
Abstract
Aberrant DNA methylation at specific genetic loci is a key molecular feature of juvenile myelomonocytic leukemia (JMML) with poor prognosis. Using quantitative high-resolution mass spectrometry, we identified RASA4 isoform 2, which maps to chromosome 7 and encodes a member of the GAP1 family of GTPase-activating proteins for small G proteins, as a recurrent target of isoform-specific DNA hypermethylation in JMML (51% of 125 patients analyzed). RASA4 isoform 2 promoter methylation correlated with clinical parameters predicting poor prognosis (older age, elevated fetal hemoglobin), with higher risk of relapse after hematopoietic stem cell transplantation, and with PTPN11 mutation. The level of isoform 2 methylation increased in relapsed cases after transplantation. Interestingly, most JMML cases with monosomy 7 exhibited hypermethylation on the remaining RASA4 allele. The results corroborate the significance of epigenetic modifications in the phenotype of aggressive JMML.
Introduction
Juvenile myelomonocytic leukemia (JMML) is a highly aggressive myeloproliferative malignancy of early childhood. The main variables used for prognostic assessment include age at diagnosis, platelet count, level of fetal hemoglobin (HbF), and extent of DNA hypermethylation.Citation1-Citation4 The molecular basis of JMML involves deregulation of the Ras pathway. Hyperactive Ras signaling can be linked to canonical mutations in PTPN11, KRAS, NRAS, NF1 or CBL in > 80% of JMML cases.Citation5-Citation9 Although these mutations are thought to represent the cardinal genetic feature of JMML, it is still unclear if and how they determine treatment resistance.Citation10-Citation12 Additional genetic changes frequently observed in JMML include chromosomal aberrations, most notably monosomy 7, which is detected in approximately 25% of cases.Citation1,Citation13 We recently reported that the strongest prognostic factor on the molecular level is neither Ras pathway mutation nor karyotype but the presence of DNA hypermethylation at several target genes,Citation4 suggesting an important role of epigenetic events in the pathobiology of resistant JMML.
We had previously identified the RASA4 locus as putative target in a search for aberrant cytosine-phospho-guanidine (CpG) island methylation within a region of chromosome 7 commonly deleted in acute myeloid leukemia (AML) of adults (unpublished data). RASA4 functions as a calcium-regulated GTPase-activating protein (GAP) for the small G proteins Ras and Rap.Citation14-Citation18 The Ras-suppressive function of RASA4 and its genetic location on chromosome 7 raised our interest in the potential role of this molecule in JMML pathophysiology. Here we report that RASA4 hypermethylation is a recurrent event in JMML that it is associated with cases at high risk of resistance to treatment and that RASA4 transcription is repressed in primary leukemia cells from children with JMML.
Results
The downstream, but not the proximal, RASA4 CpG island is frequently hypermethylated in JMML
RASA4 is encoded in a locus with three alternative transcription start sites (TSS), two of which are situated within CpG-rich regions (CpG islands) (Genome Reference Consortium human build 37 patch release 10, 2012/08/31) (). The most 5′ TSS (hereafter referred to as TSS1) gives rise to the full-length transcript of 2412 coding nucleotides spread across 21 exons (RefSeq accession number NM 006989) and a variant that skips exon 17 (NM 1079877). The second TSS (referred to as TSS2) is located in front of exon 14 and produces a shorter variant with 5 exons and a 612-nucleotide open reading frame (UCSC Genome Browser ID uc011klc.1, accessed on 2013/01/25). The third TSS (TSS3) is located in exon 16 outside a CpG-rich region. Its transcript has 5 exons with an open reading frame of 519 nucleotides (UCSC Genome Browser ID uc011kkz.2, accessed on 2013/01/25). We employed quantitative high-resolution mass spectrometry (Sequenom MassARRAY)Citation19 to analyze the level of cytosine methylation in the TSS1 and TSS2 CpG islands using granulocyte DNA from 125 children with JMML. Granulocyte DNA from 17 healthy volunteers was used as control (–).
