Abstract
Although most CpG islands are generally thought to remain unmethylated in all adult somatic tissues, recent genome-wide approaches have found that some CpG islands have distinct methylation patterns in various tissues, with most differences being seen between germ cells and somatic tissues. Few studies have addressed this among human somatic tissues and fewer still have studied the same sets of tissues from multiple individuals. In the current study, we used Restriction Landmark Genomic Scanning to study tissue specific methylation patterns in a set of twelve human tissues collected from multiple individuals. We identified 34 differentially methylated CpG islands among these tissues, many of which showed consistent patterns in multiple individuals. Of particular interest were striking differences in CpG island methylation, not only among brain regions, but also between white and grey matter of the same region. These findings were confirmed for selected loci by quantitative bisulfite sequencing. Cluster analysis of the RLGS data indicated that several tissues clustered together, but the strongest clustering was in brain. Tissues from different brain regions clustered together, and, as a group, brain tissues were distinct from either mesoderm or endoderm derived tissues which demonstrated limited clustering. These data demonstrate consistent tissue specific methylation for certain CpG islands, with clear differences between white and grey matter of the brain. Furthermore, there was an overall pattern of tissue specifically methylated CpG islands that distinguished neural tissues from non-neural.
Introduction
For over 30 years, the role of DNA methylation in mammals has been under investigation. It has been clearly demonstrated that DNA methylation is required for survival beyond early embryogenesis in mouse.Citation1,Citation2 What is not yet clear is the exact nature of this absolute requirement for DNA methylation. It is well established that DNA methylation plays a critical role in X chromosome inactivation in females,Citation3 and in allele specific expression of imprinted genes.Citation4 In addition, a large body of data has shown correlation between lack of methylation of tissue specific genes in the expressing tissues and methylation of these genes in non-expressing tissues.Citation5,Citation6 These data suggest that DNA methylation may play a critical role in mammalian development.
Of all CpG dinucleotides in vertebrate genomes, 60–90% are methylated.Citation7,Citation8 The majority of the CpG dinucleotides in the mammalian genome are scattered throughout bulk chromatin, generally heavily methylated and thought to contribute to the repression of transcription from repetitive elements such as Alus and retrotransposons.Citation9 In human DNA, approximately 15% of the CpG dinucleotides are found in CpG islands,Citation7,Citation8 defined as stretches of DNA >200 bp, with a G + C content greater than 50% and an observed to expected ratio of CpG ≥0.6.Citation10 CpG islands are often found in the promoters of both “house keeping” genes and tissue specific genes. Aside from the inactivated X chromosome and imprinted genes, the thinking has been that CpG islands are normally unmethylated, at least in germ cells,Citation7 and perhaps throughout the developing and adult organismCitation5 for most CpG islands.
Tissue specific genes with non-CpG island promoters have been shown to exhibit tissue specific methylation. However, tissue specific genes with CpG island promoters initially received less attention. Earlier studies using candidate gene approaches led to conflicting ideas about tissue specific methylation of CpG islands. One study looking at the methylation status of seven tissue specific gene promoters found little or no methylation in the promoters of four genes with CpG island-like characteristics,Citation11 supporting the idea that tissue specific genes with CpG island promoters are not methylated, regardless of developmental state, tissue or expression. Other data, however, have shown that CpG islands can indeed become methylated in normal adult tissues and this phenomenon may be related to age.Citation12 Recently a number of genomic-based studies looking at very large numbers of CpG islands in normal somatic tissues have been published that provide strong evidence that tissue-specific transcription is controlled, in part, by tissue-specific differentially methylated regions, both CpG island and non-CpG island.
Using bisulfite DNA sequencing, Eckhart et al. (2006)Citation13 reported high-resolution methylation profiles of human chromosomes 6, 20 and 22 from 12 different tissues. These data indicated that evolutionarily conserved regions are the predominant sites for differential DNA methylation and that methylation specifically within a core region surrounding the transcriptional start site is an informative surrogate for promoter wide methylation. Another study took a more genome wide approach at a lower resolutionCitation14 to generate an epigenomic map of DNA methylation, RNA polymerase II occupancy and chromatin state for 16,000 promoters in human primary fibroblasts and mature sperm. The results showed that promoter sequence and gene function are major predictors of promoter methylation states and suggested that weak CpG islands are predisposed to de novo methylation during differentiation while strong CpG island promoters are mostly unmethylated, even when inactive.
