-1 and tgr1-1 and with promoter-proximal 24 nt siRNAs was 6.5 at a P value of 0.008. This P value was much lower than a P value of 0.013 for the average expression of 3.01 which was observed for the 22 TGS downregulated genes with 24 nt siRNAs (). Genes downregulated in both mutants, with or without 24 nt siRNAs, exhibit a level of expression fold change similar to genes repressed in mop1-1 mutants alone ().
While the expression of the strongly upregulated genes that appear to be direct targets of a MOP1-mediated pathway varied significantly, the distribution of confidence of expression was higher and had a narrower range than the entire set of mop1-1 DEGs () and the average P value decreased when genes were categorized using multiple features (). This suggests that in mop1-1, expression of the subset of genes considered to be direct targets of TGS was the most strongly and consistently affected. Based on findings reported herein, and those reviewed in ref. Citation56, 24 nt siRNAs alone are not sufficient to predict gene silencing by RdDM and other potential signals and features must be considered.
Higher changes in expression fold-change observed in TGS targets with near-gene transposons and 24 nt siRNAs
Pol IV-RdDM has been hypothesized to function at gene-proximal TEs to regulate expression of the gene.Citation4 Based on our observation of enhanced expression changes at TGS targets with 24 nt siRNAs, we hypothesized that specific classes of TEs might be enriched in the regulatory regions of MOP1-regulated genes. We investigated the relationship between the presence of class II elements (Maize Transposable Element Consortium) within 2 kb flanking TSSs (+ or – 1 kb) and expression at targets of MOP1-mediated TGS. Of the genes differentially expressed in mop1-1 alone, we identified 153 upregulated and 177 downregulated genes with proximity to the conserved terminal inverted repeat sequence (TIRs) of class II transposons (; Tables S6 and S7). Of these, 76 upregulated and 98 downregulated genes also had proximal 24 nt siRNAs (). Downregulated genes displayed an expression level similar to that observed in the 413 mop1–1 downregulated genes. In comparison to downregulated genes, the average fold change expression of upregulated genes in these categories was higher, yet similar to upregulated genes without proximal RdDM features ().
At putative TGS targets, we identified 29 upregulated and 18 repressed genes with proximal TIRs (; Tables S8 and S9) and 16 upregulated and 11 downregulated with proximal TIRs and 24 nt siRNAs (). We found that putative epigenetic targets with 24 nt siRNAs and proximal DNA transposons (TIRs) include genes with the highest average expression fold change in mop1 mutants (). The average P value also decreased for genes in this subset (), suggesting that these genes respond strongly and consistently to MOP1 activity. Notably, this group of 16 genes that we expect to be directly targeted by MOP1 was not represented in our prior analyses of nucleosome position in mop1-1 mutants,Citation30 and may explain the small number of genes that we observed to be effected in that study.
In all categories examined, downregulated genes exhibited similar average expression changes irrespective of the presence of siRNAs and/or proximal TIRs (). The observed increment in fold change expression with additive RdDM-associated features at putative epigenetic TGS targets demonstrates that multiple signals are utilized to confer MOP1 target specificity. To date, no consensus sequence exists for Pol IV and Pol V transcriptional targets (reviewed in reference Citation57) so it is difficult to identify direct targets of Pol IV-RdDM. Herein, we demonstrate that the effect of MOP1 on gene expression is enhanced at genes with proximal TIRs and homologous siRNAs. This trend may apply to only a subset of MOP1-mediated targets, but provides evidence that direct targets of MOP1 are best predicted by overlaying multiple indicating factors.
In summary, the chromatin accessibility profile established in mop1-1 immature ears is consistent with direct and secondary consequences of misregulation of gene expression, related to alterations in global chromatin structure in mop1-1. Consistently, we identified genes where expression increased and other genes where transcription was repressed in mop1-1 mutants. Although secondary consequences in expression are expected, a portion of upregulated genes are likely to be direct targets a MOP1-mediated transcriptional gene silencing pathway. We found that, on average, genes likely to be epigenetic TGS targets with homologous 24 nt siRNAs and proximal class II transposons included genes whose expression was more dramatically and consistently affected in mop1-1. We were unable to identify a similar subset of genes by limiting features in downregulated genes, suggesting that transcriptional repression in mop1-1 individuals may result from reduced ROS1 expression, or silencing by an alternative RdDM-TGS pathway.
