Abstract
We performed a genome-wide analysis of aberrant DNA methylation in chronic lymphocytic leukemia (CLL) using methylated CpG island amplification (MCA) coupled with a promoter microarray. We identified 280 potential targets of aberrant DNA methylation in CLL. These genes were located more frequently in chromosomes 19 (16%, p=0.001), 16 (11%, p=0.001), 17 (10%, p=0.02) and 11 (9%, p=0.02) and could be grouped in several functional networks. Methylation status was confirmed for 22 of these genes (SOX11, DLX1, FAM62C, SOX14, RSPO1, ADCY5, HAND2,SPOCK, MLL, ING1, PRIMA1, BCL11B, LTBP2, BNC1, NR2F2, SALL1, GALGT2, LHX1, DLX4, KLK10, TFAP2 and APP) in 78 CLL patients by pyrosequencing. As a proof of principle, we analyzed the expression of 2 genes, PRIMA1 and APP, in primary cells and of GALGT2, TFAP2C and PRIMA1 in leukemia cells. There was an inverse association between methylation and gene expression. This could be reversed by treatment with 5-aza-2’-deoxycytidine in cell lines. Treatment in a clinical trial with 5-azacitidine resulted in decreased methylation of LINE, DLX4 and SALL1 in the peripheral blood B-cells of patients with CLL. IgVH mutational status or ZAP-70 expression were not associated with specific methylation profiles. By multivariate analysis, methylation of LINE and APP was associated with shorter overall survival (p = 0.045 and 0.0035, respectively). This study demonstrates that aberrant DNA methylation is common and has potential prognostic and therapeutic value in CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is the result of clonal expansion of malignant B cells.Citation1,Citation2 In general, patients with CLL without indication for therapy are observed without treatment until disease progression. This is due in part to lack of molecular markers that predict clinical benefit for early therapeutic intervention. Over the last decade, research in CLL has resulted in multiple significant advances. These include the development of several new therapeutic agents, including nucleoside analogues,Citation3 monoclonal antibodiesCitation4 and chemo-immunotherapy combinations.Citation5,Citation6 In addition, several molecular alterations with prognostic values have also been identified. These include specific cytogenetic patterns,Citation7 mutational status of the immunoglobulin heavy chain variable gene (IgVH),Citation8 expression of CD38,Citation9 and ZAP-70.Citation10,Citation11 This information is of significance as it confirms at the molecular level the clinical heterogeneity of patients with CLL, and may enable the design of specific interventions for patients with CLL at different risk.
Aberrant DNA methylation of multiple promoter-associated CpG islands can result in suppression of gene expression and the functional inactivation of tumor suppressor genes.Citation12 Interest in this phenomenon is derived not only from its molecular implications, but also because it is reversible, both in vitro and in vivo, using agents which inhibit DNA methyltransferase activity. These drugs have shown clinical activity in patients with leukemia.Citation13 Data from several groups have demonstrated that DNA methylation of multiple promoter-associated CpG islands is common in lymphoid leukemia, including CLL.Citation14–Citation16 Our group has been interested in acute lymphoblastic leukemia (ALL).Citation17 In ALL, specific methylation patterns are associated with distinct prognosis.Citation18 Studies have shown that reactivation of specific genes in ALL leukemia cells resulted in selective induction of cell death in cells with specific methylation alterations.Citation19,Citation20 In CLL, specific methylation alterations have been associated with certain genetic lesions, and methylation of the DAPK1 gene has been associated with familial CLL.Citation20 More recently, large scale methylation analysis have identified methylation patterns associated with specific genetic lesions in CLL.Citation14
Based on the data discussed above regarding the prevalence of aberrant DNA methylation in ALL, lineage similarities between the ALL and CLL leukemia cells, and previous data in CLL, we performed a genome-wide methylation profile of patients with CLL using methylated CpG island amplification (MCA) coupled with promoter microarray assay. The aim of the study was to identify specific methylation alterations with potential functional and clinical relevance in CLL. Detection of multiple aberrant DNA methylation in CLL could result in the development of an epigenetic classification of the disease with prognostic and therapeutic potential.
Results
Patient characteristics.
Cryopreserved peripheral blood lymphocytes from 78 patients with CLL (Suppl. Table 2) and normal CD19+ B cells from peripheral blood of ten healthy volunteers were used in the study. All patients had a confirmed diagnosis of CLL by flow cytometry with known IgVH mutational status and ZAP-70 expression. Unmutated (≥98% homology to germline) IgVH gene was detected in 38 patients (49%) by sequencing, and ZAP-70 was positive in 26 patients (33%) by flow cytometry. Standard metaphase karyotype analysis was performed in 34 patients. Twenty-four of them had diploid cytogenetics, three had trisomy 12, four had 11q deletion and three had complex abnormalities. FISH was performed in 18 patients, one had no abnormalities, eight had 13q deletion, four had 17p deletion, three had 11q deletion and two had trisomyCitation12. The median time from diagnosis to presentation to MDACC was 14 mo (range, 1–232 mo). Sixty-seven patients were previously untreated, six had one prior treatment, two had two prior treatments, and three had three or more prior treatments. Fifty-three patients received treatment after initial presentation, and the median time to treatment was 13 mo (range, 0–76 mo). The median follow-up time for all patients was 53 mo (range, 0–83 mo), and thirteen patients had died during the follow-up period.
