Abstract
Background: Methylation of tumor suppression genes (TSGs) is common in myeloid malignancies. However, application of this as a molecular marker for risk stratification in patients with AML is limited. Design and Methods: To elucidate the impact of patterns of TSG methylation on outcome in cytogenetically normal patients, 106 samples from patients with having normal cytogenetic AML were evaluated for methylation of 12 genes by MSP. For sake of comparison, samples from patients with AML and abnormal cytogenetics (n=63) were also evaluated. Results: Methylation frequencies in the whole group (n=169) were similar to previous reports for CDH1 (31%), ER (31%), FHIT (9%), p15INK4b (44%), p73 (25%), and SOCS1 (75%). Methylation of CTNNA1 was observed in 10%, CEBP-α in16%, CEBP-δ in 2%, MLH1 in 24%, MGMT in 11% and DAPK in 2% of AML samples. We find that DNA methylation was more prevalent in patients with normal compared to karyotypically abnormal AML for most genes; CEBPα (20% vs 9%), CTNNA1 (14% vs 4%), and ER (41% vs 19%) (p<0.05 for all comparisons). In contrast, p73 was more frequently methylated in patients with karyotypic abnormalities (17% vs 38%; p<0.05), perhaps due to specific silencing of the pro-apoptotic promoter shifting p73 gene expression to the anti-apoptotic transcript. In AML patients with normal cytogenetics, TSG methylation was not associated with event free or overall survival in a multivariate analysis. Conclusions: In patients with AML, TSG methylation is more frequent in patients with normal karyotype than those with karyotypic abnormalities but does not confer independent prognostic information for patients with normal cytogenetics.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by uncontrolled proliferation of primitive hematopoietic elements and recurrent cytogenetic abnormalities.Citation1 Specific genetic abnormalities (i.e., t(8; 21), inv(16), t(15; 17)) are pathognomonic and prognostically important in patients with AML. However, most patients with AML have a normal karyotype and, in these patients, it is often difficult to predict long-term outcome.Citation2,Citation3 The prognostic role of novel molecular markers, including mutations of FLT3 or NPM1 genes and DNA methylation of p15INK4b, ER or CDH1 have been reported in AML patients.Citation4–Citation6 While the former have been adopted into widespread clinical practice, methylation of tumor suppressor genes (TSGs) has not yet proven useful as a prognostic marker, since previous studies have included heterogeneous patient populations and there has been a failure to control for the cytogenetic profile.Citation7,Citation8
Advances in chemotherapy and transplantation have substantially improved long-term survival in children with AML, but these advances have not similarly improved the outcomes for adults. Strategies to identify patients who are likely to relapse may be useful in developing new treatment algorithms.Citation3 This goal is particularly important in the large (∼50%) fraction of adult AML patients presenting with normal cytogenetics, in whom conventional treatment does not initially include bone marrow transplantation. Since these leukemias lack gross chromosomal (genetic) alterations, it is plausible that epigenetic changes may be more important in driving the pathogenesis in this setting. Previous work suggesting that methylation of TSGs may be prognostically important in patients with AML is also of interest in light of novel demethylating therapies (i.e., 5-azacytidine and 5-aza-2′-deoxycitidine) with documented efficacy in myeloid malignancies.Citation9–Citation12 However, few, if any, studies have confirmed these previous reports, suggesting the possibility of positive publication bias, and none have limited the analysis to genetically homogenous patients.
This study was designed to assess methylation of genes with previously published biologic rationale and/or prognostic significance in AML. We chose this technique, rather than a genome wide strategy, due to the small number of patients in this study and the significant risk of false discovery inherent in these broader assessments. Our focus was to validate genes known to play a role in AML rather than to identify new genes for this unique set of patients. Methylation specific PCR (MSP) was used as the technique to assess gene specific methylation since this has been shown to detect methylation which is associated with alterations in expression of associated genes. (see Suppl. Table 1 for references by gene).
In order to examine the role of DNA methylation of TSGs in providing prognostic information we (a) determined the frequency of methylation of 12 TSGs across cytogenetic and molecular (FLT3 and NPM1 mutant) risk groups, (b) compared the methylation frequencies for AML patients with normal karyotype de novo versus those with antecedent cytopenias/myelodysplastic syndrome (MDS), (c) compared methylation frequencies for patients with normal karyotype AML at the time of diagnosis to those presenting with normal karyotype AML at the time of relapse and (d) evaluated the impact of methylation of TSGs as a prognostic biomarker of clinical outcome in uniformly treated (cytarabine-based chemotherapy) AML patients with a normal cytogenetic profile. Supplementary Table 1 details the chromosomal location of the 12 genes selected, their previously published rates of methylation and the rationale for their importance in AML.