Figure 1. The second RASA4 CpG island is frequently hypermethylated in JMML. (A) Structure of the RASA4 genetic locus and composition of transcript variants. Large gray boxes represent coding exons; small gray boxes, 5′ untranslated region; arrows, transcription start sites; green boxes, CpG islands; red boxes, amplicons used for mass spectrometry-based (MassArray) quantification of CpG methylation. (B) Color-coded map of CpG methylation in RASA4 CpG islands 1 and 2. The color gradient ranges from light green (0% methylation) to dark blue (100% methylation). Columns represent CpG units and rows represent DNA samples. (C) Dot plot of average CpG methylation for RASA4 CpG islands 1 and 2. Abbreviations: TSS, transcription start site; CpG, cytosine-phospho-guanine; JMML, juvenile myelomonocytic leukemia.
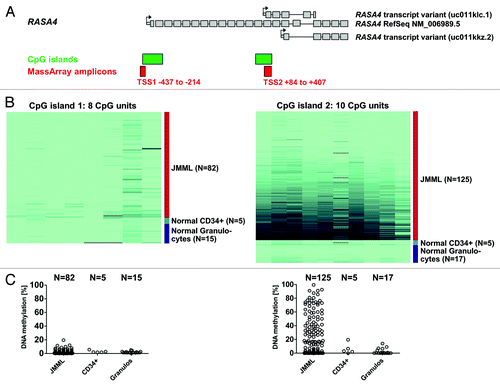
The JMML cohort included 46 cases (38%) with somatic PTPN11 mutation, 29 cases (24%) with somatic KRAS/NRAS mutation and 17 cases (14%) with a clinical diagnosis of neurofibromatosis type 1 (NF-1). Thirteen cases (11%) carried germline CBL mutations with somatic loss of heterozygosity, whereas cases with germline PTPN11 or RAS mutations (indicating Noonan syndrome) were excluded. Cytogenetically, 24 cases (20%) had monosomy 7 and 86 cases (71%) had a normal karyotype. The numbers indicate that the landscape of molecular genetic and cytogenetic aberrations in the study cohort is representative of the general JMML population.Citation3 Except for two cases, the study cohort is identical with that of a previous publication and more detailed information on clinical and hematological features can be found there.Citation4
We observed no difference in DNA methylation level between JMML and healthy granulocytes for the upstream CpG island (TSS1), which was virtually unmethylated in both leukemic and normal cells (–). In contrast, approximately half of all JMML cases exhibited increased DNA methylation in the downstream CpG island (TSS2). The median density of CpG methylation was 16.8% (range 0.0–90.1%) in JMML but only 1.4% (range 0.0–15.1%) in normal granulocytes (P < 0.01, Mann-Whitney test) (–). We defined TSS2 as hypermethylated if the average methylation level (measured across 10 CpG units, ) was greater than 16%; this threshold corresponds to the mean plus three standard deviations in 17 granulocyte samples from healthy individuals. Sixty-four of 125 JMML cases (51%) exhibited RASA4 TSS2 hypermethylation. To ensure that low RASA4 TSS2 methylation was not specific to granulocytes but representative of normal hematopoiesis, we also measured the RASA4 TSS2 methylation levels in CD34+ hematopoietic stem cells separated from BM of 5 healthy donors and found values equivalent to normal granulocytes (–). The finding that DNA hypermethylation at distinct promoter regions is a feature of some, but not all, cases of JMML confirms earlier observationsCitation4 and underscores that aberrant CpG methylation is a non-random pathophysiologic feature in a subgroup of patients with this disease.