Identification of 280 differentially hypermethylated CpG islands in patients with CLL using MCA/promoter microarray.
Because lineage similarities between CLL and ALL, we first studied the methylation characteristics of a set of genes known to be methylated in ALL, including p15, p57, p73, HOXA5 and THSB2.Citation18,Citation24–Citation27 The methylation frequency of these genes in CLL was significantly lower than previously reported in ALL, except for THSB2 gene. We also analyzed methylation characteristics of BIM, ZAP70 and ID4. Results are shown in Supplemental Table 3. Since we were unable to identify informative DNA methylation markers in CLL using this gene specific approach, we then performed a genome-wide search using a MCA/promoter microarray assay. We performed the MCA experiment by pooling DNA from two patients with 17p deletion CLL (tester) and CD19+ NBCs from two age-matched controls (driver) following standard procedure, since 17p deletion is a poor prognostic feature in patients with CLL. Using this approach, we identified 280 candidate markers that were differentially hypermethylated in patients with CLL (Suppl. Table 4). We selected only genes with two putative SmaI cutting sites, and also contained CpG islands in their promoter region based on results from Human Blat analysis (http://genome.ucsc.edu). We focused on those genes that had a normalized log2 ratio of ≥1.3 (which is equivalent to a 2.5-fold increase in signal intensity over controls) as shown by the microarray analysis. We excluded hypermethylated loci that represented unknown genes, spliced EST, mRNA or hypothetical proteins.
The 280 candidate genes were distributed across all chromosomes (Suppl. Fig. 1), but a significant number of them were localized in the following chromosomes: 25 on chromosome 11 (9%, p = 0.02), 30 on chromosome 16 (11%, p = 0.001), 27 on chromosome 17 (10%, p = 0.02) and 44 on chromosome 19 (16%, p = 0.001). We also performed interaction pathway and functional analysis of these 280 candidate genes using the Ingenuity Pathway Analysis tools 3.0. The analysis divided the 280 candidate genes into 25 functional networks, with a majority of genes falling into the top ten networks (Suppl. Table 5). The major functions of the genes involved in these networks included cell growth and differentiation, tissue and organ development, tissue morphology, cancer, cell death and cell cycle regulation.
Validation of methylation profile of 22 candidate genes in patients with CLL.
Due to the impracticability of validating all genes identified in the microarray, we focused on 22 genes to confirm their methylation status in 78 patients with CLL and also NBCs from ten healthy volunteers. These 22 genes were selected because they have higher log2 ratio and their potential function as tumor suppressors. As expected, most of the 22 genes had higher levels of methylation in patients with CLL than NBCs (), with the exception of MLL, BNC1 and SALL1 (these 3 genes all had high level of methylation in NBCs). The methylation of LINE sequences (Alu repeats of the long interspersed nucleotide elements of genomic DNA) was used as a marker for global DNA methylation. Because of concomitant methylation of multiple genes is common in leukemia, we analyzed the number of genes methylated out of the 22 genes in these 78 patients. We found that majority of the patients had between 8 and 11 of the genes methylated, and no patient had less than five genes methylated or more than 13 genes methylated (Suppl. Fig. 2). This was consistent with the hypermethylator phenotype reported in other leukemia, including ALL and acute myeloid leukemia.Citation25,Citation28 The higher level of methylation for these genes in the two testers compared with the two drivers further confirmed the validity of this assay.
Expression of target genes and response to epigenetic manipulation.
To study the relationship between methylation and gene expression, we analyzed the mRNA levels of two genes (PRIMA1 and APP) in the 78 patients with CLL. We found an inverse correlation between methylation and gene expression for both PRIMA1 (p = 0.0001) and APP (p = 0.0166) ( and B). We then studied the in vitro effect of epigenetic modulation with a DNA methyltransferase inhibitor with or without histone deacetylase inhibitor. We used Raji and HL60 acute leukemia cell lines in the study due to lack of wellestablished CLL cell lines. We analyzed mRNA expression of three genes (GALGT2, TFAP2C and PRIMA1) before and after treatment with 5-aza-2′-deoxyctidie and/or trichostatin A in these cells. We observed a significantly increased expression of all three genes after treatment with 5-aza-2′-deoxyctidine. An additive/synergistic effect on gene expression was also observed after pretreatment of cells with 5-aza-2′-deoxyctidine and then followed by trichostatin A ( and D). These data indicated that promoter methylation of these genes is associated with suppressed gene expression in leukemia cells and this can be reversed by treatment with DNA methyltransferase inhibitors, and more potently, in combination with histone deacetylase inhibitors.