Results
Patient characteristics.
A total of 169 AML patient samples were collected, of which 106 had normal cytogenetics. details demographic information for the patients. Median age was 61 for both groups. Of the normal cytogenetic samples, 34 were procured at relapse and 72 were procured from patients at initial diagnosis. These 72 samples (from patients at initial AML diagnosis) were annotated for treatment received. All 72 patients received cytarabine based induction therapy, with the majority of patients receiving combination therapy with cytarabine and daunorubicin. Of the 72 patients, there were 39 with de novo AML (four received FLAM,Citation17 three received the Linker regimen,Citation18 six received standard “7 plus 3” and 26 received ACDVP16Citation19,Citation20) and 33 patients had antecedent MDS (five received FLAM and 28 received ACDVP16). No patients received epigenetic therapy with 5-azacytidine or deoxy-azacytidine prior to sample analysis. Since all 72 patients were administered a cytarabine based induction regimen, their event free and overall survival was assessed and outcomes were correlated with methylation of individual genes or numbers of genes. These results were analyzed in a multivariate model including age, total WBC count at diagnosis (greater or less than 50,000/uL as a dichotomous variable), antecedent cytopenias, NPM1 mutations and FLT3-ITD status. Age was assessed as a continuous variable. ()
Methylation frequency by gene.
The observed methylation frequencies for our AML samples was similar to that in previous studies (see Suppl. Table 1) for CDH1 (31%; 53/169), ER (31%; 52/169), FHIT (9%; 16/169), p15INK4b (44%; 74/169), p73 (25%; 42/169) and SOCS1 (75%; 127/169). Methylation of CTNNA1 was observed in 10% (17/169), CEBP-α in 16% (27/169), MLH1 in 24% (41/169) and MGMT in 11% (19/169) of AML samples; these genes have rarely been reported in the literature previously. Methylation of CEBP-δ (2%; 3/169) and DAPK (2%; 2/109) was rare. displays methylation frequencies by karyotypic risk group. SOCS1 and p15 were the most frequently methylated genes, followed by p73 and CDH1. No significant differences were seen in methylation frequency between different (low, medium, high) cytogenetic risk groups.
Comparisons between methylation patterns by epigenetic and genetic alterations.
While the previous analysis had suggested no distinct differences between varying cytogenetics risk groups, we sought to determine whether the overall level of genetic alterations were associated with any differences in the frequency DNA methylation. To do so, we compared all normal karyotype patients to those with any karyotypic abnormality. demonstrates methylation frequencies for the patients with normal cytogenetics (n = 106) and those with any karyotypic abnormality (n = 63), consisting predominantly of patients with complex karyotype (n = 30). TSG methylation was more frequent (p < 0.05) in patients with normal cytogenetics than those with a karyotypic abnormality for CEBP-α, (20% vs. 9%), CTNNA1 (13% vs. 4%) and ER (41% vs 19%) (). In contrast, only p73 methylation was more common in AML samples with a cytogenetic abnormality (38% versus 17% in normal karyotype; p < 0.05). Methylation of this gene was particularly common in samples with a complex karyotype, with 41% of these leukemias having p73 methylation. It is also noted that frequency of methylation of p73 was 70% in the good risk cytogenetic group, of which five were APL from the total of ten samples examined.
Methylation frequency in FLT3 and NMP1 mutated AML.
We next examined whether the differences in DNA methylation observed for gross genetic changes, that is cytological rearrangements, would also be observed with smaller genetic alterations. The two most common mutational changes in AML involve FLT3 and NPM1. and B demonstrate the relative frequency of methylation by gene for patients with and without these abnormalities. No significant differences were found in the frequency of any TSG methylation according to the presence of FLT3-ITD or NPM1 mutations, suggesting that the altered DNA methylation patterns observed with chromosomal rearrangements did not also include these individual gene mutations.
Methylation frequency by antecedent hematological diagnosis in normal karyotpe AML.
In patients with AML, the presence of an antecedent hematologic condition (i.e., MDS, myeloproliferative disorder or cytopenias >6 months prior to diagnosis) portends a worse prognosis.Citation21 Furthermore, such an antecedent diagnosis has been associated with an increased rate of TSG methylation.Citation11 Within our group of patients uniformly treated with high-dose chemotherapy, no significant differences were observed between the rates of TSG methylation in normal karyotype AML patients with (n = 33) and without (n = 39) a history of antecedent cytopenias/MDS ().
Methylation frequency in normal karyotype aml: diagnostic versus relapsed samples.
No significant differences in methylation were observed between normal karyotype samples obtained from patients at the time of diagnosis (n = 72) and those obtained from patients at the time of AML relapse (n = 34). A trend towards increased methylation of p73 was appreciated in the relapsed samples (13% vs. 26%) ().
