
Abstract
Cellular identity in both normal and disease processes is determined by programmed epigenetic activation or silencing of specific gene subsets. Here, we have used human cells harboring epigenetically silent GFP-reporter genes to perform a genome-wide siRNA knockdown screen for the identification of cellular factors that are required to maintain epigenetic gene silencing. This unbiased screen interrogated 21,121 genes, and we identified and validated a set of 128 protein factors. This set showed enrichment for functional categories, and protein-protein interactions. Among this set were known epigenetic silencing factors, factors with no previously identified role in epigenetic gene silencing, as well as unstudied factors. The set included non-nuclear factors, for example, components of the integrin-adhesome. A key finding was that the E1 and E2 enzymes of the small ubiquitin-like modifier (SUMO) pathway (SAE1, SAE2/UBA2, UBC9/UBE2I) are essential for maintenance of epigenetic silencing. This work provides the first genome-wide functional view of human factors that mediate epigenetic gene silencing. The screen output identifies novel epigenetic factors, networks, and mechanisms, and provides a set of candidate targets for epigenetic therapy and cellular reprogramming.
Introduction
Epigenetic processes direct heritable changes in gene expression without changes in DNA sequence.Citation1 “Epigenetic gene silencing” describes the transcriptional shutoff of specific genes during development and cellular differentiation.Citation2,Citation3 Such epigenetic regulation can be viewed as a layered process, with external signals initiating the silencing of specific genes.Citation4 At the chromatin level, epigenetic control is mediated by proteins that modify DNA and chromatin, and remodel nucleosomes. Specifically, the active or silent gene states are controlled by enzymatic placement and removal of a complex set of chemical modifications on DNA and histones.Citation5-Citation7 The modifications include DNA methylation, and a variety of post-translational histone modifications (acetylation, methylation, phosphorylation, and others), which are placed by members of large families of enzymes. These chromatin modifications are in turn recognized by protein complexes that ultimately control the active or silent transcriptional gene states.Citation8 Epigenetic gene silencing can be divided into three processes: initiation of gene silencing, maintenance of the repressive chromatin state, and inheritance through S-phase and mitosis.Citation4,Citation9,Citation10 In addition to these processes, recent studies have demonstrated the functional importance of spatial chromatin organization in epigenetic control.Citation11,Citation12
Although the general models for epigenetic control are well supported, numerous complexities in the placement and reading of epigenetic modifications on nucleosomal histones and DNA have been detected: context dependence, combinatorial effects, temporal patterns with respect to gene activity, rapid dynamics, and histone modification-DNA methylation crosstalk.Citation13-Citation16 Such complexities have made it difficult to identify key drivers of epigenetic control. Further, a consequence of these complexities is the potential for misregulation. The epigenetic gene-silent state is of particular interest as errors in placement of epigenetic silencing marks are associated with cancer and other epigenetic diseases.Citation17,Citation18 However, a prominent feature of epigenetic control is reversibility. Unlike fixed genetic errors, epigenetic errors such as occur in cancer may therefore be reversible at the chromatin level. This reversibility led to the concept, now in practice, of “epigenetic therapy,” whereby the reactivation of epigenetically silent tumor suppressor genes may restore growth control in cancer cells.Citation17-Citation21 Currently, the major focus of epigenetic therapy efforts is to develop and refine inhibitors of two families of epigenetic silencing factors, the histone deacetylases (HDACs) and DNA methyltransferases (DNMTs).Citation17-Citation21 There is intense interest in identifying additional therapeutic targets for reactivation of epigenetically silent genes. It is therefore critical to identify the entire complement of human factors and networks that mediate epigenetic gene silencing. While much is known about epigenetic marking systems, there have been no studies that provide an unbiased genome-wide functional view of the epigenetic factors and pathways in human cells. Understanding the entire spectrum of epigenetic mechanisms that regulate gene silencing is important with respect not only to cancer therapeutics, but to fundamental mechanisms of cellular differentiation and reprogramming.22
We have developed and validated a robust high-throughput cell-based GFP reporter system to identify human factors and pathways that maintain epigenetic gene silencing. Here we describe the results of an unbiased genome-wide siRNA screen in which 21,121 human genes were interrogated to reveal roles in epigenetic gene silencing. The set of 128 factors identified in this screen, as described herein, was derived through a combination of biological readout, technical validation, and statistical analyses. The results of this screen demonstrate that siRNA knockdown of numerous individual factors, including known epigenetic repressors, is sufficient for release of epigenetic gene silencing. The factor set showed enrichment for functional categories, known protein-protein interactions, and also included 18 previously uncharacterized factors thereby identifying them as novel epigenetic silencing mediators.
Results
siRNA screen for epigenetic silencing factors and pathways
We previously described a cell-based reporter system that consists of a HeLa cell population harboring epigenetically silenced GFP-reporter genes under control of the CMV promoter.Citation23 This system is based on the principle that epigenetic gene silencing is reversible, and that a silent reporter gene can be reactivated by inhibition of key epigenetic silencing factors (). The reporter cell population harbors chromosomally dispersed, epigenetically silent GFP genes, thus favoring detection of multiple classes of silencing mechanisms, rather than idiosyncratic, locus-specific mechanisms. This reporter cell system was used previously to carry out an siRNA knockdown screen with a biased epigenetic factor set (200 genes/factors).Citation23 The GFP signal after target gene knockdown was used as readout, providing a quantitative measure of reporter gene reactivation (). We showed that it is possible to reactivate silent reporter genes by siRNA-mediated knockdown of epigenetic factors involved directly in gene silencing (e.g., HDACs and DMNTs).Citation23 In the current study, we use this system to perform an unbiased genome-wide, gene-by-gene siRNA screen. The unbiased nature of the screen provided the potential to detect proximal chromatin silencing factors, upstream intracellular pathways, and novel, unstudied epigenetic factors.
Figure 1. A Genome-wide siRNA screen identifies genes involved in epigenetic gene silencing. (A) A GFP reporter-based system for identification of cellular factors involved in epigenetic gene silencing. (B) Outline and summary of the two-step genome-wide siRNA screen. (C) A schematic view of the primary screen (top) and the validation screen (bottom). Bracket over wells indicates placement of alternating positive and negative controls. (D, E) Gene Ontology (GO) analysis of factors identified in the screen: subcellular localization (D) and pathway analysis (E). In Panel E, the colored slices indicate the 67 factors enriched in GO categories, as represented in .