To further study the functional role of these genes and their response to epigenetic therapy, we analyzed the in vivo effect on the methylation status of target genes in the DNA samples from peripheral blood of two patients treated in a clinical trial with 5-azacitidine (). Only two patients with CLL have been enrolled and treated in this study so far. In the first patient, we observed decreased LINE methylation at day 7 of treatment, and also a 40% decrease in SOX11 methylation at day 49 of treatment. In the second patient, we observed a significant decrease in SXO11 methylation during both cycle 1 and cycle 2 of treatment, and also a 70% decrease in DLX4 methylation during cycle 1 of treatment.
Association between gene methylation and prognostic factors in CLL.
To further characterize the clinical relevance of methylation profile of the genes analyzed here, we performed hierarchic cluster analysis of DNA methylation data. In general, we did not observe any difference in the methylation profile of the 22 genes regardless of IgVH mutational status or ZAP-70 expression in this group of patients (). These data suggested that DNA methylation might be a secondary event in the evolution of CLL. On further analysis, we found that unmutated IgVH status was associated with increased methylation levels of LINE (p < 0.0001) and SALL1 (p = 0.0008). In contrast, mutated IgVH was associated with increased methylation levels of RSPO1 (p = 0.001), GALGT2 (p = 0.002), APP (p = 0.003) and PRIMA1 (p = 0.01). Expression of ZAP-70 was associated with increased methylation of RSPO1 (p = 0.004) and APP (p = 0.04); while negative ZAP-70 expression was associated with increased methylation of LINE (p < 0.0001) and SALL1 (p = 0.048) ().
Association between patient characteristics, methylation status and overall survival.
We performed univariate Cox proportional hazard model analysis and found that laboratory parameters associated with overall survival in this group of patients included absolute lymphocyte count (HR = 1.008, p = 0.05), hemoglobin (HR = 0.66, p = 0.025), and β-2 microglobulin (HR = 1.42, p = 0.001) (Suppl. Table 6). In addition, unmutated IgVH status predicted for shorter survival and an almost 6-fold increase in the risk of death (HR = 5.83, p = 0.022), while expression levels of ZAP-70 were not predictive for overall survival in this patient group. Patient who received prior treatment also had a shorter overall survival (HR = 8.33, p = 0.0001). We also explored the associations between methylation status of the 22 candidate genes and overall survival using the same model. We identified that increased methylation of LINE, APP, SALL1 and PRIMA1 was correlated with shorter overall survival. In the multivariate Cox proportional hazards model, higher β-2 microglobulin, increased methylation of LINE and APP predicted for shorter overall survival. After adjusting for LINE and APP, 1 unit increase of β-2 microglobulin increased the risk of death by approximately 54% (p = 0.0015); after adjusting for β-2 microglobulin and APP, 1% increase of LINE methylation increased the risk of death by 19% (p = 0.019); and after adjusting for β-2 microglobulin and LINE, 1% increase of APP methylation increased the risk of death by 8% (p = 0.001). On further analysis, the dichotomized methylation levels of LINE and APP using the optimal cut-points at 58.8 and 17% were significantly associated with overall survival with the adjusted p values of 0.045 and 0.0035, respectively ( and C and ).
Discussion
In this study, we have performed a genome-wide methylation analysis of patients with CLL, and identified 280 potential new targets of aberrant DNA methylation. Methylation of at least 25 of these genes (WNT9A, DLX11, GAD1, GBX2, INHA, CYP1B1, FAM62C, RSPO11, HAND1, SLC22A3, HOXA9, NPM2, LBX1, MYOD11, THY1, MDK, TBX3, FGF14, PDX26, PRIMA1, SSTR1, REC8L1, SALL1, SPAG5 and KLK10) has been previously reported in other malignancies, but rarely in CLL. These 280 genes can be clustered into specific functional networks involving in multiple molecular processes and signaling pathways. We mapped these genes to their chromosomal locations, and observed there was a bias towards chromosomes 11, 16, 17 and 19. Since we initially used DNA from patients with 17p deletion, we were particularly interested in genes that were located in that area or might have interaction with p53. There were 27 genes mapped to chromosome 17 and four of them (PRIMA1, TFAP4, SIRT2, TP53INP2) have been reported to functionally interact with p53. Importantly, alterations of chromosome 17 and 11 have been associated with poor prognosis in CLL.Citation7
We validated methylation of 22 candidate genes using bisulfite pyrosequencing because it was impractical to analyze all genes identified in the current study. Consistent with previously described hypermethylator phenotype in acute leukemia,Citation25,Citation28 we observed a significant fraction of patients were characterized by the concordant methylation of multiple genes. Furthermore, methylation of these genes was functionally relevant as it was inversely associated with gene silencing. Also, treatment of leukemia cells with epigenetic modulators resulted in increased gene expression. These observations were of clinical relevance, since methylation of several of these genes was associated with important genetic and prognostic factors in this patient population, including IgVH mutational status and ZAP-70 expression. For instance, increased LINE and SALL1 methylation was associated with unmutated IgVH, a marker of poor prognosis in CLL.Citation9 Increased methylation of RSPO1 and APP was associated with positive expression of ZAP-70, also a poor prognostic marker in CLL.Citation11 Several genes were associated with shorter overall survival by univariate analysis, including increased methylation levels of LINE, APP, SALL1 and PRIMA1. In multivariate analysis, increased methylation of LINE and APP were associated with worse outcome. Amplification of APP gene (amyloid precursor protein) has been described in acute myeloid leukemiaCitation29 and maybe involved in Notch signaling,Citation30 an important pathway in ALL. But the role of APP in CLL is currently unknown. SALL1 gene encodes a zinc finger protein and mutations of this gene have been associated with the Townes-Brocks syndrome. Besides our recent report of SALL1 methylation in CLL, little is known of the function of this gene in leukemia. Methylation of LINE is a marker of global DNA methylation that has been shown to be associated with microsatellite instability.Citation31 Prior report has indicated a relationship between IgVH mutational status and global gene methylation.Citation32 In this current study, methylation profiles were not different in these patients regardless of their IgVH mutational status or ZAP-70 expression, which indicates that these two events maybe independent upon methylation status.