Implication of p73 methylation frequency in samples with cytogenetic abnormalities.
Of the 12 genes examined, only p73 was more frequently methylated in samples with an abnormal rather than a normal karyotype, and this difference was statistically significant (p < 0.04). The abnormal karyotype was frequently noted to be associated with the complex cytogenetic profile rather than a monosomy alteration. One explanation for this is the complex transcripts generated from the p73 gene, depicted in demonstrating the two different transcripts, highlighting the promoter regions for each and the location of the CpG island. The long transcript,originating from the 5′ promoter, TAp73, is transcribed from a promoter within a CpG island.Citation16 DNA methylation of this promoter results in silencing the pro-apoptotic TAp73 gene transcript, while potentially allowing retained expression of the anti-apoptotic DNp73 transcript. Previous studies only demonstrated loss of the longer or full-length transcriptCitation16 since dual promoter usage was not known at the time of identification of silencing of p73. To test this, we examined several cell lines to determine if methylation of p73 results in loss of the TAp73 transcript with preserved DNp73 expression.
KG1a and U937 leukemia cell lines have complete promoter region methylation of p73, and silencing of the TAp73 gene transcript ( and C). In KG1a there was continued expression of the DNp73 gene transcript, while in U937 this transcript was also silenced. The HL60 leukemia cell line demonstrated hemimethylation of p73 with a corresponding decrease in the TAp73 gene product, but with significant expression of the DNp73 gene product. ML1 was entirely unmethylated and had normal levels of both TAp73 and DNp73. Treatment of KG1a and U937 cell lines with 1 uM 5-azacytidine for 72 h resulted in reversal of methylation of the p73 gene () and re-expression of the TAp73 transcript (). Treatment with 300 nM suberoylanilide hydroxamic acid (SAHA) did not reverse methylation nor did this result in re-expression of the TAp73 gene product.
TSG methylation as a prognostic marker.
Methylation of the 12 TSGs analyzed did not correlate with outcome in our uniformly treated, newly diagnosed patients with normal karyotype. and B demonstrate multivariate hazard ratios for relapse and death by gene in this group. Methylation of p73 and MLH1 demonstrate a trend towards an adverse impact upon disease free survival, however this finding was not statistically significant. We examined the impact of methylation of multiple TSGs in concert and found no statistically significant impact of methylation of multiple genes on outcome in this sample of uniformly treated patients. Furthermore, there was no correlation between primary refractory disease status after high-dose induction chemotherapy and methylation of individual TSG or groups of TSGs (data not shown).
Discussion
Over 50% of patients diagnosed with AML have normal cytogenetics. In these patients, current prognostic indicators do not predict those patients at higher risk of relapse who would benefit from initial therapy with an experimental regimen or a bone marrow transplant. Methylation of TSGs has been reported to predict poor outcome in patients with AML.Citation5 Our data suggest that methylation of TSGs is more frequent in AML patients with normal karyotype than those with karyotypic abnormalities; however, we found no evidence that methylation of any one of the 12 genes (or group of genes) correlated with outcome in AML patients with normal cytogenetics that were uniformly treated with standard cytarabine based induction chemotherapy. Our findings may differ from previous reports because we studied a more uniform patient population. Specifically, previous reports have been limited by sample selection and heterogeneity of both disease and treatment.Citation5,Citation7,Citation12 Our study carefully details the methylation of potentially important TSGs in a uniformly treated population of patients within the recognized paradigm of cytogenetic and molecular risk groups in AML.
Comparison of normal karyotype patients to those with structural abnormalities was performed with the rationale that structural/genomic chromosomal changes may “trump” the biological impact of epigenetic gene silencing. Methylation frequency of CEBP-α, CTNNA1, ER and p73 differed (p < 0.05) between patients with normal cytogenetics and those with karyotypic abnormalities. For all genes except p73, TSG methylation was more common in patients with a normal karyotype than in those with any cytogenetic abnormality (). These differences suggest that patients with a normal karyotype (e.g., lacking large genetic changes) may have a greater degree of epigenetic alteration(s).
Although there are reports documenting the acquisition of promoter hypermethylation at the time of relapse for individuals sampled serially we found no difference in the rates of methylation between normal karyotype samples from the time of AML diagnosis versus those obtained from patients at the time of relapse.Citation22 Since we evaluated two different patient populations (one group at diagnosis and one at relapse) this may explain the difference in our results from the previous literature. The heterogeneity between individual patients may be greater than the differences in methylation frequency that occur for individual genes from the time of diagnosis to relapse. Furthermore, it may suggest that it is important to evaluate the change in methylation status (i.e., unmethylated to methylated) rather than frequency of methylation for each gene.