Materials and Methods
Plant materials/genetic stocks
Genetic stocks were obtained from J. Dorweiler and are the result of the mop1-1 mutation (Dorweiler et al. 2000) introgressed in the B73 reference genome, and backcrossed for seven generations, at which stocks are considered near isogenic to the recurrent parent. The genotypes of the parental lines were as mop1-1 parent (K55: B’, Pl, r-g, mop1-1, and B73 recurrent parent: b, pl-sr, r-r, Mop1. 3–5 cm ear shoots were harvested and immediately frozen in liquid nitrogen and stored at –80 °C until use. Homozygous wild type (WT) and homozygous mop1 mutant (mop1-1) individuals were used. Plants were genotyped under the following PCR conditions: 94 °C for 5 min (1×); 95 °C for 30 s, 56 °C for 45 s, 72 °C for 45 s (30×); 72 °C for 10 min (1×); hold for 4 °C (as needed); using the following primers: KM384: (5′-TCTCCACCGC CCACTTGAT-3′); KM385: (5′-CCCAAGAGCT GTCTCGTATC CGT-3′); KM386: (5′-CTTCATCTCG AAGTAGCGCT TGTTGTCC-3′).
Nuclei isolation and cleavage with micrococcal nuclease (MNase)
Maize 3–5cm ear shoots were harvested and immediately flash frozen in liquid nitrogen. Flash frozen tissue was ground into a fine powder in liquid nitrogen. Nuclei were isolated from mop1–1 mutants and WT immature ears using a nuclei isolation buffer modified from Steinmüller and Apel, 1986. Frozen, ground tissue was added to Nuclei Isolation Fix Buffer (NIFB: 20 mM TRIS-HCl pH 7.8; 250 mM Sucrose; 5 mM MgCl2; 5 mM KCl; 40% Glycerol, 0.1% β-mercaptoethanol [βME]; 0.1 mM PMSF and 1% Formaldehyde). Nuclei were fixed for 10 min at room temperature (RT) after which formaldehyde was quenched by adding Glycine to 125 mM for 5 min at RT. Tissue was collected by centrifugation (1000 × g, 10 min, 4 °C) and resuspended in Nuclei Isolation Buffer with Triton (NITB: 20 mM TRIS-HCl pH 7.8; 250 mM Sucrose; 5 mM MgCl2; 5 mM KCl; 40% Glycerol, 0.1% BME; 0.1 mM PMSF; 1% Triton X-100). The suspension was filtered through 2 layers of nylon mesh and nuclei were collected by centrifugation (1000 × g, 10 min, 4 °C). Nuclei were gently resuspended in ice-cold Nuclei Resuspension Buffer/MNase Digestion Buffer (NRBT/MDB: 50 mM TRIS-HCl pH 7.5, 320 mM sucrose, 4 mM MgCl2, 1 mMCaCl2, 1% Triton, 0.1 mM PMSF), flash frozen, and stored until use. Nuclei (equivalent to 25 µg DNA) were subjected to digestion with limiting amounts of micrococcal nuclease (MNase: 0.1, 0.75, and 0.25 units) (Worthington Biochemical Corp) for 5mins at 37 °C and the reaction stopped with EGTA. Digested nuclei were decrosslinked at 65 °C overnight in the presence of 1% SDS and 0.2 mg/mL Proteinase K. DNA was extracted via phase extraction using phenol: chloroform and precipitated with NaOAc and ethanol and treated with RNaseA. DNA fragments were separated on a 1% agarose TAE gel. DNA was isolated from the respective gel fragments: MNase-resistant (high-molecular weight [HMW] DNA, undigested by MNase) and MNase-sensitive (low-molecular weight MNase digested DNA ladder, 150 bp to ~4000 bp representing DNA fragments readily accessible to digestion by limiting amounts of MNase). Each biological replicate consists of two ear shoots pooled together. The data represents the average of three biological replicates.