The data presented here are of significance for several reasons. First, we provided a large unbiased list of potential targets of DNA methylation in human leukemia. It is possible that several of these genes could have potential tumor suppressor properties and the study of these genes may provide further information regarding CLL and other leukemia. Second, from a translational perspective, methylation of several of these genes was associated with important clinical and prognostic characteristics. It is therefore possible that systematic analysis of these genes may help in the development of new prognostic biomarkers in CLL and may guide therapeutic timing in these patients. Third, it is known that DNA methyltransferase inhibitors act in part through reversing aberrant DNA methylation and restoring gene expression. The data presented here raised the possibility that the use of drugs like 5-azacitidine or decitabine may have clinical activity in this disease, and we observed decreased methylation of several of these genes in CLL patients treated with 5-azacitidine. Ongoing phase II trials are evaluating this possibility at our center.
Several groups have already presented data in the analysis of DNA methylation in CLL. Using RLGS, Rush et al. identified 193 potential targets of methylation in CLL.Citation33 Of interest is that only seven of these genes (CYP1B1, PPRN2, TBX3, FGF14, BASP1, NKX2.3 and NS3ST2) were commonly identified by both genome-wide approaches. Other genes shown to be frequently methylated in CLL include ID4Citation34 and TWIST2.Citation35 Recently, Raval et al. reported on the methylation of DAPK1, and its interactions with HOXB7, in both sporadic and familial CLL.Citation20 We detected HOXB7 in our array and follow up studies of epigenetic regulation of HOXB7 gene in CLL are ongoing. It is of interest that there is relatively little concordance between our findings and those of these other reports. One likely explanation is our focus on patients with alteration of chromosome 17. Therefore the results presented here should be considered as potential. Furthermore, and in contrast with the data of Kanduri et al.,Citation14 we could not identify any associations between specific genetic alterations and methylation patterns. This could also be explained due to our focus on chromosome 17 as well as technical differences between the assays used in these studies.
There are several limitations to the data presented here. First, the sensitivity and specificity of the MCA/promoter microarray is not fully known, but it has been shown to have a high reproducibility by both our group and others.Citation21,Citation22 Furthermore, we have compared our results in CLL and that in ALL using the same technique and found that only 10% of the genes were shared between them.Citation36 It should also be noted that in the MCA assay, we used DNA from two patients with 17p deletion because it confers poor prognosis in CLL. It is thus possible that performing additional experiments of other patient subgroups with different genetic features could result in the identification of other valid targets. Second, there are different techniques to perform large-scale methylation analysis and is probable that our study may only provide a partial view of the epigenome of the CLL. Indeed one limitation of our study is the fact that we only used DNA from two patients to perform the MCA experiment. This was illustrated by the differences of our results from those of Rush et al.Citation33 It is most likely both sets of data are complementary. Most importantly, the in vivo methylation and expression data suggested a functional relevance for at least some of the genes. Finally, the impact of these epigenetic alterations in the setting of CLL therapy was not studied as the samples used here were collected in patients not treated uniformly in clinical trials. It will be important to analyze the clinical impact of methylation of some of these genes. It should also be pointed out that the clinical and prognostic associations observed in this study should be considered as exploratory and needs to be confirmed in larger prospective studies.
In conclusion, the MCA/promoter microarray technique is a useful tool to study the genome-wide DNA methylation profile in leukemia or other cancer types. We identified 280 potential targets of aberrant DNA methylation in patients with CLL that carry 17p deletion. Methylation status of several of these genes may have significant prognostic and therapeutic values in CLL. Further studies are needed to characterize the functional roles of these genes and molecular pathways involved that may impact on prognosis and response to therapy in patients with CLL.