A comparative genomic approach identified evolutionarily conserved tissue specific CpG island methylation between mouse and human. Such CpG islands identified in a genome wide scan of DNA from various mouse tissuesCitation15 were studied in a candidate gene approach in human tissues.Citation16 Of the 14 mouse CpG islands with tissue specific methylation, six were found to conserve similar patterns of methylation in the human orthologs. Five of these correlated with gene expression patterns, providing further evidence that human gene expression could be regulated by tissue specific CpG island methylation-mediated gene silencing. Multiple additional groups have recently applied various genome wide CpG island methylation approachesCitation17–Citation20 to confirm and broaden the ideas that CpG island methylation plays an important role in cellular identity and tissue specific gene expression and these regions mostly have a wide range of CpG densities.
Most studies have not addressed the question of inter individual variability of tissue specific CpG island methylation in human tissues. Only a few studies used the same tissues from multiple individuals at the candidate gene level,Citation16,Citation18,Citation20 but none of these address the issue by using genomic approaches on the same tissues from multiple individuals to determine if tissue specific methylation of the CpG islands was consistent. More recent genomics based approaches have addressed this issue and shown that CpG island methylation patterns in normal human tissues are more similar among the same tissue studied from multiple individuals than tissues from the same individual compared to each other.Citation21 Another study found that inter-individual differences in DNA methylation patterns could be correlated to age and environmental exposures such as tobacco use.Citation22
Focusing on three regions of the brain, a recent study addressed the issue of inter-individual differences in tissue specific methylation.Citation23 This study determined the quantitative DNA methylation levels of 1,505 CpG sites across 76 brain samples representing cerebral cortex (n = 35), cerebellum (n = 34) and pons (n = 7). A set of 156 loc showed statistically significant methylation differences among brain regions, and unsupervised hierarchical analysis showed strong clustering of the cerebra distinct from the clustering of the cerebella, and pons. Their data suggest that DNA methylation signatures distinguish brain regions, are consistent across many individuals and may help account for region-specific functional specialization.
In the present study, we have analyzed an average of approximately 1500 NotI sites by Restriction Landmark Genomic Scanning (RLGS), from 12 different tissues obtained from seven individuals. Each tissue was obtained from a minimum of two individuals and a maximum of five. Our data clearly show the existence of tissue specific CpG island methylation among regions of the brain as well as among other tissue types and that many of these methylation differences are preserved among different individuals. Furthermore, we demonstrate that differences in tissue specific methylation not only exists in different regions of the brain, as previously reported,Citation23 but also between the grey and white matter of the same brain region. Despite differences among brain regions, neural tissues were clearly distinguished from non-neural.
Results
Tissue specific methylation differences found in multiple individuals.
RLGS profiles were prepared for DNA from 12 tissues collected at autopsy from seven patients. Distinct brain regions were collected including the basal ganglia, the cerebellum and the frontal and temporal lobes of the cerebrum. Both the cerebellum and the frontal lobe of the cerebrum were separated into grey matter and white matter. Non-brain tissues included kidney, lung, prostate, spleen, stomach and thyroid. All profiles from a single patient were compared against each other, and differences in the two dimensional patterns were noted. Not all tissues were collected from or produced high quality DNA from each patient. shows the tissues for which RLGS gels were analyzed from each patient. A total of 33 RLGS profiles were analyzed in this way. Profiles between the seven patients were not directly compared in order to avoid noting pattern differences due to genetic polymorphism. However, differences noted within a single patient's set of profiles were studied in all 33 RLGS profiles from all tissues in each patient.
The RLGS fragments are named according to the numbering system for our master RLGS profile previously described.Citation29 A total of 141 RLGS spots showed tissue specific methylation differences within the tissues of at least one patient. Of these, 82 RLGS spots were present, or only partially methylated, in at least one tissue from every patient, therefore ruling out genetic polymorphism as an explanation for RLGS spot loss. It is important to note that we made an exception for patient 3. The spots that were present in at least one tissue in all patients but not present in patient 3 (only 19 such spots), were still considered non-polymorphic for this patient because we only have data from two brain tissues of this patient. These 82 spots, listed in Supplemental Table 1, comprise our set of non-polymorphic RLGS spots showing differential methylation among normal tissues.
Of these 82 RLGS spots, we have identified the sequence represented by these spots for 34 by various RLGS spot cloning approaches.Citation24,Citation28 shows the methylation patterns for each of those 34 RLGS spots in each tissue in each patient, with black boxes indicating methylation. The genomic location and gene homology information for each of the 34 RLGS spots is shown in . Supplemental Table 1 shows the data for all 82 non-polymorphic RLGS spots (including those whose genomic location has not been identified) that exhibited tissue specific methylation differences.