Microarray design, labeling and hybridization/MNase-chip
NimbleGen HD2 12-plex custom designed maize genome microarrays were used in these experiments. Each subarray contains ~134 300 unique, tiled and isothermal oligonucleotide probes that span the maize genome at a ~14 kb spacing designed to span all 10 maize chromosomes. The NimbleGen 12-plex slide ID is: 100428_ZMB73_JD_CGH_HX12. 500–1000 ng of isolated DNA was labeled using the NimbleGen Dual-Color Labeling Kit (NimbleGen) according to manufacturer’s protocol. Resistant/Inaccessible DNA (test) was labeled with Cy3 and Sensitive/Accessible DNA (reference) was labeled with Cy5. Twenty micrograms each (Cy3 and Cy5) labeled DNA were combined and used for hybridization. Arrays were hybridized for ~72 h at 42 °C after which slides were washed according to manufacturer’s protocol. Slides were scanned using a NimbleGen MS 200 Microarray Scanner and images were aligned using NimbleGen software.
Expression microarrays
Total RNA was isolated from maize ear shoots harvested and immediately flash frozen in liquid nitrogen and stored at –80 °C until use. Frozen tissue was ground into a fine powder in liquid nitrogen. Total RNA isolation was performed using TRI reagent according to manufacturer’s instructions (Molecular Research Center, Cat No. TR118). RNA was purified using Zymo RNA Clean and ConcentratorTM Kit (Zymo; Cat. No. R1017), according to manufacturer’s instructions. cDNA was synthesized using SuperScript TM III Reverse Transcriptase (Invitrogen Cat. No.18080-051). Briefly, 10 µg of RNA was reverse transcribed according to the manufacturer’s instructions using Oligo(dT)20 primers. cDNA synthesis was performed at 50 °C for 50 min. The reaction was terminated at 85 °C for 5 min. RNA was removed using 10 units RNaseH (Invitrogen) and 4 µg RNaseA (Qiagen) at 37 °C for 30 min. cDNA was purified using the QIAquick PCR Purification Kit (Qiagen). cDNA was labeled using the NimbleGen Dual-Color Labeling Kit (NimbleGen) according to manufacturer’s protocol. WT cDNA was labeled with Cy3 and mutant cDNA was labeled with Cy5. Six micrograms each labeled cDNA were combined and hybridized to a 385 K array. Arrays we hybridized overnight at 42 °C after which slides were washed according to manufacturer’s protocol. Slides were scanned using a NimbleGen MS 200 Microarray Scanner and images were aligned using NimbleGen software. NimbleGen gene expression microarrays designed by S. Kaeppler and R. Buell as described by Sekhon RS et al.Citation41 The NimbleGen Array ID is: 090319_Zea_KR_ExpTil. Normalized using RMA (Robust Multi-chip Average). RMA normalized expression valuesCitation58 were log2 transformed and fold change was analyzed using DNASTAR Arraystar® 4 software (http://www.dnastar.com/t-sub-products-genomics-arraystar.aspx). P values were calculated using the Student t test, one-tail.
Maize microarray expression annotation and gene ontology analysis
Maize gene annotations were obtained using NCBI standalone BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) by searching Arabidopsis TAIR10 (http://www.arabidopsis.org) and Rice Genome Annotation Project version 7.0 (http://rice.plantbiology.msu.edu/) databases. Top results were used as annotations for maize genes and e-value was set to 0.001. For upregulated and downregulated genes (2-fold cutoff), gene ontology (GO) analysis performed using BLAST2GO.Citation59 Briefly, sequences were identified through a local Blast search of the NCBI nr database and imported into BLAST2GO. This program was used to extract GO terms associated to each of the obtained hits and returned an evaluated GO annotation for the query sequences. If there were no meaningful BLAST searches, the program returned N/A.