Materials and Methods
Human specimens.
Cryopreserved peripheral blood lymphocytes from 78 patients with CLL were obtained from the CLL Research Consortium Tissue Bank. All patients provided informed consent at the time of sample collection according to institutional guidelines. DNA and RNA were extracted using Tri-zol reagents from Invitrogen (Carlsbad, CA, USA) following standard protocols. DNA and RNA were also obtained from normal CD19+ B cells (NBCs) from 10 healthy volunteers with a median age of 59 years (range, 32–70 years). NBCs were isolated using Human B Cell Isolation Kit from Miltenyi Biotec (Auburn, CA, USA) according to manufacturer's protocol.
Human leukemia cell lines.
HL60 and Raji human leukemia cell lines from American Type Culture Collection (Mansssas, VA, USA) were used in the current study due to lack of well established CLL cell lines. Cells were cultured in RPMI 1640 medium from Gibco BRL (Grand Island, NY, USA) supplemented with 10% fetal bovine serum and penicillin-streptomycin.
DNA bisulfite treatment and bisulfite pyrosequencing.
Bisulfite treatment of genomic DNA and bisulfite pyrosequencing were performed as previously described.Citation18 For pyrosequencing, a two-step PCR reaction was performed using specially designed forward and reverse primers (Suppl. Table 1). Sequencing primer was designed to analyze at least 3–4 CpG sites in the amplified promoter region. Primers were designed to be located in the proximity of transcription start sites using Pyrosequencing Assay Design Software from Biotage (Uppsala, Sweden). Quantification of cytosine methylation was performed using the PSQ HS96A 1.2 software package. A gene was considered as methylated if the methylation density was ≥10%.
MCA/promoter microarray.
The principles underlying MCA and the use of the Agilent promoter microarray have been recently described by several studies.Citation21–Citation23 MCA involves amplification of closely spaced Sma I sites (CCCGGG) to enrich for methylated CpG islands in the promoter region. The MCA experiment was performed following standard procedure,Citation23 using pooled genomic DNA (5 µg) from two CLL patients with chromosome 17p deletion (tester), and from NBCs of two age and sex (male) matched healthy volunteers (driver). We selected patients with 17p deletion because or their poor prognosis in patients with CLL. Thus, genes indentified in this assay could be associated with distinct prognosis.
PCR products from MCA experiment were labeled using CGH labeling kit from Invitrogen according to manufacturer's protocol. Briefly, 2 µg of PCR product was mixed with random primer solution and water to a final volume of 75 µl. The mixture was incubated at 95°C for 5 min and immediately transferred to an ice-water bath. After that, dUTP nucleotide mix, Klenow enzyme and either Cy5 (tester amplicon) or Cy3 (driver amplicon) were added to the mixture and incubated at 37°C for 3 hours in the dark. The reaction was terminated using a stopping buffer. Samples were washed using CGH column from Invitrogen. Approximately 5 µg each of Cy5- and Cy3-labeled samples were mixed in 150 µl diluted water. After that, 50 µl human Cot-1 DNA, 50 µl Agilent blocking agent and 250 µl Agilent hybridization buffer were added in the mixture in that order. The mixture was incubated at 95°C for 3 min, 37°C for 30 min and then hybridized to Agilent human proximal promoter microarray slides (44K array) from Agilent Technologies (Santa Clara, CA, USA) according to the Agilent aCGH array hybridization protocol. The hybridization reaction was carried out in a rotisserie hybridization oven at 65°C, 10 rpm for 40 h. Slides were subsequently washed with Agilent Oligo aCGH wash buffers, acetonitrile, stabilization and drying solutions from Agilent Technologies in the dark. After the final wash, slides were kept in dark and scanned within 30 min using Agilent G2565AA scanner. Data were analyzed using Agilent CHIP Analytics 1.2 software package. The MCA/microarray experiments were performed in duplicate.
Interaction networks and functional analysis of microarray data.
Interaction network and functional analysis of microarray data were performed using online version of the Ingenuity Pathway Analysis tools 3.0 from Ingenuity System Inc., (Redwood City, CA, USA).
Analysis of gene expression using real-time RT-PCR.
Gene expression was analyzed using conventional real-time RT-PCR assays as previously published.Citation21
Epigenetic modulation of target genes.
To study the in vivo effect of the DNA hypomethylating agent 5-azacitidine, we performed sequential bisulfite pyrosequencing analysis of selected genes using DNA isolated from peripheral blood samples of two CLL patients treated with 5-azacitidine in a clinical trial at M.D. Anderson Cancer Center (MDACC). Consent for sample collection at different time points during treatment was obtained following institutional guidelines.