RASA4 undergoes transcript-specific gene silencing in JMML
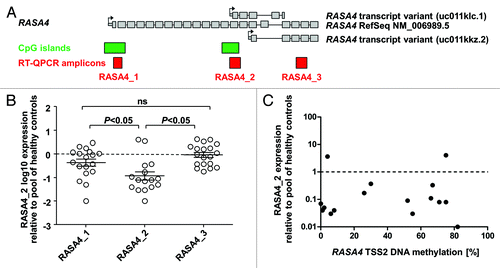
We next investigated whether the expression of different RASA4 transcripts was altered in primary leukemia cells from children with JMML. We designed three amplicons for quantitative reverse-transcription PCR (): Amplicon 1 bridges the first intron of the full-length variant and thus detects only transcripts originating at TSS1. Amplicon 2 covers the 5′ untranslated region at TSS2 and the adjacent exon, making it specific for transcripts starting at TSS2. Amplicon 3 bridges intron 18 of the full-length transcript or intron 3 of transcripts off TSS3 and hence detects the full-length variant and the TSS3 variant but not the one starting at TSS2. We measured the specific expression of RASA4 transcript variants relative to three housekeeping genes (HPRT, ACTB, GAPDH) in MNC RNA from 16 JMML patients, using pooled MNC RNA from 5 healthy donors as a reference (). The expression of RASA4 TSS2 transcripts was markedly decreased in most JMML samples (median 0.08 relative to control RNA, interquartile range 0.04–0.29), whereas the expression level of RASA4 TSS1 transcripts in JMML was essentially normal (median 0.6 relative to control RNA, interquartile range 0.17–1.18). There was no difference between JMML and healthy donors for amplicon 3 (median, 0.95; interquartile range 0.37–2.06). Remarkably, RASA4 TSS2 transcription was diminished in all but 2 JMML samples, irrespective of DNA hypermethylation at RASA4 TSS2 (). Taken together, the results indicate that RASA4 TSS2 transcription is specifically repressed in most cases of JMML but, at the same time, suggest that aberrant DNA methylation at RASA4 TSS2 is not the primary mechanism behind this downregulation.
Figure 2. The second RASA4 transcript is repressed in JMML. (A) Structure of the RASA4 genetic locus and composition of transcript variants. Large gray boxes represent coding exons; small gray boxes, 5′ untranslated region; arrows, transcription start sites; green boxes, CpG islands; red boxes, amplicons used for RT-QPCR. (B) Expression level of three RASA4 transcripts in JMML mononuclear cells. Expression for each transcript was measured by three RT-QPCRs normalizing to HPRT, ACTB and GAPDH, respectively, and results were averaged. Expression in pooled blood cell RNA from 5 healthy individuals was used as calibrator and set to 1.0. P values were computed using analysis-of-variance across the groups (transcripts) and Tukey’s multiple comparison test for pairwise comparisons. (C) Expression levels of the RASA4 variant 2 transcript in mononuclear cells from 15 JMML patients in relation to CpG island methylation. Abbreviations: CpG, cytosine-phospho-guanine; RT-QPCR, reverse-transcriptase quantitative polymerase chain reaction.
RASA4 hypermethylation is associated with poor prognosis in JMML and is increased in resistant cases
To study whether RASA4 TSS2 hypermethylation correlates with clinical or hematological parameters in JMML, the cohort of 125 patients was grouped into tertiles according to the level of RASA4 TSS2 methylation (). There was a strong association between high methylation and older age at diagnosis. The median age at diagnosis was 0.6 y in the lowest tertile, 1.3 y in the middle tertile and 3.5 y in the highest tertile (P < 0.01, Kruskal-Wallis test). Hypermethylation was also associated with the level of HbF. Forty-seven percent of patients in the lowest tertile of RASA4 methylation had normal HbF (adjusted for age) whereas HbF was elevated in 85% of patients in the highest tertile (P = 0.01, chi-square test). Higher-methylation cases had more severe thrombocytopenia (median 85 x 109/l in the lowest methylation tertile vs. 48 x 109/l in the highest tertile; P = 0.05, Kruskal-Wallis test). Since all these parameters are predictive of outcome in JMML,Citation1 we then performed survival analyses in the three methylation groups. The probability of 5-y overall survival in 41 cases constituting the lowest tertile of methylation was 0.65 (95% confidence interval [CI] 0.46–0.84), whereas it was 0.55 (95% CI 0.37–0.73) in the middle tertile and only 0.36 (95% CI 0.14–0.58) in the highest tertile (lowest vs. highest: P = 0.08, log-rank test) (). Allogeneic hematopoietic stem cell transplantation (HSCT) had been performed in 100 of the 125 children. Among these transplanted patients, the 5-y cumulative incidence of relapse was 0.16 (95% CI 0.07–0.35) in the lowest tertile of methylation, 0.26 (95% CI 0.15–0.46) in the middle tertile and 0.51 (95% CI 0.36–0.73) in the highest tertile (lowest vs. highest: P < 0.01, log-rank test) (). A multivariate Cox analysis demonstrated that RASA4 methylation had a stronger prognostic influence than age or platelet count: RASA4 methylation, P < 0.01; age, P = 0.14; platelets, P = 0.19; as expected, all three factors were highly correlated. The relative risk of relapse after HSCT between the highest and lowest methylation tertiles was 4.0 (95% CI 1.5–10.9). HbF was excluded from the multivariate model because values were missing in 28 cases.