mop1 siRNA alignment
mop1 siRNA data from Nobuta et al. (2009) was downloaded from NCBI using the following GEO accession numbers: GSM306487 (WT) and GSM306488 (mop1-1). siRNA sequences were aligned to the B73 reference genome (v2) with Bowtie2Citation60 using default parameters. Small RNAs were grouped into four categories: ≤20 nt, 21–22 nt, 23–24 nt, and ≥25 nt using R. Herein, we used the 23–24 nt class of siRNAs for further analysis. BEDToolsCitation61 was used to calculate siRNA counts for the maize filtered gene set (FGS) and at transcription start sites (TSSs) of mop1-1 differentially expressed genes (DEGs). In-house Python and R scripts were applied to organize results and make a pipeline for analysis.
TGR1 gene expression analysis
Total RNA was isolated and pooled from immature ears of 20 Tgr1 non-mutant and 20 tgr1 mutant plants. RNA isolation was performed using TRI reagent according to manufacturer’s instructions (Molecular Research Center, Cat No. TR118). The libraries were prepared using NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (New England Biolabs). The quality of total RNA were measured by a Bioanalyzer and then subjected to RNA-Sequencing using an Illumina HiSeq2000 single-end sequencing instrument. Raw reads were trimmed by FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) and aligned to B73 reference genome (AGPv2) using Tophat/Bowtie.Citation62 Differential gene expression analysis was performed using the Cufflink v2.1.1 software package adopting default settings.Citation63
| Abbreviations: | ||
| TGS | = | transcriptional gene silencing |
| RdDM | = | RNA-directed DNA methylation |
| siRNAs | = | small interfering RNAs |
| Pol IV | = | RNA polymerase IV |
| nt | = | nucleotide |
| MOP1 | = | mediator of paramutation1 |
| MNase | = | micrococcal nuclease |
| TSSs | = | transcription start sites |
| WT | = | wild type |
| DEGs | = | differentially expressed genes |
| TEs | = | transposable elements |
| RDR | = | RNA-dependent RNA polymerase |
Additional material
Download Zip (1.8 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr Steve Miller (FSU Microarray Facility) for technical assistance with labeling and hybridizations of microarrays; Daniel Vera and Drs Justin Fincher, Hank Bass, and Jonathan Dennis for accessibility array design and analysis. We also wish to thank Dr Jane Dorweiler for mop1–1 B73 genetic stocks, Dr Brooke Druliner for assistance with the chromatin accessibility assay, and Amy Sloan for sharing tgr1 expression data. This work was funded by the National Science Foundation through awards IOS-PGRP-1025954 and MCB- 0952579. The authors would like to thank the anonymous reviewers for their helpful comments and suggestions.
References
- Eamens A, Wang MB, Smith NA, Waterhouse PM. RNA silencing in plants: yesterday, today, and tomorrow. Plant Physiol 2008; 147:456 - 68; http://dx.doi.org/10.1104/pp.108.117275; PMID: 18524877
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 2010; 11:204 - 20; http://dx.doi.org/10.1038/nrg2719; PMID: 20142834
- Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A 2010; 107:8689 - 94; http://dx.doi.org/10.1073/pnas.1002720107; PMID: 20395551
- Gent JI, Ellis NA, Guo L, Harkess AE, Yao Y, Zhang X, Dawe RK. CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res 2013; 23:628 - 37; http://dx.doi.org/10.1101/gr.146985.112; PMID: 23269663