To further study the in vitro effect of DNA methylation and/or histone acetylation on gene expression, we treated HL60 and Raji leukemia cells with the selective DNA methyltransferase inhibitor, 5-Aza-2′-deoxycytidine from Sigma (St. Louis, MO, USA) with or without histone deacetylase inhibitor (HDACI) trichostatin A from ICN Biomedicals (Shelton, CT, USA). Briefly, leukemia cells were cultured in media supplemented with 5-aza-2′-deoxycytidine at 1 µM for 4 days alone, or in combination of 5-aza-2′-deoxycytidine at 1 µM for 4 days and then trichostatin A at 500 nM for the last 24 h, or trichostatin A at 500 nM for the last 24 hours alone without pre-treatment with 5-aza-2′-deoxycytidine. Medium was changed daily with fresh drugs. The mRNA was isolated from leukemia cells at the end of treatment and gene expression was analyzed using real-time RT-PCR assay.
Statistical analysis.
Association between categorical variables was assessed via cross-tabulation and Fisher's exact test. Wilcoxon rank sum test was used to assess the difference in continuous variables between groups. Spearman correlation method was used to determine the correlations between age and DNA methylation levels. Kaplan-Meier product-limit method was used to generate the survival curves, where the overall survival is defined as the time between referral to MDACC and last follow-up. Overall survival was calculated from time to referral at MDACC, and not from initial diagnosis, because samples were collected at the time of referral to MDACC and we do not know whether methylation patterns change with time. Log-rank test was subsequently used to assess the difference in survival between groups to study the effects of exploratory covariates on overall survival. Cox proportional hazards regression models were used to assess the hazard ratio and the goodness-of-fit of the models were evaluated by martingale residual plots. In univariate Cox regression, the effects of variables were modeled individually. All variables with p values less than 0.15 were included into an initial multivariate Cox proportional hazards model, and a stepwise model selection procedure was performed with equal entering and staying probabilities of 0.1. At the end of model selection, variables with p values less than 0.05 were retained. A recursive partitioning technique was also applied to find the optimal cut-points for methylation of genes with respect to overall survival.
All computations were carried out on DELL PC using the Windows NT operating system in SAS software from SAS Institute (Cary, NC, USA) and S-plus 2000 software package from Insightful Corp. (Seattle, WA, USA). All reported p values were 2-sided, and p < 0.05 was considered statistically significant.
Figures and Tables
Figure 1 Heatmap representations of methylation profiles of 22 candidate genes. Methylation was measured in patients with CLL (n = 78) and NBCs (n = 10) using bisulfite pyrosequencing. Green indicates a methylation density of <10%, yellow 10–49% and red ≥50%. White indicates lack of data due to failure of the assay for this particular sample. Tester includes the profile of the two patients with CLL and 17p deletion, driver includes the NBCs from two healthy volunteers whose DNA was used to perform the original MCA experiment. The methylation profiles of the two testers and two drivers are shown at the top of the figure.
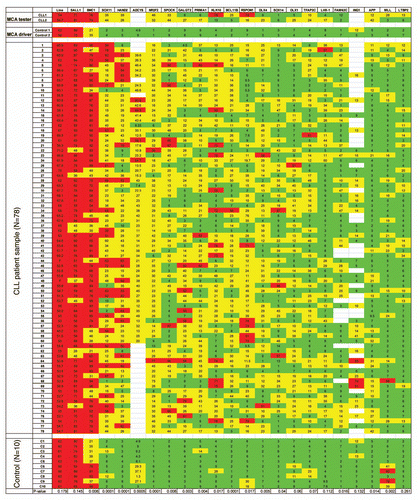
Figure 2 (A and B) Correlation between methylation (%) and gene expression (dCTt) of two selected genes, PRIMA1 and APP. Methylation was measured by bisulfite pyrosequencing and gene expression was measured by real-time RT-PCR assay. (C and D) Epigenetic modulation of GALGT2, TFAP2C and PRIMA1 gene expression in Raji and HL60 leukemia cell lines. Leukemia cells were treated with 5-aza-2′-deoxycytidine (DAC) at 1 µM for 4 days alone, or DAC at 1 µM for 4 days and then trichostatin A (TSA) at 500 nM for the last 24 h, or TSA at 500 nM for the last 24 h alone without pre-treatment with DAC. Medium was changed daily with fresh drugs, and gene expression was measured by real-time RT-PCR assay.
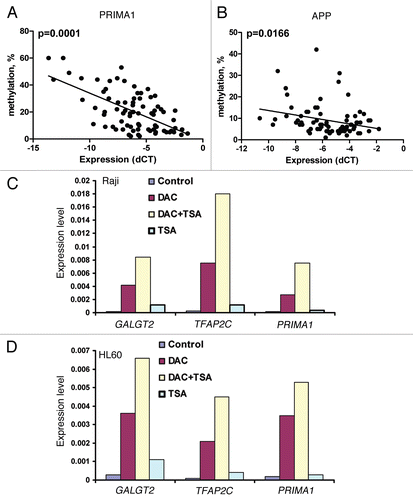
Figure 3 (A) In vivo effect of 5-azacitidine in patients with CLL. Changes in methylation of LINE, SOX11 and DLX4 genes were measured in two patients with CLL who were treated in a clinical trial with 5-azacitidine. DNA was isolated from peripheral blood mononuclear cells and the methylation was measured by bisulfite pyrosequencing at each time point indicated. C, cycle number; D, days of treatment. (B and C) Methylation of LINE, APP and overall survival. Kaplan-Meyer survival analysis showing both increased methylation of LINE (p = 0.045) and APP (p = 0.0035) were associated with shorter overall survival using optimal cut-points at 58.8 and 17%, respectively.