shows an example of the RLGS analysis of all 12 tissues, with brain tissues from patient 1 and non-brain tissues from patient 2. The RLGS fragment 3D24 (see arrow, ) is present in the cerebellar white matter (C. White) profile, but is clearly absent from the cerebellar cortex (grey matter) (C. Cort). As indicated in and seen as varying intensity of the 3D24 fragment in , certain other nervous system tissues were also scored as showing methylation. We estimate our ability to score methylation by RLGS to be constrained by a lower detection limit of 35–45% fragment intensity loss, depending on the quality of the DNA.Citation29 Subsequent cloning of the 3D24 RLGS fragment (NotI-EcoRV fragment) and use of this fragment as a hybridization probe for Southern blot analysis in demonstrates partial methylation in all tissues. Quantitation of the methylation was accomplished by comparing the intensity of the methylated band to the combined intensities of the methylated and unmethylated bands to determine the estimated percent methylation for each tissue (). These data show that the expected sensitivity of RLGS analysis fits well with the subsequent measurement of tissue specific methylation by quantitative Southern blot analysis. Moreover, these data demonstrate high level methylation of an RLGS spot only in cerebellum cortex, but only low level methylation in other tissues, including the white matter of the cerebellum.
The most dramatic and consistent methylation differences across multiple patients occurred between brain regions and even between grey and white matter of the same region. Although many loci showed methylation specifically in a limited number of tissues, such as 3D24 described above, which was methylated in four out of five patients' cerebellar cortex (), other loci showed a lack of methylation consistently in one tissue. For example, RLGS spot 3B07, which is found at the 3′ end of the CNPY1 gene, is specifically unmethylated in each case of cerebellar cortex (n = 5), but methylated in most other brain regions including the white matter of the cerebellum. shows representative RLGS gel regions demonstrating the presence of the spot in the cerebellar cortex gels of three patients, and absence of the spot in other brain regions.
We performed hierarchical cluster analysis on the total RLGS data set, shown in , to look for patterns of methylation. We found that while some tissues tended to cluster together based on methylation patterns, such as C. cort, more striking was that neural tissues strongly clustered distinctly from nonneural tissues. The cluster analysis used a “manhattan” distance (absolute distance) between samples and the “Ward” method for clustering. In short, the “manhattan” distance between two samples is equal to the number of loci that show methylation differences while the “Ward” method for clustering attempts to minimize the sum of squares of any two (hypothetical) clusters than can be formed at each step in the clustering algorithm. From examining the dendogram, it appears there are two major groups, one group consists of the mesoderm and endoderm derived tissues while the other group consists of only the brain tissues, which are ectoderm derived.
shows representative RLGS profiles pointing out spot 3D46 for six ectoderm derived tissues from three patients. The spot is absent or barely detectable in all six profiles. However, this spot is present in mesoderm and endoderm derived tissues including stomach of patients 2 and 9 and spleen of patient 5, lung from patients 2 and 5 and the thyroid of patient 8 (). Given the limited number of mesoderm or endoderm derived tissues in the study and that the only ectoderm derived tissues used are neural, we cannot distinguish whether or not the differences in methylation pattern are related to the germ layer from which the tissues are derived, or more specifically are related to differences between neural and non-neural cell lineages.
MAQMA confirms tissue and neural specific methylation.
The RLGS data presented above provide information about the methylation status of the NotI restriction sites and relies upon a subjective determination of spot loss, which can vary in degree due to partial methylation (). In order to verify the RLGS results and to obtain more extensive methylation information around the area of the NotI site with quantitative data we used bisulfite sequencing by Mass Array Quantitative Methylation Analysis (MAQMA) on the Sequenome platform.Citation31,Citation32 MAQMA gives a quantitative value of percent methylation at each informative CpG dinucleotide. A diagrammatic representation of the quantitative data is shown in for RLGS spot 1F22, which generally confirms the RLGS results shown in , with the most methylation seen in the cerebellar cortex and the frontal lobe white matter. In order to get a single value for comparative purposes between tissue types or the neural or non-neural nature of the tissues, the average percent methylation of all the CpG dinucleotides within the sequenced CpG island was calculated and the results were plotted ().
These data confirmed tissue specific methylation differences for the spots 1F22, 3B07, 3D24 and 3D46. For all four loci, the cerebellar cortex showed the differential methylation with 3B07 being the only case of less methylation. For all four loci, the consistency of methylation level in all five cerebellar cortex samples is striking. For RLGS spot 1F22 (), the cerebellar cortex has an average of 78% methylation (n = 5), while the white matter from the same region exhibited only 35% methylation (n = 3). RLGS spot 3B07 (), showed hypomethylation in grey matter with an average of 31%, while white matter averaged 52%. In this case, even the cerebellar white matter had slightly less methylation than any of the other tissues, which averaged 62% methylation (n = 24). By quantitative Southern blot analysis ( and C) spot 3D24 exhibited a high level of methylation in cerebellar cortex but only a moderate methylation level in all other tissues and this is confirmed again in .