Methylation of TSG predicts poor outcome in patients with AML.Citation23 We examined the impact of methylation of these genes on survival in the entire group and, in contrast to prior reports from other groups, found no evidence that methylation of any one gene correlated with outcome.Citation7 Previous reports in the literature are limited by sample selection and heterogeneity of both disease and treatment.Citation5,Citation7,Citation12 We therefore examined a significant number of AML patients with normal cytogenetics, all of whom had undergone standard induction chemotherapy at a single institution. Even within this more uniform population, methylation of the selected genes did not confer a statistically significant impact upon event free or overall survival.
Among the 12 genes examined, only p73 was more frequently methylated in AML samples with an abnormal karyotype. Although this finding does not directly implicate methylation of p73 in the development of chromosomal abnormalities, the association is particularly interesting since two different isoforms of p73 (TAp73 and DNp73) appear to affect the resulting phenotype. The long transcript (TAp73) includes exons 1 and 2 of the gene and is transcribed from an upstream promoter possessing a typical CpG island, while the short transcript (DNp73) is transcribed from a distinct promoter near exon 3. These two isoforms appear to have pro and anti-apoptotic effects respectively, and loss of the long isoform, TAp73 results in a high incidence of spontaneous tumors and genomic instability in TAp73 deficient mice.Citation24 This is consistent with the critical role of p73 in caspase-independent mitotic death,Citation25 suggesting that with loss of pro-apoptotic TAp73, cells would survive abnormal mitosis and accumulate genomic alterations. The phenotype observed in KG1a and HL60 cell lines, loss of the TAp73 transcript and preservation of DNp73 transcription, may produce an environment permissive for genomic instability and provides an explanation for our observation that samples with cytogenetic abnormalities are more likely to have methylation of p73. While p73 methylation is a relatively uncommon event in all patients with AML, the frequency of 41% of patients with a complex karyotype suggests that this may contribute to the acquisition of additional genomic abnormalities.
Our study has certain limitations. Although we did not identify epigenetic changes that predicted outcome in AML patients with normal cytogenetics, it is possible that methylation of genes not yet identified may be associated with adverse outcome in these patients. Improved technologies for identifying genetic and epigenetic alterations (HELLP assay and microarray studies) may help to identify markers that can be used for risk stratification of this population in the future.Citation26 Of published studies, ours is one of the most comprehensive analysis of TSG epigenetic changes by MSP within the recognized paradigm of cytogenetic and molecular risk groups in AML. Nevertheless, it is limited by the small size of our patient population.
Although none of the genes examined in our study were associated with a statistically significant impact on prognosis, there was a trend towards adverse outcome for patients with p73 and MLH1 methylation, perhaps suggesting that functional loss of these genes may provide leukemia cells with the ability to tolerate DNA damage and thus confer chemotherapy resistance. Further investigation into the impact of p73 methylation on genetic stability is necessary to verify this hypothesis.
Design and Methods
Cell lines.
Cell lines (DKO, HL60, KG1a, ML1 and U937) were purchased from American Type Culture Collection and grown in recommended media (ATCC, Manassas, VA, USA). Pelleted cells from each cell line were used for RNA and DNA extraction. DNA was extracted and bisulfite treated for analysis as described below.
Patients, diagnosis and treatment.
One hundred and sixty nine freshly frozen bone marrow or peripheral blood samples collected between February 1998 and January 2008 from patients with AML were obtained from the Johns Hopkins Leukemia bank. The diagnosis of AML was made by standard criteriaCitation13 in 91 men and 78 women with a median age of 61 years (). All clinical samples and clinical data were obtained in accordance with HIPAA regulations as part of a protocol approved by the Institutional Review Board at the Johns Hopkins Hospital, and all patients signed informed consent according to Health and Human Services guidelines and the Declaration of Helsinki. Samples were annotated for patient age and white blood cell count (WBC) at diagnosis, cytogenetics by karyotype and FISH analysis, induction therapy and overall and event free survival. Clinical data for overall and event free survival was obtained for the 72 treated patients with normal cytogenetics and outcomes were correlated with methylation status for the genes in our panel.
DNA extraction.
Freshly frozen bone marrow or peripheral blood samples were mixed with an equal volume of lysis buffer (50 mM Tris, 50 mM EDTA, 2% sodium dodecyl sulfate, 10 mg/mL protein kinase) and incubated overnight at 55°C. One half volume of lysis buffer was added daily until the sample had been completely digested. Once digestion was complete, DNA was extracted with phenol:chloroform:isoamyl alcohol (25:24:1), ethanol precipitated and re-suspended in 50–500 uL of double distilled water (ddH2O). DNA was quantified using the NanoDrop® ND-1,000 Spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA).