Table 1.RASA4 TSS2 methylation and clinical or hematological parameters in JMML
Figure 3.RASA4 TSS2 hypermethylation is associated with poor prognosis in JMML. (A) Outcome of 125 JMML patients divided into tertiles by RASA4 TSS2 methylation. The probability of survival from diagnosis, irrespective of disease status, is plotted. Patients alive at last follow-up were censored. Death was considered as event. Numbers indicate the probability of 5-y survival and the 95% confidence interval. (B) Cumulative incidence of relapse of 100 patients who had received HSCT. Numbers indicate the 5-y cumulative incidence of relapse and the 95% confidence interval. Relapse incidence was defined as the probability of JMML relapse at a given time. Death without relapse was considered a competing event. (C)RASA4 TSS2 methylation levels in four JMML patients at initial diagnosis and at relapse after HSCT. Abbreviation: HSCT, hematopoietic stem cell transplantation.
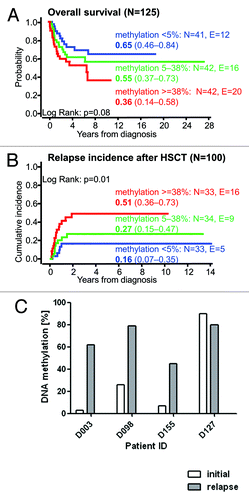
We also compared the level of RASA4 TSS2 methylation at initial presentation to that at relapse after HSCT in four cases where longitudinal DNA samples were available (). All cases except one (in which methylation was already 90% at diagnosis) exhibited a dramatic increase in RASA4 methylation after relapse. In conclusion, epigenetic modification at RASA4 TSS2 is found in JMML patients with poor prognostic features and identifies cases at high risk of treatment failure.
Hypermethylation of RASA4 is associated with specific genetic subgroups of JMML
JMML is characterized by genetic mutations of the Ras signal transduction cascade, which result in hyperactive signaling. We asked how hypermethylation in the RASA4 TSS2 CpG island was associated with different genetic subgroups (; ). When the 125 JMML cases were separated into thirds by their level of RASA4 TSS2 methylation, PTPN11-mutant cases were vastly overrepresented in the third with the highest methylation (). Across the entire cohort, the median RASA4 methylation was 37.6% in 46 patients with PTPN11 mutation but only 4.5% in 74 cases with wild type PTPN11 (including cases with other mutation or no identified mutation; 5 cases were uninformative with respect to mutation status) (P < 0.01; Mann-Whitney test) (). The association of PTPN11 mutation with RASA4 TSS2 methylation is compatible with the observation that PTPN11-mutant cases tend to run a more aggressive clinical course.Citation11,Citation12 Conversely, there was only one patient with CBL alteration among 42 in the highest methylation tertile, but 8 CBL-mutant cases out of 42 in the lowest tertile (P = 0.04, chi-square test). A recent exome-wide sequencing study identified mutations in SETBP1 and JAK3 as genetic lesions in JMML with probable prognostic impact, but information on these mutations was unavailable in our cohort, precluding a comparison with RASA4 methylation.Citation20
Figure 4. Association between RASA4 TSS2 methylation and genetic features in JMML. (A) Proportion of cases with NF1, PTPN11, KRAS, NRAS or CBL gene mutation or no identified mutation in 41 cases with lowest RASA4 TSS2 methylation level (black bars) or 42 cases with highest RASA4 TSS2 methylation level (gray bars) after dividing the entire cohort of 125 cases by methylation tertile. (B)RASA4 TSS2 methylation levels in 74 cases with wild type PTPN11 and 46 cases with PTPN11 mutation. Horizontal lines represent median values. (C)RASA4 TSS2 methylation levels in 25 cases with monosomy 7, 83 cases with normal cytogenetics and 11 cases with other chromosomal aberrations. Horizontal lines represent median values. Abbreviations: -7, monosomy 7; CGN, cytogenetically normal.