Additionally, these data demonstrate neural vs. non-neural methylation specificity for RLGS spots 1F22, 3D24 and 3D46, regardless of whether or not samples of cerebellar cortex were included in the analysis. For example, shows the methylation status of 3D46. All the brain tissues have a higher level of methylation (51–78%) than the non-neural tissues (both the endoderm derived tissues (18–48%) and the mesoderm derived tissues (17–38%). Statistical analysis demonstrated that there is significantly (p << 0.001) more methylation in the brain tissues than non-neural tissues (n = 17, and 15, respectively). Furthermore, even when the cerebellar cortex samples were removed from the analysis, the difference was still highly significant (p << 0.001). Similar neural vs. non-neural methylation specificity is seen for 1F22, and to a lesser extent 3D24. This was not the case for 3B07, which shows hypomethylation of the CpG island only in cerebellar cortex.
Discussion
The data presented in this article provide a global assessment of CpG island methylation differences in normal adult human somatic tissues. Our data demonstrate tissue specific CpG island hypermethylation, confirmed by multiple approaches, for three RLGS loci: 3D24 (pseudogene of AP1S2), 3D46 (KIF19) and 1F22 (LHX2), as well as tissue specific CpG island hypomethylation of 3B07 (CNPY1). We further demonstrate that not only do different regions of the human brain have differential methylation patterns, perhaps related to their specific functions as suggested by the study of Laad-Acosta et al. (2007),Citation23 but also there are clear differences between grey matter and white matter within the same brain region. This was most clearly demonstrated comparing cerebellar cortex and white matter for RLGS spot 1F22. We propose that methylation differences between the cortex and the white matter reflect differences in the predominant cell types from which the DNA is extracted—the nuclei of neurons in the cortex and of oligodendricytes and astrocytes in the white matter. We also provide evidence suggestive of germ layer specificity in the methylation patterns, with the confirmed examples of the RLGS loci 3D46 (KIF19) and for 1F22 (LHX2). Ectoderm derived (brain) tissues had the highest levels of methylation for these two spots, while the mesodermal tissues and endodermal tissues showed low levels of methylation. A caveat to this interpretation, however, is that we have only neural tissues from ectoderm to compare. Therefore, it is possible that the differences in methylation we demonstrate have more to do with neural vs. non-neural differentiation than the germ layer from which the tissues are derived. Similar conclusions about the relationship between NotI site methylation patterns and developmental similarity of the cell types have been postulated by Sakamoto et al.Citation34 in mouse tissues.
A finding of particular interest is the high percentage of methylation specifically in the cerebellar cortex for the gene LHX2 (LIM/homeobox protein LH2) compared to other regions of the brain. The gene LHX2 acts as a transcriptional regulator and has been recently reported to play the role of a classic selector gene in determining cortical identity during brain development in mouse.Citation35 We found that the LHX2 gene (RLGS spot 1F22) remains highly methylated in the cerebellar cortex (range 76–81%; n = 5) compared to other regions in adult human brain (range 26–56%; n = 12). As shown in Supplemental Figure 1, the Allen Brain Atlas Project found very low expression of Lhx2 in mouse cerebellum by in situ hybridization, but high expression in some forebrain structures that include the frontal and temporal lobes (used in our study). This fits well with our observations that LHX2 is highly methylated only in the cerebellum. In addition, LHX2 is believed to play a role in mesoderm (mesenchyme) development in mouse.Citation36 In our study, the Lhx2 gene shows minimal methylation in mesodermal tissues like kidney, prostate and spleen.
Due to lack of availability of RNA from the autopsy samples we could not directly study gene expression in our study. However, from the GNF expression database we could correlate methylation and expression patterns of some of the genes. In agreement with our methylation findings and the in situ findings from the Allen Brain Atlas Project, the GNF expression database indicates that the Lhx2 gene is highly expressed in most brain regions in both mouse and human except the cerebellum where its expression is comparatively low (Suppl. Fig. 2). Interestingly, this gene is not expressed in most non-brain tissues including the ones used in our study, yet we did not detect DNA methylation in these tissues by either RLGS analysis or bisulfite sequencing. This suggests that DNA methylation is not required for silencing of the gene in some tissues. These observations suggest that DNA methylation may only be required to silence this gene in neural cells, while non-neural cells do not have the ability to express LHX2 regardless of the methylation status.