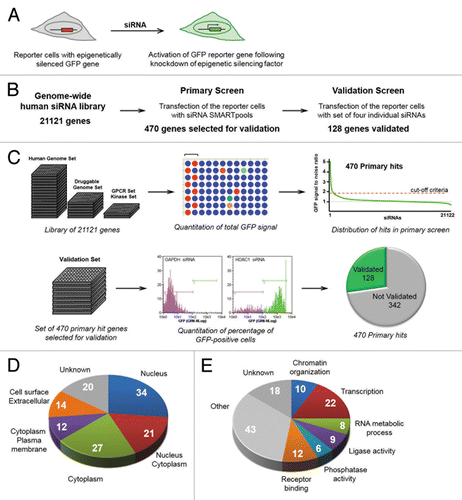
The overall design of the genome-wide screen is outlined in . The Dharmacon siRNA SMARTpool genome-wide library was used, which targets 21,121 human genes. The primary screen consisted of testing each gene/factor individually with siRNA SMARTpools containing a mix of four individual siRNAs. The details of the genome-wide siRNA screening procedure are outlined in , and are described in the Methods section. The assay was optimized for siRNA transfection in a 96-well plate format. SiRNAs targeting GAPDH and HDAC1 were used as negative and positive controls, respectively. The primary screen was performed using the complete genome-wide siRNA library. The reporter cells were transfected with siRNAs, and 96 h post-transfection, the level of GFP reporter gene reactivation was quantitated using a plate reader. Results of the primary genome-wide screen are shown as distribution of the ratio of GFP fluorescence relative to the negative control (). A distinct set of 470 factors with high signal-to-noise ratio were selected for validation (see Methods section for details).
Validation testing of the 470 primary hits was performed using an siRNA library of deconvoluted pools of four single siRNAs per target. Reporter cells were transfected with individual siRNAs and reactivation of GFP reporter genes was measured by quantification of the percentage of GFP-positive cells using flow cytometry. Shown are flow cytometry profiles of GFP signal intensities, and gates, of cells transfected with siRNAs targeting the negative and positive controls, GAPDH and HDAC1, respectively (). A gene/factor was considered validated if at least two individual siRNAs produced GFP reactivation according to the parameters outlined in the Methods section. A pie chart indicates that 128 genes from the 470 primary hit set were validated with two or more individual siRNAs (). The complete distribution of the number of individual siRNAs from each pool of four that promoted reactivation is shown in Figure S1. The overall validation rate for each primary hit fits well with previous studies that used the same siRNA library to study other biological processes.Citation24 Thus, results of the validation step indicate that the overall screen output is of the expected quality for this siRNA library. Table S1 provides comprehensive data from both the primary and validation screens, and Table S2 provides a list of the 128 validated genes with descriptions.
Data analysis and factor hit representation
The set of 128 validated factors were organized according to the Gene Ontology (GO) database, for subcellular localization () and significantly enriched functional categories (). Interestingly, factors in this set showed a broad spectrum of localization within the cell, indicating layers of epigenetic control.Citation4 However, the validated targets were significantly enriched for factors localizing to the nucleus (2.2-fold more than expected by chance alone, P = 0.0002), and such factors are implicated as having a direct role in epigenetic gene silencing. Simple inspection of the gene list revealed that among these nuclear factors were those previously known to play a role in epigenetic gene silencing, including HDAC1, SETDB1/KTM1E, ATF7IP/MCAF, and CHAF1A. Furthermore, several of these factors were detected in our previous biased screenCitation23 (see Discussion). Therefore, the screen output appeared to validate our unbiased approach, as both proximal and upstream factors were detected. We next set out to systematically analyze the screen output.
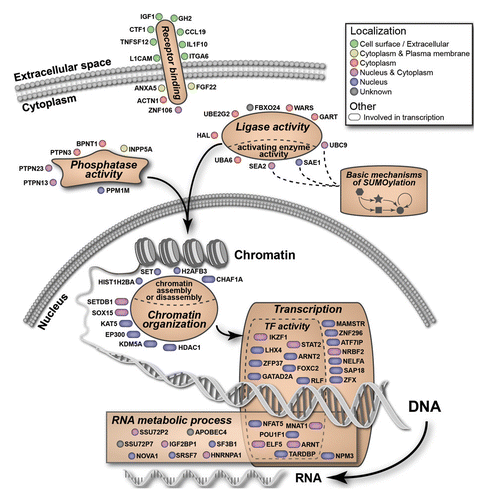
We tested the list of validated targets for statistical enrichment of GO categories and pathways, using the initial 21,121 screened genes/factors as a reference (Table S3). A total of 67 of the 128 factors were members of 6 enriched GO functional categories: “chromatin organization,” “transcription,” “RNA metabolic process,” “ligase activity,” “phosphatase activity,” and “receptor binding” (). The detailed enrichment analysis for the 128 hits is summarized in Table S3. The 67 members of the enriched categories are illustrated in , highlighting subcellular localization and refined annotated functions. In addition to organization based on enriched categories, we performed an unbiased analysis of the 128 factor set using EMBL String,Citation25 a database of known protein interactions. The set was found to be enriched for protein-protein interactions compared with a genome background (P = 0.0001). The network display map is provided in Figure S4, and will be referred to below to highlight specific features of the 128 factor set. In general, the output shows a number of interactions, and features a “core” factor set that includes histone modifiers and factors involved in transcription.
Figure 2. A summary of factors detected in the screen along with their membership in enriched GO categories. See Figure S2 for additional details.