Bisulfite treatment.
Bisulfite modification of 1–2 ug of DNA per sample was performed using the Zymo Research EZ DNA methylation kit™ (Orange, CA, USA) as per manufacturer's instructions. Bisulfite treated DNA was re-suspended in 40–80 uL of ddH2O.
Methylation-specific polymerase chain reaction.
Methylation-specific PCR primers for CTNNA1, CEBP-α, CEBP-δ, DAPK, CDH1, ER, FHIT, MLH1, MGMT, p15INK4b, p73 and SOCS1 have been published previously and were purchased from Integrated DNA Technologies (Coralville, IA, USA). All primers appear in Supplementary Table 2. MSP was carried out in 25 uL reaction volumes containing 10x MSP buffer (16.6 mM ammonium sulfate/ 67 mM Tris, pH8.8/6.7 mM MgCl2/ 10 nM 2-mercaptoethanol), 10 mM deoxynucleoside triphosphates, 0.5 uL of a 5 mM solution of each of the methylated or unmethylated primers, 0.5 uL of JumpStart™ REDtaq® DNA polymerase (Sigma-Aldrich, St. Louis, MO, USA) and 2–4 uL of bisulfite treated DNA.Citation14 DNA was amplified with the following protocol: 95°C for 5 min, followed by 35 cycles of 95°C for 30s, 58 or 60°C for 30s (58°C for all genes except MGMT and MLH1 for which an annealing temperature of 60°C was utilized), 72°C for 30s and a final extension step of 72°C for 5 minutes. In vitro methylated DNA (IVD) was created by treating human cell line DNA with SSI-1 Methylase (New England BioLabs, Ipswich, MA, USA) as directed and used as a positive control following bisulfite treatment as described above. Normal lymphocytes collected from six healthy donors were used as a negative control. Seven and a half microliters of each amplification product were resolved on 2% agarose gel containing GelStar® Nucleic Acid Gel Stain (Lonza, Rockland, ME, USA) and visualized under ultraviolet light (UV). Genes with a methylation frequency less than 5% were excluded from our survival analysis due to a lack of statistical power.
FLT3-ITD mutational analysis.
For qualitative analysis of FLT3-ITD mutations, genomic DNA was isolated as described above. PCR was performed using QuantiTect SYBR Green PCR Master Mix (Qiagen, Valencia, CA, USA), with primers flanking the juxtamembrane coding region as previously published (Sense 5′-GCA ATT TAG GTA TGA AAG CCA GC-3′, anti-sense 5′-CTT TCA GCA TTT TGA CGG CAA CC-3′).Citation15 The following protocol was used: 95°C for 5 min, followed by 35 cycles of 94°C for 30s, 55°C for 30s, 72°C for 1 min, and a final extension cycle of 72°C for 10 min. 10 µL of each amplification product was resolved on 2.5% agarose gel containing GelStar® Nucleic Acid Gel Stain (Lonza, Rockland, ME, USA) and visualized under UV.
NPM1 mutational analysis.
DNA corresponding to exon 12 on NPM1 was amplified by PCR using the following primers: NPMF 5′-TTA ACT CTC TGG TGG TAG AAT GAA-3′, NPMR 5′-CAA GAC TAT TTG CCA TTC CTA AC-3′. 25 µL reactions were prepared for each sample using 12.5 uL of QuantiTect SYBR Green PCR Master Mix (Qiagen, Valencia, CA, USA), 7.5 µL of ddH2O, 0.5 µL of 5 mM solution of each primer and 4 µL (50–100 ng) of genomic DNA. Samples were amplified using the following protocol: 94°C for 10 min, 35 cycles of 94°C for 30s, 58°C for 30s, 72°C for 1 min and a final extension cycle of 72°C for 10 min. PCR amplification products were mixed with an equal volume of 2x formamide loading buffer (90% [v:v] deionized formamide, 1X sodium borate, 0.4% [w:v] each of xylene cyanol and bromophenol blue), incubated at 100°C for 3 min and loaded onto a 4.5% nondenaturing polyacrylamide sodium borate gel. Electrophoresis was performed at 100V for 4–6 h at 4°C. Gels were stained with ethidium bromide and PCR products visualized under UV.
RNA extraction and RT-PCR.