The CNPY1 gene shows exactly the opposite pattern to LHX2. While LHX2 is important in forebrain development, CNPY1 is important in hindbrain development. The Canopy 1 gene was found to be expressed in the midbrain-hindbrain boundary in zebra fish and plays a major role in the development of the midbrain tectum and the cerebellum.Citation37 The Allen Brain Atlas Project found that mouse Cnpy1 is highly expressed in the cerebellum, but not in most other brain regions (Suppl. Fig. 1). The GNF expression database shows a similar result with a high level of expression only seen in the cerebellum (Suppl. Fig. 2). Our data show that this gene (RLGS spot 3B07) is highly methylated in all tissues except cerebellum (), and is thus consistent with its methylation playing a role in silencing of the gene in noncerebellar tissues.
Taken together, these data indicate that a limited number of CpG islands display tissue specific methylation and demonstrate a remarkable amount of specificity not only comparing one brain region to another, but also among different regions in the same neuroanatomical structure. A more comprehensive microarray or high-throughput sequencing based approach would certainly identify additional such CpG islands, but the proportion would be expected to be the same. We suggest that these tissue specific methylation patterns are a critical aspect of the regulatory mechanisms of tissue-specific gene expression during different phases of development. Furthermore, the fact that different regions of the brain have differential patterns of methylation supports the notion that DNA methylation is a major determinant of the functional specializations of specific brain regions, and perhaps even its cellular composition. Additionally, our finding of neural specific methylation patterns suggests that methylation of some CpG islands may be determined early in development. This may occur in association with some of the epigenetic differences that have been demonstrated at loci that bind polycomb repressive complex 2 (PRC2) as stem cells differentiate along different lineages.Citation38
Materials and Methods
Normal tissue samples.
Normal tissues were obtained from seven patients at autopsy performed within 4–8 h of death. Tissues were immediately frozen in liquid nitrogen and stored at −70°C. Tissues were obtained from the following patient donors: Patient 1 was a 54-year-old Caucasian female; Patient 2 was an 80-year-old Caucasian male; Patient 3 was a 38-year-old Caucasian male; Patient 4 was a 69-year-old Caucasian female; Patient 5 was a 60-year-old male; Patient 6 was a 58 year old male; Patient 9 was a 40 year old male. For all patients there was no evidence of neoplastic or autoimmune disease at the time of death. Tissues from patients 1–4 were collected with approval of the institutional review board of The Ohio State University, which determined that the specimens used in this research were from non-living persons and therefore, according to the United States Code of Federal Regulations § 46.102(f), does not constitute human subjects research. Tissues from patients 5, 6 and 9 were collected with approval by the ethics committee of the board of physicians at the Westfalian Wilhelms University in Munster Germany (IRB # 2007-254-f-S).
Restriction landmark genomic scanning and spot cloning.
The protocol for extraction of genomic DNA from tissues was described previously.Citation24 The published protocol of Dai et al. Citation25 was followed for RLGS gels. RLGS spots of interest were cloned as previously described.Citation24,Citation26–Citation28 RLGS gel analysis was performed as previously described, with visual inspection of profiles.Citation29,Citation30 We have previously demonstrated, and confirm in this study, that the this approach accurately identifies DNA methylation as loss of spot intensity relative to surrounding spots on the same gel when DNA methylation is approximately 40% or greater at the NotI site.Citation29,Citation30
Southern blot analysis.
Genomic DNAs were digested with the appropriate restriction endonucleases for 4 h and subsequently Southern blot analysis was carried out as previously described.Citation24 Southern blots were quantitated using a PhosphorImager and the Imagequant software (Molecular Dynamics, Inc.) to quantitate individual bands. In each lane, the total pixel intensity of an equal area was quantitated in the position of the both the methylated band and the unmethylated band. To represent percentage of methylation, the value for the methylated band was divided by the sum of the methylated and unmethylated band, and multiplied by 100. Since all calculation and measurements are done within each individual lane, unequal loading of the lanes is controlled for.
Sodium bisulfite treatment.
Sodium bisulfite treatment to convert unmethylated cytosine to thymidine was completed following the manufacturer's protocol EZ-96 DNA Methylation Kit (Zymo Research, Orange, CA, USA) using 750 ng in 50 µl of distilled water and M-Dilution Buffer. The treated samples were resuspended in 75 µl of M-Elution Buffer and stored at −20°C.
MassARRAY quantitative methylation analysis (MAQMA).
MassArray Quantitative Methylation Analysis (MAQMA) was performed using the MassARRAY Compact system developed by the Sequenome Company, as previously described.Citation31 Primer sequences are available upon request. This system utilizes mass spectrometry (MS) for the detection and quantitative analysis of DNA methylation. This approach has been shown to a be highly accurate and reproducible way to quantitate methylation.Citation32
Statistics.