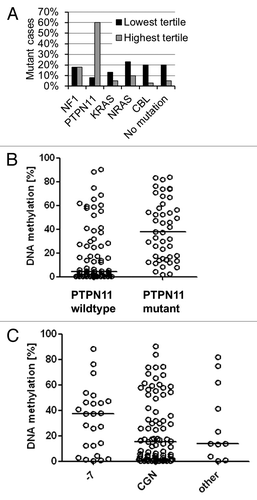
We also determined whether there were differences between cytogenetic subgroups of JMML. The overall association between RASA4 methylation tertiles and karyotype did not reach statistical significance (), but it was still noteworthy that there were only 6 cases with < 5% RASA4 methylation among 25 cases with monosomy 7. The median level of methylation was 37.5% in 25 cases with monosomy 7 as compared with 15.4% in 83 JMML cases with normal karyotype and 14.0% in 11 JMML cases with other chromosomal aberrations (cytogenetic information was missing in 6 cases) (). Although the association of monosomy 7 with RASA4 methylation may suggest a two-hit mechanism, the lack of association between reduced expression and DNA methylation does not support this concept.
RASA4 hypermethylation does not add to the prognostic power of DNA methylation at other genetic loci reported previously
Cancers typically show a high degree of concordance of methylation at different genetic loci. We therefore asked whether the prognostic significance of RASA4 methylation was independent of our previously published “predictive DNA methylation score” calculated from methylation levels at four other genes.Citation4 The Cox regression model from the previous publication was recalculated for prediction of overall survival in a training cohort of 79 patients, leaving out or including the methylation information of RASA4 TSS2. The new prognosticator was then tested in a validation cohort of 46 patients (Figure S1). However, including the RASA4 TSS2 variables as additional factors in Cox regression models for overall survival or event-free survival after HSCT did not result in improved prediction performance compared with a model containing the previously defined methylation score alone.Citation4 This suggests that DNA hypermethylation at any of the loci reflects a broader methylator phenotype in high-risk JMML and that it need not be of specific functional significance at each locus.
Discussion
Recent research has highlighted the importance of epigenetic aberrations in JMML, which have stronger prognostic significance than hematological features or genetic lesions.Citation4,Citation21 We were interested in discovering novel epigenetic targets and focused on a commonly deleted segment of chromosome 7. This identified the CpG island surrounding the second transcriptional start site of the RASA4 gene as a recurrent target of hypermethylation in JMML (64 of 125 cases, 51%). The DNA sequence was unmethylated in healthy hematopoietic nucleated cells and transcription was active, in accordance with data published in the GNF database which indicate high RASA4 expression in blood cells relative to most other human tissues.Citation22 In contrast, the transcript was silenced in 87% of JMML samples available for expression analysis. However, the downregulation was independent of the level of DNA methylation at the 5′ CpG island. These observations suggest that transcriptional inactivation of RASA4 may well have a role in JMML, but is not consistently linked with the epigenetic mechanism of promoter CpG methylation. Nevertheless, dense RASA4 methylation predicted poor outcome and a ~50% relapse risk after HSCT, reinforcing earlier observations that aberrant DNA methylation in JMML is associated with cases at high risk of treatment failure.Citation4
Assuming that the Ras-antagonistic activity of RASA4 observed in non-leukemic biochemical assaysCitation14,Citation18 is relevant also to JMML, our findings would be compatible with a model where selective pressure favors JMML clones which have acquired a competitive advantage by removing the inhibitory activity of RASA4 on Ras. This downregulation appears to be primarily non-epigenetic. High-risk cases then might go on to hypermethylate RASA4 TSS2, “locking in” a silent state of the promoter. Several observations support this model: 1) The extent of RASA4 methylation in many non-high-risk JMML cases was higher than that observed in granulocytes of healthy subjects but not dense enough to immediately explain the 10-fold downregulation of the transcript; 2) RASA4 hypermethylation was clearly associated with unfavorable clinical (older age, thrombocytopenia) and genetic (PTPN11 mutation) features; 3) RASA4 methylation levels increased with disease progression.