Detection of the SUMO pathway as a mediator of epigenetic gene silencing
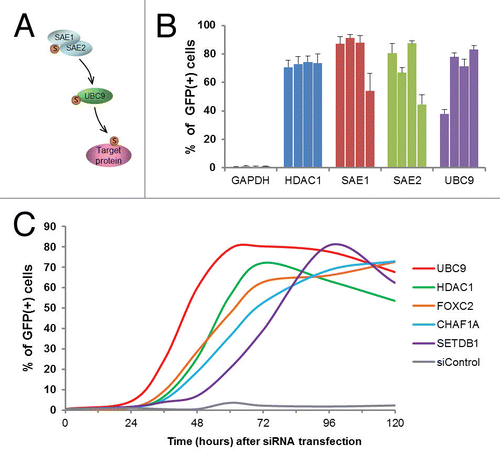
The screen revealed a significant role for the small ubiquitin-like modifier (SUMO) pathway in maintaining epigenetic silencing (, Fig. S4). The SUMO pathway includes an E1-E2-E3 enzymatic cascade similar to the ubiquitin pathway, as well as de-sumoylating enzymes.Citation26 Hundreds of cellular proteins are subject to SUMO modifications, which result in altered activities or localization.Citation27 The screen detected three key sumoylation pathway factors: two comprising the E1 enzyme complex (SAE1 and SAE2/UBA2) and the sole human E2 enzyme, UBC9/UBE2I (). With respect to the representation of SUMO pathway factors within the entire library, the detection of SAE1, SAE2/UBA2, and UBC9/UBE2I represented a greater than 30-fold enrichment (Table S3). Overall, the siRNA pool deconvolution step performed during validation generally revealed a broad distribution in the number of positive siRNAs among the validated pools (i.e., from 2 to 4 siRNAs). Strikingly, all four siRNAs from each of the SAE1, SAE2/UBA2, and UBC9/UBE2I pools were positive (), providing high confidence in the obtained results. As the E1 and E2 enzymes act in cascade, the identified roles of both upstream (SAE1, SAE2/UBA2) and downstream (UBC9/UBE2I) factors provide compelling evidence for an essential role of this pathway in epigenetic gene silencing.
Figure 3. Detection of role for SUMO pathway components in epigenetic gene silencing. (A) A schematic representation of the SUMO modification pathway. (B) Reactivation of GFP reporter genes after knockdown of the indicated SUMO pathway components. The reporter cells were transfected with the indicated siRNAs targeting the SUMO pathway (four individual siRNAs per tested gene), and the percent of GFP-positive cells was measured 96 h post transfection. SiRNA pools targeting GAPDH and HDAC1 were used as quadruplicate negative and positive controls, respectively. Error bars indicate standard deviation, n = 6. (C) A time course of GFP reporter gene reactivation after siRNA knockdown of indicated genes. siControl, negative control siRNA.
Temporal analysis of reactivation after factor knockdown
The roles of factors identified by siRNA knockdown may be either direct, i.e., on chromatin, or through indirect mechanisms, such as upstream triggers of nuclear events. A rapid release of GFP gene silencing after siRNA knockdown could indicate a direct role in the maintenance of epigenetic silencing for the tested factor. Although the biological effect produced by siRNA knockdown is dependent largely on the half-life of the protein after the target mRNA is destroyed, a rapid effect is more readily interpretable. That is, a delay in reactivation could reflect either an indirect role for the factor, or simply the prolonged presence of the protein after mRNA knockdown. In contrast, very rapid reactivation would suggest both efficient depletion of the protein, as well as a more direct role for the factor, i.e., not involving a cascade of events.
To identify factors that might be acting directly to maintain epigenetic silencing, we performed analyses for a set of 20 factors to measure the rapidity of the response by monitoring GFP reactivation profiles (Fig. S3). Shown in are representative GFP reactivation profiles produced after knockdown of selected factors. The factor knockdown profiles extracted from Figure S3 (UBC9, HDAC1, FOXC2, CHAF1A, SETDB1) were chosen to represent members of different functional protein families (SUMO pathway, histone deacetylase, transcription factors, chromatin remodeling factors and histone methyltransferase, respectively). The knockdown of UBC9/UBE2I resulted in the most rapid reactivation of the GFP reporter gene among the 20 factors tested, with a maximum reached at 56 h post-siRNA addition (, Fig. S3). In contrast, treatment with HDAC1 siRNA, targeting a known “direct” chromatin silencing factor, showed a GFP maximum at 72 h. Therefore, the very rapid reactivation of GFP genes after UBC9/UBE2I knockdown suggests a direct role for this protein in epigenetic gene silencing (see Discussion). Regardless of the difficulties in interpreting these results at the mechanistic level (as outlined above), these temporal analyses provide important relative measurements of the potency of factor knockdown.
Reactivation of epigenetically silent cellular genes
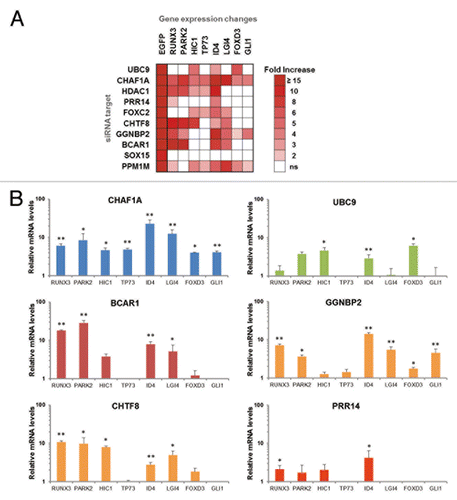
We next investigated whether factors identified in the GFP reactivation screen function in epigenetic silencing of cellular genes. Eight developmental and tumor suppressor genes that are epigenetically repressed in HeLa cells were tested for reactivation after knockdown of a functionally diverse group of 10 factors from the 128 factor set (). HeLa GFP reporter cells were transfected with the indicated siRNAs and, at 72 h posttransfection, mRNA levels were analyzed by qRT-PCR. shows a heat map of genes that displayed an increase in mRNA expression, with the reactivation criteria being 2-fold or greater (with a p-value < 0.05) compared with the siRNA negative control. GFP mRNA reactivation was monitored as a positive control. Knockdown of CHAF1A resulted in reactivation of all eight cellular genes tested (). CHAF1A functions in the inheritance of repressive chromatin marks through S-phase,Citation28 and was detected in our earlier biased siRNA screen using the same reporter cells.Citation23 Of the ten factors tested, only SOX15 knockdown failed to reactivate any of the cellular genes. Knockdown of other factors resulted in reactivation of at least four genes, but in each case, a unique set was reactivated. These results suggest that the screen identified epigenetic silencing factors that act through distinct mechanisms. Notably, UBC9/UBE2I knockdown resulted in reactivation of only several genes, implying that the role of the SUMO pathway in epigenetic silencing is not universal ().