For each CpG site locus, a Wilcoxon Rank Sum test was used to compare the mean percentage of methylation as measured by MAQMA across tissue types. Significance was assessed by univariate p-values less than 0.05 (). The heat map and dendogram shown in were produced using the “image” function in R with a “manhattan” distance (absolute distance) between samples and the “Ward” method for clustering.Citation33 In short, the “manhattan” distance between two samples is equal to the number of loci that show methylation differences while the “Ward” method for clustering attempts to minimize the sum of squares of any two (hypothetical) clusters than can be formed at each step in the clustering algorithm. The R computing language was used to create and perform all statistical testing.Citation33
Abbreviations
| HAT | = | histone acetyltransferase |
| HDAC | = | histone deacetylase |
Figures and Tables
Figure 1 An example of tissue specific methylation identified by RLGS analysis of 12 tissues and quantitated by Southern blot analysis. (A) Cutouts of the full RLGS analysis on all 12 tissues with arrows indicating fragment 3D24. (B) Quantitative Southern blot analysis using the 3D24 NotI-EcoRV fragment as hybridization probe. Lane one contains cerebellar cortex DNA digested with EcoRV only. Lanes 2–13 are all double digested with EcoRV and NotI. The DNAs in lanes 2–13 are loaded in the same order as indicated in (A) with peripheral nerve replacing kidney in lane 8. Depending on DNA availability, 0.5 µg (peripheral nerve) to 10 µg (cerebellar cortex) were loaded in each lane. Bands indicating methylation or no methylation are indicated. (C) Quantitation of the percent methylation detected by the Southern blot in (B). For lanes 2–13, percent methylation was determined as described in the Materials and Methods. *, 13 kb EcoRV sequence without a NotI site that is homologous to the probe. BLAST analysis with the probe sequence identified a BAC clone explaining this band.Citation29 (D) Representative RLGS profilesections showing spot 3B07 and demonstrating consistent spot presence (lack of methylation) in C. cort of multiple patients, but spot absence (methylation) in other brain regions from multiple patients. Abbreviations: B. gang, basal ganglia; C. cort, Cerebellar cortex; C. white, Cerebellar white matter; F. cort, Frontal lobe cortex; F. white, Frontal lobe white matter; T. cort, Temporal lobe cortex.
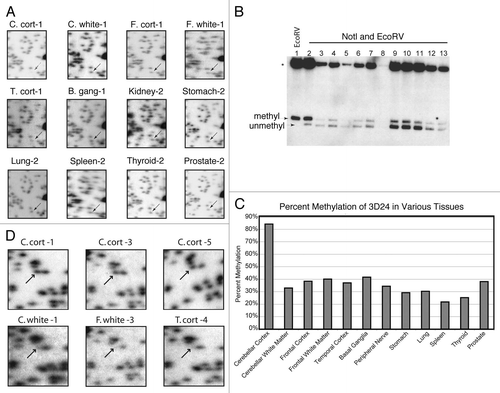
Figure 2 Hierarchical clustering of RLGS data. The heat map and dendogram (top) show the “manhattan” distance (absolute distance) between samples using the “Ward” method for clustering. The RLGS spots are arranged on the Y-axis. Black boxes indicate RLGS spots that were positive for methylation in the indicated tissue. Tissues are arranged on the X-axis. Abbreviations: B. gang, basal ganglia; C. cort, Cerebellar cortex; C. white, Cerebellar white matter; F. cort, Frontal lobe cortex; F. white, Frontal lobe white matter; T. cort, Temporal lobe cortex; Ecto, ectoderm derived tissue; Meso, mesoderm derived tissue; Endo, endoderm derived tissue.
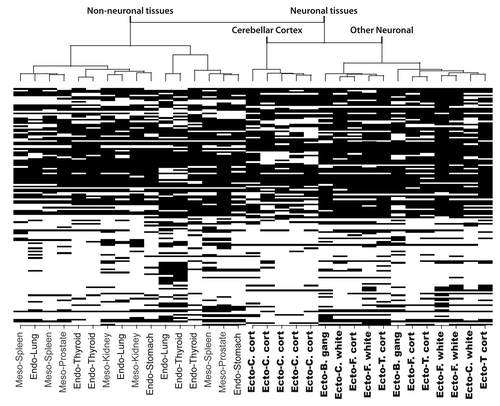
Figure 3 RLGS profiles of spot 3D46 showing neural vs. non-neural tissue specificity. (A) The spot is absent (top) or faintly present (bottom) in ectodermal tissues (C. cortex, T. cortex and B. ganglia). (B) In mesodermal and endodermal tissues (stomach, spleen, lung and thyroid) the spot is clearly present, hence unmethylated. Abbreviations: B. gang, basal ganglia; C. cort, cerebellar cortex; C. white, cerebellar white matter; F. white, frontal lobe white matter; T. cort, temporal lobe cortex.