RNA was extracted from cell lines using the RNeasy Mini Kit from Qiagen (Valencia, CA, USA) according to manufacturer's instructions. cDNA was made from RNA using Oligo dT(20) and the Superscript™ III RT enzyme (Invitrogen, Carlsbad, CA, USA). RT reactions were carried out with and without the RT enzyme to exclude the possibility of non-coding occult DNA sequences within the extracted samples. RT-PCR primers for p73 (TAp73) have been published previouslyCitation16. Primers for the internal short transcript of p73 (DNp73) were designed and validated by the authors. Primers for TAp73 were 5′-CGG GAC GGA CGC CGA TG-3′ (sense, exon 1) and 5′-GAA GGT CGA AGT AGG TGC TGT CTG G-3′ (antisense, exon 3). Those for DNp73 were 5′-ACT AGC TGC GGA GCC TCT C-3′ (sense, exon 3) and 5′-TGC TAG CAG ATT GAA CTG G-3′ (antisense, exon 5). β-Actin was used as a positive control for gene expression. PCR reactions were optimized and carried out in 25 µL reaction volumes using 12.5 µL of QuantiTect SYBR Green PCR Master Mix (Qiagen, Valencia, CA, USA), 7.5 µL of ddH2O, 0.5 µL of 5 mM solution of each primer and 4 µL (50–100 ng) of genomic DNA. Reaction conditions were 95°C for 15 min, 25 to 40 cycles of 95°C for 30 s (35 cycles for TAp73, 40 cycles for DAp73 and 25 cycles for β-Actin), 60°C for 30s, 72°C for 30s, and a final extension cycle of 72°C for 10 min. 8 µL of each amplification product were resolved on 2.5% agarose gel containing GelStar® Nucleic Acid Gel Stain (Lonza, Rockland, ME, USA) and visualized under UV.
Statistical analysis.
All patient samples were obtained from the Johns Hopkins Leukemia Tumor bank. Clinical information was collected from the electronic patient record. Associations between individual gene markers and variables such as NPM1 mutation, FLT3 mutation, antecedent disease status and white blood cell count at diagnosis were assessed by means of logistic regression analysis. Two-sided p values <0.05 were considered to indicate statistical significance. Univariate and multivariable Cox proportional hazards regression models were utilized to assess the independent effect of DNA methylation on disease recurrence and mortality. Results of all regression models are reported as hazard ratios with corresponding 95% confidence intervals. To control for multiple testing, Bonferroni adjustment was used to correct for family-wise error rate. All statistical calculations were performed with the use of STATA 10 statistical package (College Station, TX, USA).
Conflict of Interest
Dr. James Herman is a consultant to and receives research support from OncoMethylome Sciences. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies. All the other authors have no conflicts of interests to disclose.
Figures and Tables
Figure 1 (A) Methylation rates by gene for each of the genes examined across the recognized cytogenetic risk groups. The good risk group includes 10 patients with t(8; 21), inv(16) or t(15:17) mutations, the intermediate group includes 106 patients with normal karyotype leukemia and 14 patients with <3 karyotypic abnormalities, the adverse group includes 30 patients with complex karyotype and 9 patients with abnormalities involving chromosomes 5, 7 or 11q23. No significant differences were observed between each of these cytogenetic risk groups. (B) When patients with normal karyotype were compared to those with a structural karyotype abnormality, differences in methylation frequency were observed. Methylation of CEBP-α, CTNNA1, ER and p73 (*) were significantly different between the two groups (p < 0.05).
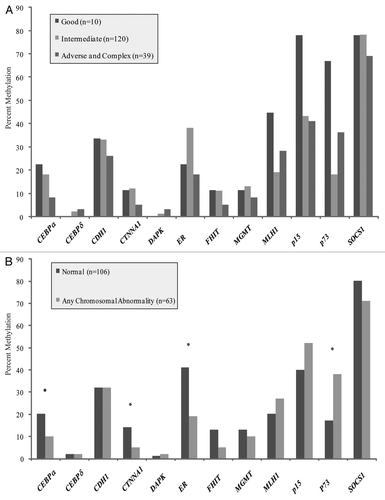
Figure 2 (A) No significant differences were observed between rates of methylation for normal karyotype samples with and without a FLT3-ITD mutation. (B) No significant differences in rates of methylation were observed between samples with and without a NPM1 mutation.
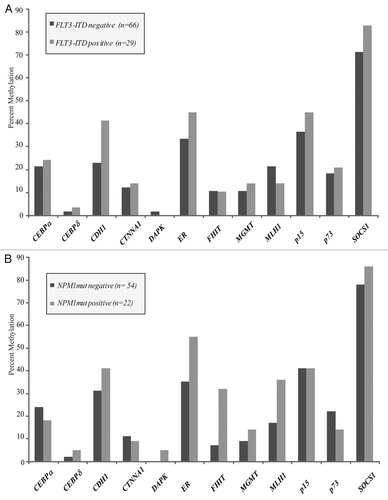
Figure 3 (A) No significant differences were seen between samples obtained from patients with de novo leukemia and those presenting with a history of an antecedent hematologic abnormality. Patients in the latter group were older, with a median age of 69 years vs. 52 years in the de novo group, and all had a diagnosis of AML at the time of sample acquisition. (B) There were no differences in methylation between normal karyotype samples obtained at the time of diagnosis as compared to those obtained at the time of AML relapse.