Figure 4. Knockdown of factors identified in the genome-wide screen results in reactivation of epigenetically silent cellular genes. (A) GFP-silent reporter cells were transfected with selected siRNAs. Levels of the indicated cellular mRNAs were measured 72 h post siRNA transfection by real-time PCR. The heat map shows the fold-increase relative to siControl-transfected cells, P < 0.05. (B) RNA levels of selected cellular genes after transfection with indicated siRNAs. Error bars indicate standard deviation, n = 3, with *p-value < 0.05 and **p-value < 0.01.
Discussion
Here we describe the results of a functional screen to identify human factors that maintain the epigenetically silent gene state. Our approach was built upon studies from our lab and others, which showed that epigenetically silent reporter genes could be reactivated by inhibitors of HDACs and DNMTs.Citation23,Citation29,Citation30 These findings led to the development of a system in which silent GFP reporter genes could be reactivated in response to siRNA-based depletion of single factors. This system was then used to identify specific epigenetic silencing factors. Our initial biased siRNA screen targeting 200 candidates, identified a core set of 15 chromatin-based epigenetic silencing factors, each of which was critical to maintain the silent state.Citation23 Here, we describe the results of an unbiased, genome-wide siRNA screen, which revealed novel epigenetic silencing factors and pathways. Using the Dharmacon siGENOME library, we identified 128 factors (, Fig. S2, S4) that included epigenetic silencing factors identified in our earlier biased 200-factor screen (CHAF1A, HDAC1, SETDB1/KMT1E, and others).
The essence of gene-by-gene siRNA screens is that knockdown of a single factor produces the tested phenotype. In the case of epigenetic silencing factors, this is of critical importance, as such single factors could thereby serve as potential targets for epigenetic therapy or cellular reprogramming. It is likely that our screen did not detect many factors that contribute to epigenetic silencing due to redundancies in their functions or the need for combinational depletion. We suggest that factors identified in this screen, are thereby non-redundant or serve as cofactors or scaffolds for other factors.
Bioinformatics analyses showed that 67 factors of the 128 factor set are members of enriched GO functional categories (, Fig. S2, and Table S3). Among these, we identified ten factors in the category of “chromatin organization.” Included are enzymes, for example, histone modifiers that place or remove of histone marks (e.g., acetylation and methylation; HDAC1, JARID1A/KDM5A, SETDB1/KMT1E, EP300), as well as factors from the “chromatin assembly and disassembly” category (CHAF1A, SET). Within this group of ten factors, we highlight several that have been identified earlier as potent epigenetic repressors: SETDB1/KMT1E is a histone methyltransferase that mediates placement of the repressive histone mark, histone 3 lysine 9 di- and tri-methyl (H3K9me2/3);Citation31 HDAC1 is a well described histone deacetylase that responsible for removing histone acetylation thus maintaining the chromatin repressive state;Citation32 the JARID1A/KDM5A is a histone lysine demethylase that removes the histone H3K4me3 mark that serves as a mark of active promoters.Citation33 In contrast to these histone modifiers, CHAF1A is a histone chaperone that forms a complex with SETDB1/KMT1E to properly maintain the H3K9me2/3 repressive mark during chromatin replication.Citation28 An unexpected hit in the screen was EP300 (p300), which is a prominent histone acetyltransferase (HAT) enzyme associated with enhancers and active genes. We speculate that the detection of p300 may indicate a silencing role, as previously described,Citation34 and this interpretation is consistent with the detection of SUMO pathway components as hits in the screen.Citation34 Collectively, the factors described above are responsible for maintenance of epigenetic histone marks during interphase (e.g., HDAC1), and also for proper inheritance of such marks during chromatin replication in S-phase and thereby in daughter cells (CHAF1A).
Two other enriched GO functional categories identified were “transcriptional regulation” and “RNA metabolic process.” It is not unexpected that transcription-related factors may act as epigenetic repressors, for example, by recruiting silencing factors.Citation35 ATF7IP/MCAF1, a member of the transcriptional regulation group, is required for placement of the repressive H3K9me3 modification in its role as a binding partner and cofactor for the histone modifying enzyme SETBD1/KMT1E, a factor also detected in the screen. SAP18 and HDAC1 are components of the Sin3 complex,Citation36 and knockdown of SAP18 was shown to activate silent cellular genes.Citation37 More unusual and unexpected roles for transcription factors in epigenetic gene silencing are possible. For example, the transcription factor ThPOK/cKrox was recently reported to promote gene silencing through its function in chromatin organization.Citation38
Beyond the direct roles for epigenetic silencing factors at the local chromatin level, we identified potential “upstream” GO functional categories: “receptor binding,” “phosphatase activity” and “ligase activity” (for the ubiquitin-like family of protein modifiers). The latter two groups contain members that may modulate epigenetic silencing factor activity through posttranslational modifications. One of the most striking findings of the screen was the identification of a role for the core enzymes of the SUMO modification pathway (SAE1, SAE2/UBA2, UBC9/UBE2I) (, Fig. S4). In general, SUMO modifications may function to modulate protein subcellular localization, protein-protein interactions, or affect other secondary protein modifications.Citation27 Consequently, there are multiple mechanisms through which the SUMO pathway could be involved in maintenance of epigenetic gene silencing. Human HDAC1 has been shown to be sumoylated, and is a direct substrate for the E2 enzyme UBC9/UBE2I.Citation39 Furthermore, sumoylation has been shown to increase HDAC1 activity.Citation39 Our previous work,Citation23 and results described here, demonstrate that HDAC1 is a key factor in epigenetic silencing in our system. Sumoylation of HDAC1 may therefore contribute to epigenetic silencing, and knockdown of factors participating in the sumoylation pathway (i.e., SAE1, SAE2/UBA2, UBC9/UBE2I) would result in loss of epigenetic silencing. To further test this idea, we used the LLO peptide of the bacterium Listeria monocytogenes, which has been reported to inhibit the sumoylation pathway by inducing degradation of UBC9/UBE2I.Citation40 Treatment of the GFP reporter cells with the LLO peptide resulted in robust reactivation of the GFP reporter (data not shown). Thus, interruption of the SUMO modification pathway by siRNA knockdown of enzymes, or by microbial protein-induced degradation, both lead to reactivation.