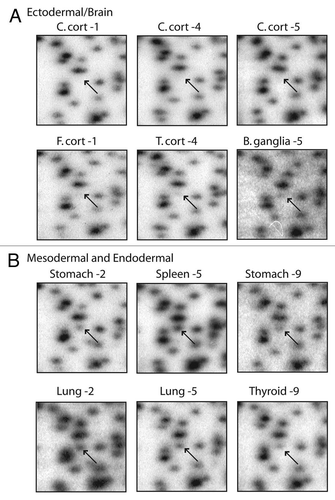
Figure 4A and B Confirmation of RLGS data using MAQMA. (A) MAQMA data for the 1F22 CpG island on all normal human tissues. Each line represents a single sample as labeled on the left indicating the patient number and tissue. In the beads-on-a string diagram, each bead represents a CpG dinucleotide. The percent of methylation is indicated by the grey scale shading as shown by the key at the top of the figure with dark grey representing 100% methylation. The two lung samples at the bottom failed to PCR amplify. To test for bisulfite PCR bias we used peripheral blood lymphocyte (PBL) DNA as a 0% methylation control, and an in vitro methylated (IVM) aliquot of the same DNA as a 100% methylated control. These DNAs were mixed at appropriate ratios to generate 75, 50 and 25% methylated controls and all the controls were bisulfite treated and used as template for bisulfite PCR and MAQMA analysis and shown at the top of the figure. (B–E) The average level of methylation detected by MAQMA across the sequenced fragment of the CpG islands for each samples is shown on the y-axis. This value comes from taking the average of MAQMA values for each CpG dinucleotide sequenced for each sample for each of the four RLGS spots indicated at the top of each figure. The samples are divided up categorically along the x-axis by tissue type on the left, and by neural vs. non-neural tissue on the right. Each symbol represents a single sample. The black, filled in circles indicate the cerebellar cortex samples. p values were determined using the Wilcoxon rank sum testCitation39 to determine if there were differences in mean methylation levels between neural and non-neural. Values are given for the Wilcoxon rank sum test performed both with and without the cerebellar cortex samples.
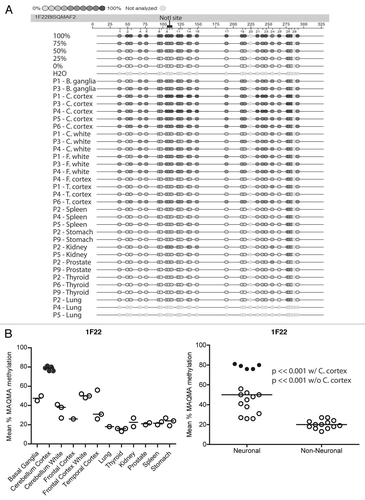
Figure 4C–E Confirmation of RLGS data using MAQMA. (B–E) The average level of methylation detected by MAQMA across the sequenced fragment of the CpG islands for each samples is shown on the y-axis. This value comes from taking the average of MAQMA values for each CpG dinucleotide sequenced for each sample for each of the four RLGS spots indicated at the top of each figure. The samples are divided up categorically along the x-axis by tissue type on the left, and by neural vs. non-neural tissue on the right. Each symbol represents a single sample. The black, filled in circles indicate the cerebellar cortex samples. p values were determined using the Wilcoxon rank sum testCitation39 to determine if there were differences in mean methylation levels between neural and non-neural. Values are given for the Wilcoxon rank sum test performed both with and without the cerebellar cortex samples.
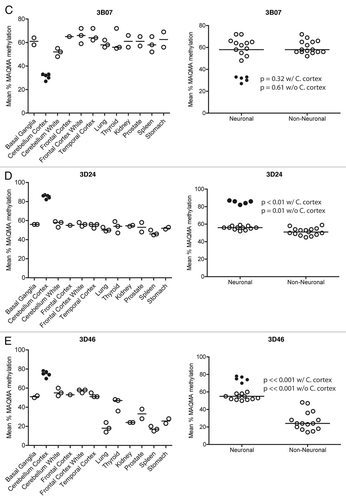
Table 1 Tissue specific methylation of cloned RLGS spots
Table 2 Genomic context of RLGS spots
Additional material
Download Zip (280.5 KB)Acknowledgements
This work was supported in part by the National Cancer Institute through the Roswell Park Cancer Center Support Grant, [CA 16056] (D.J.S., S.G., J.C.M.).