Apart from RASA4, four other genes have previously been identified whose DNA methylation status predicts the clinical course of JMML.Citation4 However, the picture of epigenetic modification in JMML is still incomplete because the analysis of aberrant CpG island methylation in JMML has so far been based on candidate-gene approaches and the results cover just a tiny minority of the ten thousands of promoter CpG islands in the human genome. Even though some of the targets of hypermethylation in JMML are prominent cancer genes such as CDKN2B,Citation23 their functional contribution to more aggressive clinical behavior remains so far obscure. While RASA4 dysregulation is of interest because of its potential interference with the principal proliferative mechanism in JMML, it cannot be ruled out that aberrant RASA4 methylation in JMML might simply be a collateral effect of a broader hypermethylation phenotype. With this question in mind, future analyses of genome-wide DNA methylation data in JMML will be instrumental in understanding if epigenetic modifications follow a defined pattern in refractory JMML or if they are merely the manifestation of global and chaotic loss of control over epigenetic processes in the leukemic cell.
Materials and Methods
Patient cell samples
Clinical cell samples from 125 children with JMML were collected in the context of the European Working Group of MDS in Childhood (EWOG-MDS) studies EWOG-MDS98 and EWOG-MDS2006 (registered with the National Institutes of Health; www.clinicaltrials.gov identifiers NCT00047268 and NCT00662090) after obtaining informed consent from parents or legal guardians in accordance with the Declaration of Helsinki and approval from institutional review committees at each participating center. Diagnostic samples included bone marrow (BM) in 69 cases and peripheral blood (PB) in 56 cases.
Nucleic acid extraction
Genomic DNA was isolated from BM or PB granulocytes using the Puregene (Qiagen, Hilden, Germany), Transfast (Peqlab, Erlangen, Germany) or DNeasy Blood and Tissue (Qiagen) kits. Total RNA was isolated from BM or PB mononuclear cells (MNC) using Trizol reagent (Invitrogen Life Technologies, Darmstadt, Germany).
Bisulfite conversion and quantitative DNA methylation analysis (MassARRAY)
Genomic DNA was bisulfite-modified using the EZ DNA methylation kit (Zymo Research, Freiburg, Germany). Primer sequences for PCR amplification are available upon request. The MassArray EpiTYPER assay (Sequenom, Hamburg, Germany) was performed as described before.Citation4 Briefly, the PCR products were transcribed in vitro, cleaved by RNase A and subjected to matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Methylation standards (0%, 20%, 40%, 60%, 80%, and 100% methylated genomic DNA) were used for data normalization.
Reverse-transcriptase quantitative polymerase chain reaction
The QuantiTect Reverse Transcription kit (Qiagen) was used, which included a procedure to remove genomic DNA. Quantitative PCR was performed on a Mastercycler EP Realplex (Eppendorf, Hamburg, Germany) using the ABsolute QPCR SYBR Green reaction mix (Thermo Scientific, Dreieich, Germany). Primer sequences are available on request. HPRT, ACTB and GAPDH were used as internal controls. RT-PCR reactions were performed in triplicate, including no-template controls. Relative expression was calculated using the comparative CT method.