- Regulski M, Lu Z, Kendall J, Donoghue MT, Reinders J, Llaca V, Deschamps S, Smith A, Levy D, McCombie WR, et al. The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res 2013; 23:1651 - 62; http://dx.doi.org/10.1101/gr.153510.112; PMID: 23739895
- Nuthikattu S, McCue AD, Panda K, Fultz D, DeFraia C, Thomas EN, Slotkin RK. The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21-22 nucleotide small interfering RNAs. Plant Physiol 2013; 162:116 - 31; http://dx.doi.org/10.1104/pp.113.216481; PMID: 23542151
- Zhang H, Zhu JK. RNA-directed DNA methylation. Curr Opin Plant Biol 2011; 14:142 - 7; http://dx.doi.org/10.1016/j.pbi.2011.02.003; PMID: 21420348
- Dorweiler JE, Carey CC, Kubo KM, Hollick JB, Kermicle JL, Chandler VL. mediator of paramutation1 is required for establishment and maintenance of paramutation at multiple maize loci. Plant Cell 2000; 12:2101 - 18; http://dx.doi.org/10.1105/tpc.12.11.2101; PMID: 11090212
- Hollick JB, Chandler VL. Genetic factors required to maintain repression of a paramutagenic maize pl1 allele. Genetics 2001; 157:369 - 78; PMID: 11139517
- Arteaga-Vazquez MA, Chandler VL. Paramutation in maize: RNA mediated trans-generational gene silencing. Curr Opin Genet Dev 2010; 20:156 - 63; http://dx.doi.org/10.1016/j.gde.2010.01.008; PMID: 20153628
- Erhard KF Jr., Stonaker JL, Parkinson SE, Lim JP, Hale CJ, Hollick JB. RNA polymerase IV functions in paramutation in Zea mays. Science 2009; 323:1201 - 5; http://dx.doi.org/10.1126/science.1164508; PMID: 19251626
- Hale CJ, Stonaker JL, Gross SM, Hollick JB. A novel Snf2 protein maintains trans-generational regulatory states established by paramutation in maize. PLoS Biol 2007; 5:e275; http://dx.doi.org/10.1371/journal.pbio.0050275; PMID: 17941719
- Kanno T, Mette MF, Kreil DP, Aufsatz W, Matzke M, Matzke AJ. Involvement of putative SNF2 chromatin remodeling protein DRD1 in RNA-directed DNA methylation. Curr Biol 2004; 14:801 - 5; http://dx.doi.org/10.1016/j.cub.2004.04.037; PMID: 15120073
- Greenberg MV, Ausin I, Chan SW, Cokus SJ, Cuperus JT, Feng S, Law JA, Chu C, Pellegrini M, Carrington JC, et al. Identification of genes required for de novo DNA methylation in Arabidopsis. Epigenetics 2011; 6:344 - 54; http://dx.doi.org/10.4161/epi.6.3.14242; PMID: 21150311
- Alleman M, Sidorenko L, McGinnis K, Seshadri V, Dorweiler JE, White J, Sikkink K, Chandler VL. An RNA-dependent RNA polymerase is required for paramutation in maize. Nature 2006; 442:295 - 8; http://dx.doi.org/10.1038/nature04884; PMID: 16855589
- Sidorenko L, Dorweiler JE, Cigan AM, Arteaga-Vazquez M, Vyas M, Kermicle J, Jurcin D, Brzeski J, Cai Y, Chandler VL. A dominant mutation in mediator of paramutation2, one of three second-largest subunits of a plant-specific RNA polymerase, disrupts multiple siRNA silencing processes. PLoS Genet 2009; 5:e1000725; http://dx.doi.org/10.1371/journal.pgen.1000725; PMID: 19936058
- Stonaker JL, Lim JP, Erhard KF Jr., Hollick JB. Diversity of Pol IV function is defined by mutations at the maize rmr7 locus. PLoS Genet 2009; 5:e1000706; http://dx.doi.org/10.1371/journal.pgen.1000706; PMID: 19936246
- Madzima TF, Mills ES, Gardiner JM, McGinnis KM. Identification of epigenetic regulators of a transcriptionally silenced transgene in maize. G3 (Bethesda) 2011; 1:75 - 83; http://dx.doi.org/10.1534/g3.111.000232; PMID: 22384320