References
- Jaenisch R. DNA methylation and imprinting: why bother?. Trends Genet 1997; 13:323 - 329
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69:915 - 926
- Panning B, Jaenisch R. RNA and the epigenetic regulation of X chromosome inactivation. Cell 1998; 93:305 - 308
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature 1993; 366:362 - 365
- Brandeis M, Ariel M, Cedar H. Dynamics of DNA methylation during development. Bioessays 1993; 15:709 - 713
- Eden S, Cedar H. Role of DNA methylation in the regulation of transcription. Curr Opin Genet Dev 1994; 4:255 - 259
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature 1986; 321:209 - 213
- Ng HH, Bird A. DNA methylation and chromatin modification. Curr Opin Genet Dev 1999; 9:158 - 163
- Turker MS, Bestor TH. Formation of methylation patterns in the mammalian genome. Mutat Res 1997; 386:119 - 130
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987; 196:261 - 282
- Walsh CP, Bestor TH. Cytosine methylation and mammalian development. Genes Dev 1999; 13:26 - 34
- Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998; 58:5489 - 5494
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 2006; 38:1378 - 1385
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457 - 466
- Song F, Smith JF, Kimura MT, Morrow AD, Matsuyama T, Nagase H, et al. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc Natl Acad Sci USA 2005; 102:3336 - 3341
- Kitamura E, Igarashi J, Morohashi A, Hida N, Oinuma T, Nemoto N, et al. Analysis of tissue-specific differentially methylated regions (TDMs) in humans. Genomics 2007; 89:326 - 337
- Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol 2008; 6:22
- Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, Graf S, et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res 2008; 18:1518 - 1529
- Schilling E, Rehli M. Global, comparative analysis of tissue-specific promoter CpG methylation. Genomics 2007; 90:314 - 323
- Shen L, Kondo Y, Guo Y, Zhang J, Zhang L, Ahmed S, et al. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet 2007; 3:2023 - 2036
- Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, et al. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet 2009; 18:4808 - 4817
- Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009; 5:1000602
- Ladd-Acosta C, Pevsner J, Sabunciyan S, Yolken RH, Webster MJ, Dinkins T, et al. DNA methylation signatures within the human brain. Am J Hum Genet 2007; 81:1304 - 1315
- Smiraglia DJ, Fruhwald MC, Costello JF, McCormick SP, Dai Z, Peltomaki P, et al. A new tool for the rapid cloning of amplified and hypermethylated human DNA sequences from restriction landmark genome scanning gels. Genomics 1999; 58:254 - 262
- Dai Z, Zhu WG, Morrison CD, Brena RM, Smiraglia DJ, Raval A, et al. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum Mol Genet 2003; 12:791 - 801
- Yu L, Liu C, Bennett K, Wu YZ, Dai Z, Vandeusen J, et al. A NotI-EcoRV promoter library for studies of genetic and epigenetic alterations in mouse models of human malignancies. Genomics 2004; 84:647 - 660
- Zardo G, Tiirikainen MI, Hong C, Misra A, Feuerstein BG, Volik S, et al. Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors. Nat Genet 2002; 32:453 - 458
- Smiraglia DJ, Kazhiyur-Mannar R, Oakes CC, Wu YZ, Liang P, Ansari T, et al. Restriction Landmark Genomic Scanning (RLGS) spot identification by second generation virtual RLGS in multiple genomes with multiple enzyme combinations. BMC Genomics 2007; 8:446
- Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet 2000; 24:132 - 138
- Smiraglia DJ, Rush LJ, Fruhwald MC, Dai Z, Held WA, Costello JF, et al. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum Mol Genet 2001; 10:1413 - 1419
- Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci USA 2005; 102:15785 - 15790
- Coolen MW, Statham AL, Gardiner-Garden M, Clark SJ. Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry: critical evaluation and improvements. Nucleic Acids Res 2007; 35:119
- Computing TRFfS. The R Statistical Computing Language 2005; www.r-project.org
- Sakamoto H, Suzuki M, Abe T, Hosoyama T, Himeno E, Tanaka S, et al. Cell type-specific methylation profiles occurring disproportionately in CpG-less regions that delineate developmental similarity. Genes Cells 2007; 12:1123 - 1132
- Mangale VS, Hirokawa KE, Satyaki PR, Gokulchandran N, Chikbire S, Subramanian L, et al. Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science 2008; 319:304 - 309
- Kolterud A, Wandzioch E, Carlsson L. Lhx2 is expressed in the septum transversum mesenchyme that becomes an integral part of the liver and the formation of these cells is independent of functional Lhx2. Gene Expr Patterns 2004; 4:521 - 528
- Hirate Y, Okamoto H. Canopy1, a Novel Regulator of FGF Signaling around the Midbrain-Hindbrain Boundary in Zebrafish. Current Biology 2006; 16:421 - 427
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006; 125:301 - 313
- Welch BL. On the comparison of several mean values: an alternative approach. Biometrika 1951; 38:330 - 336