Methyl-binding domain protein 1 (MBD1) sumoylation has also been implicated in epigenetic silencing through promoting interactions with the SETDB1/KMT1E cofactor ATF7IP/MCAF1. Both MBD1 and ATF7IP/MCAF1 were identified as epigenetic repressors previously,Citation41,Citation42 as well as in the current study. Interruption of sumoylation has been shown to cause the dissociation of ATF7IP/MCAF1 and heterochromatin protein 1 (HP1) from MBD1-containing foci, along with the loss of the H3K9me3 heterochromatin mark.Citation43 Lastly, it has been reported that histone H4 is a direct target for sumoylation by UBC9/UBE2I, and this modification results in a repressive chromatin state.Citation44,Citation45 The rapid reactivation of the silent GFP gene after UBC9/UBE2I knockdown () implicates a relatively direct role of the SUMO pathway in silencing in this system.
Of the 128 factor set, eight (ARNT, C20orf112, EP300, IKZF1, LHX4, POU1F1, SEPT9, SET) are encoded by genes that have been associated with recurrent chromosomal aberrations in cancer according to the Mitelman Database (Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (2014). Mitelman F, Johansson B and Mertens F (eds.), http://cgap.nci.nih.gov/Chromosomes/Mitelman). The enrichment for this class of genes is 1.9-fold (P = 0.08) among the hits, and detailed descriptions of the eight genes are provided in Table S2. Of note, the C20orf112 gene encodes a 436 amino acid protein with no known function. A fusion between the C20orf112 and RUNX1 genes has been identified in acute myeloid leukemia (AML), and a PAX5-C20orf112 gene fusion was detected in acute lymphoblastic leukemia (ALL). IKZF1 is a widely expressed transcriptional regulator, interacts with HDAC1, and functions in lymphoid development.Citation46 The IKZF1 gene promoter is translocated to the BCL6 locus in B-cell non-Hodgkin lymphoma, and the IKZF1 gene is deleted in other lymphoid diseases.Citation47 The SEPT9 gene encodes a member of the septin gene family of filamentous structural proteins.Citation48 The SEPT9 gene is involved in a translocation with the MLL gene in myelodysplastic syndrome (MDS).Citation49 Lastly, the screen identified SET, a known epigenetic silencing factor that appears to function by blocking acetylation of histones.Citation50 A SET-NUP214 translocation has been identified in both AML and ALL.Citation51 Whether the putative epigenetic silencing roles for these eight factors are related to the described gene aberrations remains to be investigated.
The unbiased nature of the siRNA screen led to the detection of non-nuclear factors that are candidates for having indirect roles in epigenetic gene silencing. In particular, factors that participate in the integrin-adhesome complex were detected (, Fig. S4). Transmembrane integrin molecules on the cell surface attach to the extracellular matrix (ECM), and the integrin-adhesion complexes mediate associations with the actin cytoskeleton. One function of adhesions is to sense mechanical stimuli in the cellular environment and translate these stimuli into a broad range of responses, including changes in cell shape or cellular identity. These external stimuli may affect gene expression via physical connectivity between the ECM and chromatin, through the cytoskeleton and the nuclear lamina.Citation52 Our screen detected five factors that are components of the integrin-adhesome complex (ACTN1, BCAR1, ITGA3, ITGA6, PPM1M),Citation53 representing a 1.93-fold enrichment (P = 0.19) (Fig. S4). More strikingly, 2 of the 18 human α integrins, α3 and α6 (encoded by the ITGA3 and ITGA6 genes, respectively) were detected in the screen. These results suggest that loss of integrin signaling can lead to reversal of epigenetic gene silencing in this system.
Implicit in the findings described here is that our reporter system is able to detect a wide range of cellular factors that impact epigenetic gene silencing. Clearly, the protein set includes general factors, rather than those unique for maintaining silencing of the CMV promoter. Indeed, several core cellular mechanisms have been shown previously to contribute to CMV promoter silencing, including histone modification and DNA methylation.Citation54 Our initial biased siRNA screen had detected roles for the histone modifying enzyme SETDB1/KMT1E and DNMT3A,Citation23 and these two factors were also detected in the current screen among the primary hits. As DNMT3A had been detected in the earlier screen,Citation23 it was not included for validation in the current study. Our experience with this reporter system is that detection of DNA methylation-based silencing is less robust, presumably due to the requirement for passive loss of DNA methylation after knockdown of DNMTs.Citation55 The focus of our current study was to use an unbiased approach to extend our understanding beyond the core silencing mechanisms. As such, we have not pursued technical improvements in the reporter system that might enhance the detection of components of the DNA methylation machinery in particular.
This screen also revealed potential roles in epigenetic silencing for previously uncharacterized or poorly characterized factors. We have investigated in detail the function of the factor, PRR14.Citation56 It was shown recently that the nuclear lamina plays an important role in maintaining an epigenetically silent heterochromatin compartment at the nuclear periphery.Citation57,Citation58 We demonstrated that PRR14 functions to tether heterochromatin to nuclear lamina, thus maintaining the peripheral nuclear heterochromatin compartment. Also, we showed that PRR14 may play a role in mitotic specification of H3K9me3-marked heterochromatin positioning at the nuclear lamina.Citation56,Citation59Interestingly, PRR14 had not been identified as a component of the nuclear lamina or heterochromatin using other techniques, but was revealed in our functional screen for epigenetic silencing factors. To date, 18 factors detected in the screen remain uncharacterized (Table S4). Further study of these factors could provide new insights into mechanisms of epigenetic gene silencing. For example, detection of the PRR14 nuclear lamina component along with integrin-adhesion factors suggests a functional linkage of the ECM to chromatin, via the nuclear lamina. This potential mechanism remains to be investigated in greater detail.
In summary, the findings described here have provided a broad functional view of human epigenetic silencing factors. The screen identified known, potent epigenetic repressors, as well as other factors not previously connected with epigenetic gene silencing. In addition, a set of uncharacterized factors was detected. We showed that one such factor, PRR14, functions in the maintenance of heterochromatin.Citation56 Future work may identify roles in biological processes, such as developmental gene regulation for novel factors discovered here, while other factors might be targetable for epigenetic therapy.
Methods
Reporter cells
The HeLa GFP-reporter cell line used for the screen was described previously.Citation23,Citation60 Cells were grown in 10% fetal bovine serum (FBS) Dulbecco's modified Eagle's medium (DMEM) media supplemented with antibiotics.