- McGinnis KM, Springer C, Lin Y, Carey CC, Chandler V. Transcriptionally silenced transgenes in maize are activated by three mutations defective in paramutation. Genetics 2006; 173:1637 - 47; http://dx.doi.org/10.1534/genetics.106.058669; PMID: 16702420
- Nobuta K, Lu C, Shrivastava R, Pillay M, De Paoli E, Accerbi M, Arteaga-Vazquez M, Sidorenko L, Jeong DH, Yen Y, et al. Distinct size distribution of endogeneous siRNAs in maize: Evidence from deep sequencing in the mop1-1 mutant. Proc Natl Acad Sci U S A 2008; 105:14958 - 63; http://dx.doi.org/10.1073/pnas.0808066105; PMID: 18815367
- Woodhouse MR, Freeling M, Lisch D. Initiation, establishment, and maintenance of heritable MuDR transposon silencing in maize are mediated by distinct factors. PLoS Biol 2006; 4:e339; http://dx.doi.org/10.1371/journal.pbio.0040339; PMID: 16968137
- Lisch D, Carey CC, Dorweiler JE, Chandler VL. A mutation that prevents paramutation in maize also reverses Mutator transposon methylation and silencing. Proc Natl Acad Sci U S A 2002; 99:6130 - 5; http://dx.doi.org/10.1073/pnas.052152199; PMID: 11959901
- Hultquist JF, Dorweiler JE. Feminized tassels of maize mop1 and ts1 mutants exhibit altered levels of miR156 and specific SBP-box genes. Planta 2008; 229:99 - 113; http://dx.doi.org/10.1007/s00425-008-0813-2; PMID: 18800226
- Jia Y, Lisch DR, Ohtsu K, Scanlon MJ, Nettleton D, Schnable PS. Loss of RNA-dependent RNA polymerase 2 (RDR2) function causes widespread and unexpected changes in the expression of transposons, genes, and 24-nt small RNAs. PLoS Genet 2009; 5:e1000737; http://dx.doi.org/10.1371/journal.pgen.1000737; PMID: 19936292
- Stam M, Belele C, Dorweiler JE, Chandler VL. Differential chromatin structure within a tandem array 100 kb upstream of the maize b1 locus is associated with paramutation. Genes Dev 2002; 16:1906 - 18; http://dx.doi.org/10.1101/gad.1006702; PMID: 12154122
- Haring M, Bader R, Louwers M, Schwabe A, van Driel R, Stam M. The role of DNA methylation, nucleosome occupancy and histone modifications in paramutation. Plant J 2010; http://dx.doi.org/10.1111/j.1365-313X.2010.04245.x; PMID: 20444233
- Fransz P, de Jong H. From nucleosome to chromosome: a dynamic organization of genetic information. Plant J 2011; 66:4 - 17; http://dx.doi.org/10.1111/j.1365-313X.2011.04526.x; PMID: 21443619
- Woodcock CL, Ghosh RP. Chromatin higher-order structure and dynamics. Cold Spring Harb Perspect Biol 2010; 2:a000596; http://dx.doi.org/10.1101/cshperspect.a000596; PMID: 20452954
- Ha M. Understanding the chromatin remodeling code. Plant Sci 2013; 211:137 - 45; http://dx.doi.org/10.1016/j.plantsci.2013.07.006; PMID: 23987819
- Labonne JD, Dorweiler JE, McGinnis KM. Changes in nucleosome position at transcriptional start sites of specific genes in Zea mays mediator of paramutation1 mutants. Epigenetics 2013; 8:8; http://dx.doi.org/10.4161/epi.24199; PMID: 23538550
- Takebayashi S, Dileep V, Ryba T, Dennis JH, Gilbert DM. Chromatin-interaction compartment switch at developmentally regulated chromosomal domains reveals an unusual principle of chromatin folding. Proc Natl Acad Sci U S A 2012; 109:12574 - 9; http://dx.doi.org/10.1073/pnas.1207185109; PMID: 22807480