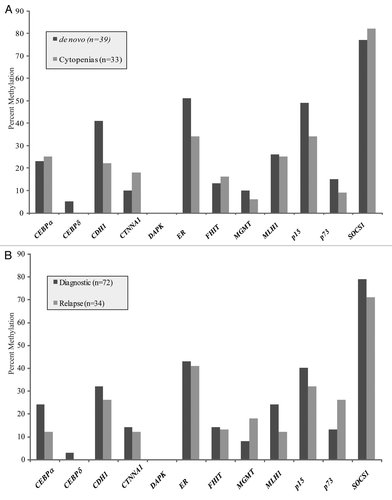
Figure 4 (A) Schematic of p73 gene demonstrating its 14 exons and the location of our MSP and RT-PCR primers. The TAp73 transcript includes exons 1–14 and has a dense CpG island upstream of the transcription start site. The DNp73 transcript includes exons 3–14. (B) Methylation Specific PCR reactions for leukemia cell lines KG1a, U937, HL60 and ML1. KG1a and U937 demonstrate complete methylation, HL60 is hemimethylated for p73, ML1 is unmethylated. NL is a negative control for methylation in normal peripheral blood lymphocytes and IVD is a positive control for methylation. (C) RT-PCR for the long (TAp73) and short (DAp73) transcripts of p73. TAp73 exerts a pro-apoptotic and DNp73 an anti-apoptotic effect. Methylation of the p73 promoter preferentially silences the long transcript of p73 while allowing continued expression of the short transcript, favoring an anti-apoptotic phenotype. (D) Treatment for 72 h with 1 µM 5-azacytidine (5AC) reverses methylation, while untreated (mock) and SAHA treated cell lines maintain a methylated phenotype. (E) Treatment of KG1a and U937 cell lines with 5AC results in re-expression of silenced TAp73 (both) and DNp73 (KG1a).
Figure 5 Uniformly treated patients with normal karyotype AML at initial diagnosis (n = 72) (A) Multivariate hazard ratio for relapse by gene with 95% confidence intervals designated by black bars. (B) Multivariate hazard ratio for death by gene corrected for age, antecedent cytopenia(s) at diagnosis, FLT3-ITD and NPM1 mutational status and total white blood cell count at time of AML diagnosis with 95% confidence intervals designated by black bars.
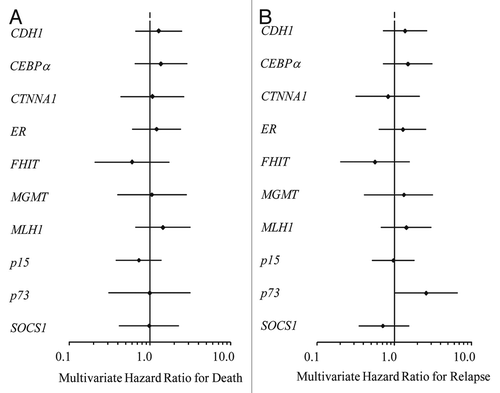
Table 1 Characteristics of the study population
Additional material
Download Zip (151.8 KB)Acknowledgements
This work was supported by The Flight Attendant Medical Research Institute (FAMRI), grant number 032053. MAM would like to acknowledge the Department of Defense (MPD510343) for support. This work was also supported by the NCI Cancer Center Support Grant (CA06793) which helped to support the Specimen Acquisition Core (SAC) Laboratory for sample collection and storage.