The high-throughput siRNA-based screen
The gene-by-gene high-throughput screening assay was designed using a robotic pipetting system for siRNA transfection, optimized in a 96-well format, with multiple readout options for measuring GFP expression. The transfection volumes and cell number were chosen according to the multi-well plate format using the manufacturer's recommendations. SiRNA and the DharmaFECT 1 (T-2001) transfection reagent were diluted in Hank's Balanced Salt Solution (HBSS), mixed, and then incubated for 20 min. Robotic pipetting was performed using the CyBi Well Vario (CyBio) liquid handler. A transfection mix containing siRNAs and transfection reagent was first dispensed into 96-well plates, followed by addition of the reporter cell suspension in complete medium without antibiotics (wet reverse transfection method). The final concentration of siRNA was 50 nM in 100 µl of the transfection solution, with a cell number of 5,000 per well. After 48 h, the transfection medium was replaced with complete growth medium, and cells were incubated for an additional 48 h. After a total of 96 h post-transfection cells were analyzed for reporter GFP gene reactivation as described below.
Data analysis
For the primary screen, data were analyzed as the ratio of sample signal to averaged negative and positive controls of the tested plate, signal-to-noise and signal-to-positive, respectively. Criterion for a positive candidate hit gene were signal to noise ratio greater than 1.8 (S/n > 1.8) and signal to positive ratio greater than 0.8 (S/P > 0.8). A total of 470 candidate genes were selected, excluding genes disconnected from the NCBI Reference Sequence (RefSeq) Database. The selected 470 candidate genes were validated with additional siRNA library containing four individual siRNAs per target genes (deconvoluted siRNA SMARTpools, Dharmacon). The validation screen data were analyzed as percentages of GFP-positive cells after transfection with single individual siRNAs. Raw values (percent GFP positive cells) for each single siRNA were averaged, and the standard deviation was calculated. A criterion for a positive single siRNA was an amount of GFP-positive cells greater than or equal to 20% after siRNA transfection with a coefficient of variation (%CV) less than 35%. This criterion was an equivalent of a fold-difference of at least 10 (F.D. > 10, P < 0.001) relative to the negative control. The background level of GFP-positive cells in negative control samples was 0.5–2.0% for the entire screen. A candidate gene was considered as validated if at least 2 out of 4 tested individual siRNAs were scored as positive. A total of 128 candidate genes were validated using these criteria. See Table S1 for overall screening results.
Bioinformatics analysis of validated hit genes
The list of 128 genes was analyzed for enrichment of Gene Ontology (GO) terms and pathways (from KEGG and BIOCARTA databases) using DAVID software.Citation61 Only results with a false discovery rate (FDR) < 20%, and enriched at least 1.5-fold, were considered significant. All significant categories are shown in Table S3 with hand-picked non-redundant categories demonstrated on the composite and S2. Any additional gene annotations were added manually using literature searching and the Genecards (www.genecards.org) database. Annotation with subcellular localization was performed using the Genecards database and GO terms. The expression of the 128 identified hit genes in HeLa cells was checked using mRNA-seq data from the Morin et al. studyCitation62 and publicly available gene expression data from the study of Zhang et al.Citation63 obtained from GEO database (data set accession GDS3581, sample GSM410912). A gene was called as expressed in HeLa cells if it had coverage of more than three tags from mRNA-seq or more than two background levels from gene expression data. A total of 108 of 128 genes were detected by one of those two platforms (see Table S2).
| Abbreviations: | ||
| GFP | = | Green Fluorescent Protein |
| SUMO | = | small ubiquitin-like modifier |
| HDAC | = | histone deacetylase |
| DNMT | = | DNA methyltransferase |
| CMV | = | Cytomegalovirus |
| siRNA | = | small interfering RNA |
| GO | = | Gene Ontology |
| AML | = | acute myeloid leukemia |
| ALL | = | acute lymphoblastic leukemia |
| MDS | = | myelodysplastic syndrome |
| FBS | = | fetal bovine serum |
| DMEM | = | Dulbecco's modified Eagle's medium |
| ECM | = | extracellular matrix |
| HBSS | = | Hank's Balanced Salt Solution |
| HAT | = | Histone acetyltransferase |
Additional material
Download Zip (5.6 MB)Acknowledgments
We thank Eti Cukierman and Christoph Seeger for critical comments, and Marie Estes for assistance in preparing this manuscript. The Fox Chase Cancer Center Translational Research and Cell Sorting Facilities were used for this work. This work was supported in whole or in part by National Institutes of Health grants: Roadmap Epigenomics DK082498 (R.A.K.), CA071515, and CA006927. This project was also funded in part, under a grant with the Pennsylvania Department of Health. The Department specifically disclaims responsibility for any analyses, interpretations, or conclusions. This work was also supported by the Fox Chase Cancer Center Keystone Program in Epigenetics and Progenitor Cells. A.P. was a recipient of an AACR Centennial Predoctoral Fellowship in Cancer Research.