- Druliner BR, Fincher JA, Sexton BS, Vera DL, Roche M, Lyle S, Dennis JH. Chromatin patterns associated with lung adenocarcinoma progression. Cell Cycle 2013; 12:1536 - 43; http://dx.doi.org/10.4161/cc.24664; PMID: 23598721
- Telford DJ, Stewart BW. Micrococcal nuclease: its specificity and use for chromatin analysis. Int J Biochem 1989; 21:127 - 37; http://dx.doi.org/10.1016/0020-711X(89)90100-6; PMID: 2663558
- Sajan SA, Hawkins RD. Methods for identifying higher-order chromatin structure. Annu Rev Genomics Hum Genet 2012; 13:59 - 82; http://dx.doi.org/10.1146/annurev-genom-090711-163818; PMID: 22703176
- He G, Chen B, Wang X, Li X, Li J, He H, Yang M, Lu L, Qi Y, Wang X, et al. Conservation and divergence of transcriptomic and epigenomic variation in maize hybrids. Genome Biol 2013; 14:R57; http://dx.doi.org/10.1186/gb-2013-14-6-r57; PMID: 23758703
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. The B73 maize genome: complexity, diversity, and dynamics. Science 2009; 326:1112 - 5; http://dx.doi.org/10.1126/science.1178534; PMID: 19965430
- Lisch D, Carey CC, Dorweiler JE, Chandler VL. A mutation that prevents paramutation in maize also reverses Mutator transposon methylation and silencing. Proc Natl Acad Sci U S A 2002; 99:6130 - 5; http://dx.doi.org/10.1073/pnas.052152199; PMID: 11959901
- Woodhouse MR, Freeling M, Lisch D. The mop1 (mediator of paramutation1) mutant progressively reactivates one of the two genes encoded by the MuDR transposon in maize. Genetics 2006; 172:579 - 92; http://dx.doi.org/10.1534/genetics.105.051383; PMID: 16219782
- Wang X, Elling AA, Li X, Li N, Peng Z, He G, Sun H, Qi Y, Liu XS, Deng XW. Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell 2009; 21:1053 - 69; http://dx.doi.org/10.1105/tpc.109.065714; PMID: 19376930
- Barber WT, Zhang W, Win H, Varala KK, Dorweiler JE, Hudson ME, Moose SP. Repeat associated small RNAs vary among parents and following hybridization in maize. Proc Natl Acad Sci U S A 2012; 109:10444 - 9; http://dx.doi.org/10.1073/pnas.1202073109; PMID: 22689990
- Sekhon RS, Lin H, Childs KL, Hansey CN, Buell CR, de Leon N, Kaeppler SM. Genome-wide atlas of transcription during maize development. Plant J 2011; 66:553 - 63; http://dx.doi.org/10.1111/j.1365-313X.2011.04527.x; PMID: 21299659
- Ng KH, Yu H, Ito T. AGAMOUS controls GIANT KILLER, a multifunctional chromatin modifier in reproductive organ patterning and differentiation. PLoS Biol 2009; 7:e1000251; http://dx.doi.org/10.1371/journal.pbio.1000251; PMID: 19956801
- Smaczniak C, Immink RG, Angenent GC, Kaufmann K. Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 2012; 139:3081 - 98; http://dx.doi.org/10.1242/dev.074674; PMID: 22872082
- Morales-Ruiz T, Ortega-Galisteo AP, Ponferrada-Marín MI, Martínez-Macías MI, Ariza RR, Roldán-Arjona T. DEMETER and REPRESSOR OF SILENCING 1 encode 5-methylcytosine DNA glycosylases. Proc Natl Acad Sci U S A 2006; 103:6853 - 8; http://dx.doi.org/10.1073/pnas.0601109103; PMID: 16624880
- Penterman J, Zilberman D, Huh JH, Ballinger T, Henikoff S, Fischer RL. DNA demethylation in the Arabidopsis genome. Proc Natl Acad Sci U S A 2007; 104:6752 - 7; http://dx.doi.org/10.1073/pnas.0701861104; PMID: 17409185
- Agius F, Kapoor A, Zhu JK. Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc Natl Acad Sci U S A 2006; 103:11796 - 801; http://dx.doi.org/10.1073/pnas.0603563103; PMID: 16864782