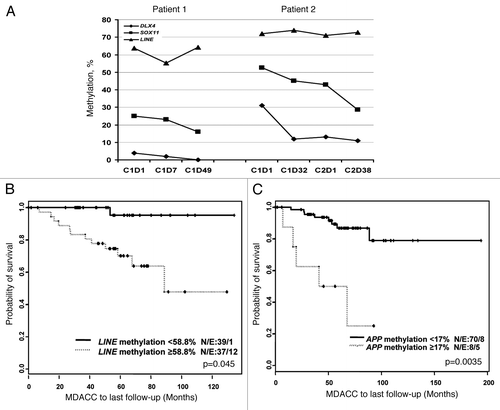
Figure 4 Hierarchic cluster analysis of methylation data and its correlation with IgVH mutational status (A) and ZAP-70 expression (B). Cluster analysis of methylation data was performed using Commoner application from National Cancer Institute (www.discover.nci.nih.gov/cimminer). Each column represents an individual patient and each row represents a gene.
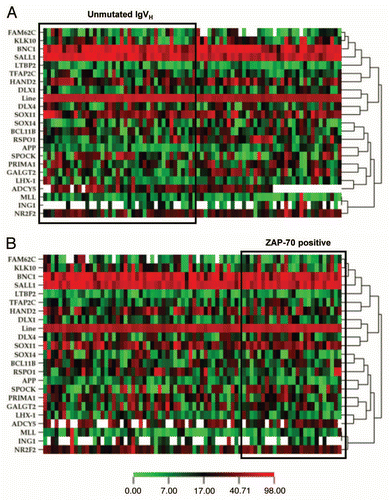
Table 1 Differences in gene methylation status by prognostic group
Table 2 Multivariable cox proportional hazard model for and survival
Additional material
Download Zip (457.2 KB)Acknowledgements
This work was supported by the CLL Global Research Foundation. G.G.-M. is also funded by grants CA100067 and CA105771 from the NCI, a Physician-Scientist Award from the Commonwealth Foundation for Cancer Research at the University of Texas M.D. Anderson Cancer Center and the Leukemia and Lymphoma Society of America. This work was also supported by a grant from Chronic Lymphocytic Leukemia Foundation to W.-G.T.
References
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. New Eng J Med 2005; 352:804 - 815
- Keating MJ, Chiorazzi N, Messmer B, Damle RN, Allen SL, Rai KR, et al. Biology and treatment of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program 2003; 153 - 175
- Rai KR, Peterson BL, Appelbaum FR, Kolitz J, Elias L, Shepherd L, et al. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. New Eng J Med 2000; 343:1750 - 1757
- O'Brien SM, Kantarjian H, Thomas DA, Giles FJ, Freireich EJ, Cortes J, et al. Rituximab dose-escalation trial in chronic lymphocytic leukemia. J Clin Oncol 2001; 19:2165 - 2170
- Byrd JC, Rai K, Peterson BL, Appelbaum FR, Morrison VA, Kolitz JE, et al. Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 2005; 105:49 - 53
- Keating MJ, O'Brien S, Albitar M, Lerner S, Plunkett W, Giles F, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol 2005; 23:4079 - 4088
- Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. New Eng J Med 2000; 343:1910 - 1916
- Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94:1848 - 1854
- Hamblin TJ, Orchard JA, Gardiner A, Oscier DG, Davis Z, Stevenson FK. Immunoglobulin V genes and CD38 expression in CLL. Blood 2000; 95:2455 - 2457
- Letestu R, Rawstron A, Ghia P, Villamor N, Leuven NB, Boettcher S, et al. Evaluation of ZAP-70 expression by flow cytometry in chronic lymphocytic leukemia: A multicentric international harmonization process. Cytometry B Clin Cytom 2006; 70:309 - 314
- Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, Weiss A, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood 2002; 100:4609 - 4614
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 692
- Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 2002; 20:2429 - 2440
- Kanduri M, Cahill N, Goransson H, Enstrom C, Ryan F, Isaksson A, et al. Differential genome-wide arraybased methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood 115:296 - 305
- Martin-Subero JI, Ammerpohl O, Bibikova M, Wickham-Garcia E, Agirre X, Alvarez S, et al. A comprehensive microarray-based DNA methylation study of 367 hematological neoplasms. PloS One 2009; 4:6986
- Rahmatpanah FB, Carstens S, Guo J, Sjahputera O, Taylor KH, Duff D, et al. Differential DNA methylation patterns of small B-cell lymphoma subclasses with different clinical behavior. Leukemia 2006; 20:1855 - 1862
- Garcia-Manero G, Daniel J, Smith TL, Kornblau SM, Lee MS, Kantarjian HM, et al. DNA Methylation of Multiple Promoter-associated CpG Islands in Adult Acute Lymphocytic Leukemia. Clin Cancer Res 2002; 8:2217 - 2224
- Shen L, Toyota M, Kondo Y, Obata T, Daniel S, Pierce S, et al. Aberrant DNA methylation of p57KIP2 identifies a cell cycle regulatory pathway with prognostic impact in adult acute lymphocytic leukemia. Blood 2003; 101:4131 - 4136