References
- Holliday R. Epigenetics: a historical overview. Epigenetics 2006; 1:76 - 80; http://dx.doi.org/10.4161/epi.1.2.2762; PMID: 17998809
- Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol 2010; 28:1079 - 88; http://dx.doi.org/10.1038/nbt.1684; PMID: 20944600
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet 2013; 14:204 - 20; http://dx.doi.org/10.1038/nrg3354; PMID: 23400093
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev 2009; 23:781 - 3; http://dx.doi.org/10.1101/gad.1787609; PMID: 19339683
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21:381 - 95; http://dx.doi.org/10.1038/cr.2011.22; PMID: 21321607
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 2006; 31:89 - 97; http://dx.doi.org/10.1016/j.tibs.2005.12.008; PMID: 16403636
- Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013; 502:472 - 9; http://dx.doi.org/10.1038/nature12750; PMID: 24153300
- Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta 2014; 1839:627 - 43; http://dx.doi.org/10.1016/j.bbagrm.2014.03.001; PMID: 24631868
- Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science 2010; 330:612 - 6; http://dx.doi.org/10.1126/science.1191078; PMID: 21030644
- Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol 2009; 10:192 - 206; http://dx.doi.org/10.1038/nrm2640; PMID: 19234478
- Bickmore WA, van Steensel B. Genome architecture: domain organization of interphase chromosomes. Cell 2013; 152:1270 - 84; http://dx.doi.org/10.1016/j.cell.2013.02.001; PMID: 23498936
- Collas P, Lund EG, Oldenburg AR. Closing the (nuclear) envelope on the genome: how nuclear lamins interact with promoters and modulate gene expression. Bioessays 2014; 36:75 - 83; http://dx.doi.org/10.1002/bies.201300138; PMID: 24272858
- Berger SL. The complex language of chromatin regulation during transcription. Nature 2007; 447:407 - 12; http://dx.doi.org/10.1038/nature05915; PMID: 17522673
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10:295 - 304; http://dx.doi.org/10.1038/nrg2540; PMID: 19308066
- Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol 2010; 28:817 - 25; http://dx.doi.org/10.1038/nbt.1662; PMID: 20657582
- Gardner KE, Allis CD, Strahl BD. Operating on chromatin, a colorful language where context matters. J Mol Biol 2011; 409:36 - 46; http://dx.doi.org/10.1016/j.jmb.2011.01.040; PMID: 21272588
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 92; http://dx.doi.org/10.1016/j.cell.2007.01.029; PMID: 17320506
- Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin 2010; 60:376 - 92; http://dx.doi.org/10.3322/caac.20085; PMID: 20959400
- Boumber Y, Issa JP. Epigenetics in cancer: what’s the future?. Oncology (Williston Park) 2011; 25:220 - 6, 228; PMID: 21548464
- Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs 2010; 19:1049 - 66; http://dx.doi.org/10.1517/13543784.2010.510514; PMID: 20687783
- Santos FP, Kantarjian H, Garcia-Manero G, Issa JP, Ravandi F. Decitabine in the treatment of myelodysplastic syndromes. Expert Rev Anticancer Ther 2010; 10:9 - 22; http://dx.doi.org/10.1016/j.stem.2009.03.005; PMID: 19341620
- Feng B, Ng JH, Heng JC, Ng HH. Molecules that promote or enhance reprogramming of somatic cells to induced pluripotent stem cells. Cell Stem Cell 2009; 4:301 - 12; http://dx.doi.org/10.1016/j.stem.2009.03.005; PMID: 19341620
- Poleshko A, Einarson MB, Shalginskikh N, Zhang R, Adams PD, Skalka AM, Katz RA. Identification of a functional network of human epigenetic silencing factors. J Biol Chem 2010; 285:422 - 33; http://dx.doi.org/10.1074/jbc.M109.064667; PMID: 19880521
- Tsui M, Xie T, Orth JD, Carpenter AE, Rudnicki S, Kim S, Shamu CE, Mitchison TJ. An intermittent live cell imaging screen for siRNA enhancers and suppressors of a kinesin-5 inhibitor. PLoS One 2009; 4:e7339; http://dx.doi.org/10.1371/journal.pone.0007339; PMID: 19802393
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013; 41:D808 - 15; http://dx.doi.org/10.1093/nar/gks1094; PMID: 23203871
- Meulmeester E, Melchior F. Cell biology: SUMO. Nature 2008; 452:709 - 11; http://dx.doi.org/10.1038/452709a; PMID: 18401402
- Wilkinson KA, Henley JM. Mechanisms, regulation and consequences of protein SUMOylation. Biochem J 2010; 428:133 - 45; http://dx.doi.org/10.1042/BJ20100158; PMID: 20462400
- Sarraf SA, Stancheva I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell 2004; 15:595 - 605; http://dx.doi.org/10.1016/j.molcel.2004.06.043; PMID: 15327775
- Raynal NJ, Si J, Taby RF, Gharibyan V, Ahmed S, Jelinek J, Estécio MR, Issa JP. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res 2012; 72:1170 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-11-3248; PMID: 22219169
- Si J, Boumber YA, Shu J, Qin T, Ahmed S, He R, Jelinek J, Issa JP. Chromatin remodeling is required for gene reactivation after decitabine-mediated DNA hypomethylation. Cancer Res 2010; 70:6968 - 77; http://dx.doi.org/10.1158/0008-5472.CAN-09-4474; PMID: 20713525
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ 3rd. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 2002; 16:919 - 32; http://dx.doi.org/10.1101/gad.973302; PMID: 11959841
- Segré CV, Chiocca S. Regulating the regulators: the post-translational code of class I HDAC1 and HDAC2. J Biomed Biotechnol 2011; 2011:690848; http://dx.doi.org/10.1155/2011/690848; PMID: 21197454
- Seward DJ, Cubberley G, Kim S, Schonewald M, Zhang L, Tripet B, Bentley DL. Demethylation of trimethylated histone H3 Lys4 in vivo by JARID1 JmjC proteins. Nat Struct Mol Biol 2007; 14:240 - 2; http://dx.doi.org/10.1038/nsmb1200; PMID: 17310255
- Girdwood D, Bumpass D, Vaughan OA, Thain A, Anderson LA, Snowden AW, Garcia-Wilson E, Perkins ND, Hay RT. P300 transcriptional repression is mediated by SUMO modification. Mol Cell 2003; 11:1043 - 54; http://dx.doi.org/10.1016/S1097-2765(03)00141-2; PMID: 12718889
- Ashburner BP, Westerheide SD, Baldwin AS Jr.. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol 2001; 21:7065 - 77; http://dx.doi.org/10.1128/MCB.21.20.7065-7077.2001; PMID: 11564889
- Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, Reinberg D. Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell 1997; 89:357 - 64; http://dx.doi.org/10.1016/S0092-8674(00)80216-0; PMID: 9150135