- Huettel B, Kanno T, Daxinger L, Aufsatz W, Matzke AJ, Matzke M. Endogenous targets of RNA-directed DNA methylation and Pol IV in Arabidopsis. EMBO J 2006; 25:2828 - 36; http://dx.doi.org/10.1038/sj.emboj.7601150; PMID: 16724114
- Li X, Qian W, Zhao Y, Wang C, Shen J, Zhu JK, Gong Z. Antisilencing role of the RNA-directed DNA methylation pathway and a histone acetyltransferase in Arabidopsis. Proc Natl Acad Sci U S A 2012; 109:11425 - 30; http://dx.doi.org/10.1073/pnas.1208557109; PMID: 22733760
- Gong Z, Morales-Ruiz T, Ariza RR, Roldán-Arjona T, David L, Zhu JK. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 2002; 111:803 - 14; http://dx.doi.org/10.1016/S0092-8674(02)01133-9; PMID: 12526807
- Otagaki S, Kasai M, Masuta C, Kanazawa A. Enhancement of RNA-directed DNA methylation of a transgene by simultaneously downregulating a ROS1 ortholog using a virus vector in Nicotiana benthamiana. Front Genet 2013; 4:44; http://dx.doi.org/10.3389/fgene.2013.00044; PMID: 23565118
- Pontier D, Picart C, Roudier F, Garcia D, Lahmy S, Azevedo J, Alart E, Laudié M, Karlowski WM, Cooke R, et al. NERD, a plant-specific GW protein, defines an additional RNAi-dependent chromatin-based pathway in Arabidopsis. Mol Cell 2012; 48:121 - 32; http://dx.doi.org/10.1016/j.molcel.2012.07.027; PMID: 22940247
- Wu L, Mao L, Qi Y. Roles of dicer-like and argonaute proteins in TAS-derived small interfering RNA-triggered DNA methylation. Plant Physiol 2012; 160:990 - 9; http://dx.doi.org/10.1104/pp.112.200279; PMID: 22846193
- Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013; 152:352 - 64; http://dx.doi.org/10.1016/j.cell.2012.10.054; PMID: 23313553
- Martínez de Alba AE, Elvira-Matelot E, Vaucheret H. Gene silencing in plants: a diversity of pathways. Biochim Biophys Acta 2013; 1829:1300 - 8; http://dx.doi.org/10.1016/j.bbagrm.2013.10.005; PMID: 24185199
- Kurihara Y, Matsui A, Kawashima M, Kaminuma E, Ishida J, Morosawa T, Mochizuki Y, Kobayashi N, Toyoda T, Shinozaki K, et al. Identification of the candidate genes regulated by RNA-directed DNA methylation in Arabidopsis. Biochem Biophys Res Commun 2008; 376:553 - 7; http://dx.doi.org/10.1016/j.bbrc.2008.09.046; PMID: 18805399
- Dalakouras A, Wassenegger M. Revisiting RNA-directed DNA methylation. RNA Biol 2013; 10:453 - 5; http://dx.doi.org/10.4161/rna.23542; PMID: 23324611
- Pikaard CS, Haag JR, Pontes OM, Blevins T, Cocklin R. A transcription fork model for Pol IV and Pol V-dependent RNA-directed DNA methylation. Cold Spring Harb Symp Quant Biol 2012; 77:205 - 12; http://dx.doi.org/10.1101/sqb.2013.77.014803; PMID: 23567894
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003; 4:249 - 64; http://dx.doi.org/10.1093/biostatistics/4.2.249; PMID: 12925520
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005; 21:3674 - 6; http://dx.doi.org/10.1093/bioinformatics/bti610; PMID: 16081474
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357 - 9; http://dx.doi.org/10.1038/nmeth.1923; PMID: 22388286
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26:841 - 2; http://dx.doi.org/10.1093/bioinformatics/btq033; PMID: 20110278
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009; 25:1105 - 11; http://dx.doi.org/10.1093/bioinformatics/btp120; PMID: 19289445
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 2013; 31:46 - 53; http://dx.doi.org/10.1038/nbt.2450; PMID: 23222703