Statistical analysis
The chi-square test was used to determine the statistical significance of a relationship between categorized variables. Nonparametric statistics were used to test continuous variables for differences between 2 subgroups (Mann-Whitney test) or more than 2 subgroups (Kruskal-Wallis test). P values < 0.05 were considered to be statistically significant. The Kaplan-Meier method was used to estimate overall survival probabilities. The 2-sided log-rank test was used to test the equality of survivorship functions in different subgroups. Curves for cumulative incidence of relapse correspond to the cause-specific hazards adjusted for the competing risk, namely transplantation-related mortality. The Gray test was used to compare cumulative incidence curves.
Additional material
Download Zip (702.1 KB)Acknowledgment
Grant support: Deutsche Forschungsgemeinschaft FL 345/4–1 (to CMN and CF), CRC 992-C05 (to CF).
References
- Niemeyer CM, Aricò M, Basso G, Biondi A, Cantù Rajnoldi A, Creutzig U, Haas O, Harbott J, Hasle H, Kerndrup G, et al, European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. Blood 1997; 89:3534 - 43; PMID: 9160658
- Locatelli F, Nöllke P, Zecca M, Korthof E, Lanino E, Peters C, Pession A, Kabisch H, Uderzo C, Bonfim CS, et al, European Working Group on Childhood MDS, European Blood and Marrow Transplantation Group. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 2005; 105:410 - 9; http://dx.doi.org/10.1182/blood-2004-05-1944; PMID: 15353481
- Niemeyer CM, Locatelli F. Chronic myeloproliferative disorders. In: Pui CH, ed. Childhood Leukemias. 3rd Edition. New York: Cambridge University Press; 2012:444-502.
- Olk-Batz C, Poetsch AR, Nöllke P, Claus R, Zucknick M, Sandrock I, Witte T, Strahm B, Hasle H, Zecca M, et al, European Working Group of Myelodysplastic Syndromes in Childhood (EWOG-MDS). Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 2011; 117:4871 - 80; http://dx.doi.org/10.1182/blood-2010-08-298968; PMID: 21406719
- Flotho C, Valcamonica S, Mach-Pascual S, Schmahl G, Corral L, Ritterbach J, Hasle H, Aricò M, Biondi A, Niemeyer CM. RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia 1999; 13:32 - 7; http://dx.doi.org/10.1038/sj.leu.2401240; PMID: 10049057
- Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hählen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 2003; 34:148 - 50; http://dx.doi.org/10.1038/ng1156; PMID: 12717436
- Flotho C, Steinemann D, Mullighan CG, Neale G, Mayer K, Kratz CP, Schlegelberger B, Downing JR, Niemeyer CM. Genome-wide single-nucleotide polymorphism analysis in juvenile myelomonocytic leukemia identifies uniparental disomy surrounding the NF1 locus in cases associated with neurofibromatosis but not in cases with mutant RAS or PTPN11. Oncogene 2007; 26:5816 - 21; http://dx.doi.org/10.1038/sj.onc.1210361; PMID: 17353900
- Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, Mullighan CG, Chen L, Bergstraesser E, Bueso-Ramos CE, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 2009; 114:1859 - 63; http://dx.doi.org/10.1182/blood-2009-01-198416; PMID: 19571318
- Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, Bunda S, Finklestein JZ, Sakamoto KM, Gorr TA, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 2010; 42:794 - 800; http://dx.doi.org/10.1038/ng.641; PMID: 20694012