- Matyash A, Singh N, Hanes SD, Urlaub H, Jäckle H. SAP18 promotes Krüppel-dependent transcriptional repression by enhancer-specific histone deacetylation. J Biol Chem 2009; 284:3012 - 20; http://dx.doi.org/10.1074/jbc.M806163200; PMID: 19049982
- Zullo JM, Demarco IA, Piqué-Regi R, Gaffney DJ, Epstein CB, Spooner CJ, Luperchio TR, Bernstein BE, Pritchard JK, Reddy KL, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 2012; 149:1474 - 87; http://dx.doi.org/10.1016/j.cell.2012.04.035; PMID: 22726435
- David G, Neptune MA, DePinho RA. SUMO-1 modification of histone deacetylase 1 (HDAC1) modulates its biological activities. J Biol Chem 2002; 277:23658 - 63; http://dx.doi.org/10.1074/jbc.M203690200; PMID: 11960997
- Ribet D, Hamon M, Gouin E, Nahori MA, Impens F, Neyret-Kahn H, Gevaert K, Vandekerckhove J, Dejean A, Cossart P. Listeria monocytogenes impairs SUMOylation for efficient infection. Nature 2010; 464:1192 - 5; http://dx.doi.org/10.1038/nature08963; PMID: 20414307
- Fujita N, Watanabe S, Ichimura T, Tsuruzoe S, Shinkai Y, Tachibana M, Chiba T, Nakao M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J Biol Chem 2003; 278:24132 - 8; http://dx.doi.org/10.1074/jbc.M302283200; PMID: 12711603
- Uchimura Y, Ichimura T, Uwada J, Tachibana T, Sugahara S, Nakao M, Saitoh H. Involvement of SUMO modification in MBD1- and MCAF1-mediated heterochromatin formation. J Biol Chem 2006; 281:23180 - 90; http://dx.doi.org/10.1074/jbc.M602280200; PMID: 16757475
- Ichimura T, Watanabe S, Sakamoto Y, Aoto T, Fujita N, Nakao M. Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J Biol Chem 2005; 280:13928 - 35; http://dx.doi.org/10.1074/jbc.M413654200; PMID: 15691849
- Nathan D, Ingvarsdottir K, Sterner DE, Bylebyl GR, Dokmanovic M, Dorsey JA, Whelan KA, Krsmanovic M, Lane WS, Meluh PB, et al. Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev 2006; 20:966 - 76; http://dx.doi.org/10.1101/gad.1404206; PMID: 16598039
- Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A 2003; 100:13225 - 30; http://dx.doi.org/10.1073/pnas.1735528100; PMID: 14578449
- Nagpal K, Watanabe KS, Tsao BP, Tsokos GC. Ikaros represses protein phosphatase 2A (PP2A) expression through an intronic binding site. J Biol Chem 2014; In press http://dx.doi.org/10.1074/jbc.M114.558197; PMID: 24692537
- Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, et al, Children’s Oncology Group. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009; 360:470 - 80; http://dx.doi.org/10.1056/NEJMoa0808253; PMID: 19129520
- Peterson EA, Petty EM. Conquering the complex world of human septins: implications for health and disease. Clin Genet 2010; 77:511 - 24; http://dx.doi.org/10.1111/j.1399-0004.2010.01392.x; PMID: 20236126
- Cerveira N, Bizarro S, Teixeira MR. MLL-SEPTIN gene fusions in hematological malignancies. Biol Chem 2011; 392:713 - 24; http://dx.doi.org/10.1515/BC.2011.072; PMID: 21714766
- Cervoni N, Detich N, Seo SB, Chakravarti D, Szyf M. The oncoprotein Set/TAF-1beta, an inhibitor of histone acetyltransferase, inhibits active demethylation of DNA, integrating DNA methylation and transcriptional silencing. J Biol Chem 2002; 277:25026 - 31; http://dx.doi.org/10.1074/jbc.M202256200; PMID: 11978794
- Quentmeier H, Schneider B, Röhrs S, Romani J, Zaborski M, Macleod RA, Drexler HG. SET-NUP214 fusion in acute myeloid leukemia- and T-cell acute lymphoblastic leukemia-derived cell lines. J Hematol Oncol 2009; 2:3; http://dx.doi.org/10.1186/1756-8722-2-3; PMID: 19166587
- Isermann P, Lammerding J. Nuclear mechanics and mechanotransduction in health and disease. Curr Biol 2013; 23:R1113 - 21; http://dx.doi.org/10.1016/j.cub.2013.11.009; PMID: 24355792
- Winograd-Katz SE, Fässler R, Geiger B, Legate KR. The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol 2014; 15:273 - 88; http://dx.doi.org/10.1038/nrm3769; PMID: 24651544
- Mehta AK, Majumdar SS, Alam P, Gulati N, Brahmachari V. Epigenetic regulation of cytomegalovirus major immediate-early promoter activity in transgenic mice. Gene 2009; 428:20 - 4; http://dx.doi.org/10.1016/j.gene.2008.09.033; PMID: 18976699
- Poleshko A, Shalginskikh N, Katz RA. Identification of epigenetic silencing factors by siRNA screening. In Epigenomics: From Chromatin Biology to Therapeutics, edited by K. Appasani (Cambridge University Press, New York, 2012), Chap. 3.
- Poleshko A, Mansfield KM, Burlingame CC, Andrake MD, Shah NR, Katz RA. The human protein PRR14 tethers heterochromatin to the nuclear lamina during interphase and mitotic exit. Cell Rep 2013; 5:292 - 301; http://dx.doi.org/10.1016/j.celrep.2013.09.024; PMID: 24209742
- Reddy KL, Zullo JM, Bertolino E, Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 2008; 452:243 - 7; http://dx.doi.org/10.1038/nature06727; PMID: 18272965
- Shevelyov YY, Nurminsky DI. The nuclear lamina as a gene-silencing hub. Curr Issues Mol Biol 2012; 14:27 - 38; PMID: 21795760
- Poleshko A, Katz RA. Specifying peripheral heterochromatin during nuclear lamina reassembly. Nucleus 2014; 5:32 - 9; http://dx.doi.org/10.4161/nucl.28167; PMID: 24637393
- Poleshko A, Palagin I, Zhang R, Boimel P, Castagna C, Adams PD, Skalka AM, Katz RA. Identification of cellular proteins that maintain retroviral epigenetic silencing: evidence for an antiviral response. J Virol 2008; 82:2313 - 23; http://dx.doi.org/10.1128/JVI.01882-07; PMID: 18094192
- Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44 - 57; http://dx.doi.org/10.1038/nprot.2008.211; PMID: 19131956
- Morin R, Bainbridge M, Fejes A, Hirst M, Krzywinski M, Pugh T, McDonald H, Varhol R, Jones S, Marra M. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. Biotechniques 2008; 45:81 - 94; http://dx.doi.org/10.2144/000112900; PMID: 18611170
- Zhang Z, Meng T, He J, Li M, Tong LJ, Xiong B, Lin L, Shen J, Miao ZH, Ding J. MT7, a novel compound from a combinatorial library, arrests mitosis via inhibiting the polymerization of microtubules. Invest New Drugs 2010; 28:715 - 28; http://dx.doi.org/10.1007/s10637-009-9303-z; PMID: 19705064