- Kuang SQ, Ling X, Sanchez-Gonzalez B, Yang H, Andreeff M, Garcia-Manero G. Differential tumor suppressor properties and transforming growth factor-beta responsiveness of p57KIP2 in leukemia cells with aberrant p57KIP2 promoter DNA methylation. Oncogene 2007; 26:1439 - 1448
- Raval A, Tanner SM, Byrd JC, Angerman EB, Perko JD, Chen SS, et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007; 129:879 - 890
- Kuang SQ, Tong WG, Yang H, Lin W, Lee MK, Fang ZH, et al. Genome-wide identification of aberrantly methylated promoter associated CpG islands in acute lymphocytic leukemia. Leukemia 2008; 22:1529 - 1538
- Estecio MR, Yan PS, Ibrahim AE, Tellez CS, Shen L, Huang TH, et al. High-throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res 2007; 17:1529 - 1536
- Toyota M, Ho C, Ahuja N, Jair KW, Li Q, Ohe-Toyota M, et al. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res 1999; 59:2307 - 2312
- Garcia-Manero G, Jeha S, Daniel J, Williamson J, Albitar M, Kantarjian HM, et al. Aberrant DNA methylation in pediatric patients with acute lymphocytic leukemia. Cancer 2003; 97:695 - 702
- Garcia-Manero G, Daniel J, Smith TL, Kornblau SM, Lee MS, Kantarjian HM, et al. DNA methylation of multiple promoter-associated CpG islands in adult acute lymphocytic leukemia. Clin Cancer Res 2002; 8:2217 - 2224
- Garcia-Manero G, Bueso-Ramos C, Daniel J, Williamson J, Kantarjian HM, Issa JP. DNA methylation patterns at relapse in adult acute lymphocytic leukemia. Clin Cancer Res 2002; 8:1897 - 1903
- Garcia-Manero G. Prognostic implications of epigenetic silencing of p15INK4B in acute promyelocytic leukemia. Leukemia 2003; 17:839 - 840
- Toyota M, Kopecky KJ, Toyota MO, Jair KW, Willman CL, Issa JP. Methylation profiling in acute myeloid leukemia. Blood 2001; 97:2823 - 2829
- Baldus CD, Liyanarachchi S, Mrozek K, Auer H, Tanner SM, Guimond M, et al. Acute myeloid leukemia with complex karyotypes and abnormal chromosome 21: Amplification discloses overexpression of APP, ETS2 and ERG genes. Proceedings of the National Academy of Sciences of the United States of America 2004; 101:3915 - 3920
- Florean C, Zampese E, Zanese M, Brunello L, Ichas F, De Giorgi F, et al. High content analysis of gammasecretase activity reveals variable dominance of presenilin mutations linked to familial Alzheimer's disease. Biochim Biophys Acta 2008;
- Estecio MR, Gharibyan V, Shen L, Ibrahim AE, Doshi K, He R, et al. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PloS One 2007; 2:399
- Lyko F, Stach D, Brenner A, Stilgenbauer S, Dohner H, Wirtz M, et al. Quantitative analysis of DNA methylation in chronic lymphocytic leukemia patients. Electrophoresis 2004; 25:1530 - 1535
- Rush LJ, Raval A, Funchain P, Johnson AJ, Smith L, Lucas DM, et al. Epigenetic profiling in chronic lymphocytic leukemia reveals novel methylation targets. Cancer Res 2004; 64:2424 - 2433
- Yu L, Liu C, Vandeusen J, Becknell B, Dai Z, Wu YZ, et al. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet 2005; 37:265 - 274
- Raval A, Lucas DM, Matkovic JJ, Bennett KL, Liyanarachchi S, Young DC, et al. TWIST2 demonstrates differential methylation in immunoglobulin variable heavy chain mutated and unmutated chronic lymphocytic leukemia. J Clin Oncol 2005; 23:3877 - 3885
- Tong W, Kuang S-Q, Wierda W, Keating M, Garcia-Manero G. Identification of Multiple Promoter Associated CpG Islands Commonly Methylated in Both Acute Lymphocytic Leukemia (ALL) and Chronic Lymphocytic Leukemia (CLL) Using Novel Genome-Wide Microarray Technique: Implications for Common Primordial Molecular Pathways in Lymphoid Leukemias. Blood 2008; 112:2263