- Flotho C, Kratz CP, Bergsträsser E, Hasle H, Starý J, Trebo M, van den Heuvel-Eibrink MM, Wójcik D, Zecca M, Locatelli F, et al, European Working Group of Myelodysplastic Syndromes in Childhood. Genotype-phenotype correlation in cases of juvenile myelomonocytic leukemia with clonal RAS mutations. Blood 2008; 111:966 - 7, author reply 967-8; http://dx.doi.org/10.1182/blood-2007-09-111831; PMID: 18182584
- Yoshida N, Yagasaki H, Xu Y, Matsuda K, Yoshimi A, Takahashi Y, Hama A, Nishio N, Muramatsu H, Watanabe N, et al. Correlation of clinical features with the mutational status of GM-CSF signaling pathway-related genes in juvenile myelomonocytic leukemia. Pediatr Res 2009; 65:334 - 40; http://dx.doi.org/10.1203/PDR.0b013e3181961d2a; PMID: 19047918
- Niemeyer CM, Strahm B, Dworzak M, de Moerloose B, Hasle H, Stary J, et al. JMML revisited: role and outcome of hematopoietic stem cell transplantation in subtypes of juvenile myelomonocytic leukemia. Blood 2012; 120:955
- Luna-Fineman S, Shannon KM, Atwater SK, Davis J, Masterson M, Ortega J, Sanders J, Steinherz P, Weinberg V, Lange BJ. Myelodysplastic and myeloproliferative disorders of childhood: a study of 167 patients. Blood 1999; 93:459 - 66; PMID: 9885207
- Lockyer PJ, Kupzig S, Cullen PJ. CAPRI regulates Ca(2+)-dependent inactivation of the Ras-MAPK pathway. Curr Biol 2001; 11:981 - 6; http://dx.doi.org/10.1016/S0960-9822(01)00261-5; PMID: 11448776
- Zhang J, Guo J, Dzhagalov I, He YW. An essential function for the calcium-promoted Ras inactivator in Fcgamma receptor-mediated phagocytosis. Nat Immunol 2005; 6:911 - 9; http://dx.doi.org/10.1038/ni1232; PMID: 16041389
- Kupzig S, Deaconescu D, Bouyoucef D, Walker SA, Liu Q, Polte CL, Daumke O, Ishizaki T, Lockyer PJ, Wittinghofer A, et al. GAP1 family members constitute bifunctional Ras and Rap GTPase-activating proteins. J Biol Chem 2006; 281:9891 - 900; http://dx.doi.org/10.1074/jbc.M512802200; PMID: 16431904
- Yarwood S, Bouyoucef-Cherchalli D, Cullen PJ, Kupzig S. The GAP1 family of GTPase-activating proteins: spatial and temporal regulators of small GTPase signalling. Biochem Soc Trans 2006; 34:846 - 50; http://dx.doi.org/10.1042/BST0340846; PMID: 17052212
- Dai Y, Walker SA, de Vet E, Cook S, Welch HC, Lockyer PJ. Ca2+-dependent monomer and dimer formation switches CAPRI Protein between Ras GTPase-activating protein (GAP) and RapGAP activities. J Biol Chem 2011; 286:19905 - 16; http://dx.doi.org/10.1074/jbc.M110.201301; PMID: 21460216
- Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A 2005; 102:15785 - 90; http://dx.doi.org/10.1073/pnas.0507816102; PMID: 16243968
- Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, Takahashi M, Kon A, Sanada M, Chiba K, Tanaka H, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 2013; 45:937 - 41; http://dx.doi.org/10.1038/ng.2698; PMID: 23832011
- Batz C, Sandrock I, Niemeyer CM, Flotho C. Methylation of the PTEN gene CpG island is infrequent in juvenile myelomonocytic leukemia: Comments on “PTEN deficiency is a common defect in juvenile myelomonocytic leukemia” [Leuk. Res. 2009;33:671-677 (Epub 2008 November 17)]. Leuk Res 2009; 33:1578 - 9, author reply 1580; http://dx.doi.org/10.1016/j.leukres.2009.04.040; PMID: 19487024
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A 2004; 101:6062 - 7; http://dx.doi.org/10.1073/pnas.0400782101; PMID: 15075390
- Hasegawa D, Manabe A, Kubota T, Kawasaki H, Hirose I, Ohtsuka Y, Tsuruta T, Ebihara Y, Goto Y, Zhao XY, et al. Methylation status of the p15 and p16 genes in paediatric myelodysplastic syndrome and juvenile myelomonocytic leukaemia. Br J Haematol 2005; 128:805 - 12; http://dx.doi.org/10.1111/j.1365-2141.2005.05392.x; PMID: 15755284