References
- Estey E, Dohner H. Acute myeloid leukaemia. Lancet 2006; 368:1894 - 1907
- Jabbour EJ, Estey E, Kantarjian HM. Adult acute myeloid leukemia. Mayo Clin Proc 2006; 81:247 - 260
- Nimer SD. Is it important to decipher the heterogeneity of “normal karyotype AML?”. Best Pract Res Clin Haematol 2008; 21:43 - 52
- Shimamoto T, Ohyashiki JH, Ohyashiki K. Methylation of p15(INK4b) and E-cadherin genes is independently correlated with poor prognosis in acute myeloid leukemia. Leuk Res 2005; 29:653 - 659
- Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P. Promoter hypermethylation of p15INK4B, HIC1, CDH1 and ER is frequent in myelodysplastic syndrome and predicts poor prognosis in early-stage patients. Eur J Haematol 2006; 76:23 - 32
- Ekmekci CG, Gutierrez MI, Siraj AK, Ozbek U, Bhatia K. Aberrant methylation of multiple tumor suppressor genes in acute myeloid leukemia. Am J Hematol 2004; 77:233 - 240
- Plass C, Oakes C, Blum W, Marcucci G. Epigenetics in acute myeloid leukemia. Semin Oncol 2008; 35:378 - 387
- Baldus CD, Bullinger L. Gene expression with prognostic implications in cytogenetically normal acute myeloid leukemia. Semin Oncol 2008; 35:356 - 364
- Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006; 106:1794 - 1803
- Pinto A, Zagonel V. 5-Aza-2′-deoxycytidine (Decitabine) and 5-azacytidine in the treatment of acute myeloid leukemias and myelodysplastic syndromes: past, present and future trends. Leukemia 1993; 7:51 - 60
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009; 10:223 - 232
- Hess CJ, Errami A, Berkhof J, Denkers F, Ossenkoppele GJ, Nygren AO, et al. Concurrent methylation of promoters from tumor associated genes predicts outcome in acute myeloid leukemia. Leuk Lymphoma 2008; 49:1132 - 1141
- Cheson BD, Cassileth PA, Head DR, Schiffer CA, Bennett JM, Bloomfield CD, et al. Report of the National Cancer Institute-sponsored workshop on definitions of diagnosis and response in acute myeloid leukemia. J Clin Oncol 1990; 8:813 - 819
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93:9821 - 9826
- Levis M, Murphy KM, Pham R, Kim KT, Stine A, Li L, et al. Internal tandem duplications of the FLT3 gene are present in leukemia stem cells. Blood 2005; 106:673 - 680
- Corn PG, Kuerbitz SJ, van Noesel MM, Esteller M, Compitello N, Baylin SB, et al. Transcriptional silencing of the p73 gene in acute lymphoblastic leukemia and Burkitt's lymphoma is associated with 5′ CpG island methylation. Cancer Res 1999; 59:3352 - 3356
- Karp JE, Smith BD, Levis MJ, Gore SD, Greer J, Hattenburg C, et al. Sequential flavopiridol, cytosine arabinoside and mitoxantrone: a phase II trial in adults with poor-risk acute myelogenous leukemia. Clin Cancer Res 2007; 13:4467 - 4473
- Linker CA, Ries CA, Damon LE, Sayre P, Navarro W, Rugo HS, et al. Autologous stem cell transplantation for acute myeloid leukemia in first remission. Biol Blood Marrow Transplant 2000; 6:50 - 57
- Geller RB, Burke PJ, Karp JE, Humphrey RL, Braine HG, Tucker RW, et al. A two-step timed sequential treatment for acute myelocytic leukemia. Blood 1989; 74:1499 - 1506
- Bolanos-Meade J, Karp JE, Guo C, Sarkodee-Adoo CB, Rapoport AP, Tidwell ML, et al. Timed sequential therapy of acute myelogenous leukemia in adults: a phase II study of retinoids in combination with the sequential administration of cytosine arabinoside, idarubicin and etoposide. Leuk Res 2003; 27:313 - 321
- Fenaux P. Myelodysplastic syndromes: From pathogenesis and prognosis to treatment. Semin Hematol 2004; 41:6 - 12
- Kroeger H, Jelinek J, Estecio MR, He R, Kondo K, Chung W, et al. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood 2008; 112:1366 - 1373
- Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, Saunthararajah Y, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 2009; 113:1315 - 1325
- Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22:2677 - 2691
- Niikura Y, Dixit A, Scott R, Perkins G, Kitagawa K. BUB1 mediation of caspase-independent mitotic death determines cell fate. J Cell Biol 2007; 178:283 - 296
- Figueroa ME, Reimers M, Thompson RF, Ye K, Li Y, Selzer RR, et al. An integrative genomic and epigenomic approach for the study of transcriptional regulation. PLoS One 2008; 3:1882
- Ongenaert M, Van Neste L, De Meyer T, Menschaert G, Bekaert S, Van Criekinge W. PubMeth: a cancer methylation database combining text-mining and expert annotation. Nucleic Acids Res 2008; 36:842 - 846
- Shimamoto T, Ohyashiki JH, Ohyashiki K. Methylation of p15(INK4b) and E-cadherin genes is independently correlated with poor prognosis in acute myeloid leukemia. Leuk Res 2005; 29:653 - 659
- Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G, et al. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8; 21) myeloid leukemia. Nat Med 2001; 7:444 - 451
- Agrawal S, Hofmann WK, Tidow N, Ehrich M, Van den Boom D, Koschmieder S, et al. The C/EBPdelta tumor suppressor is silenced by hypermethylation in acute myeloid leukemia. Blood 2007; 109:3895 - 3905
- Liu TX, Becker MW, Jelinek J, Wu WS, Deng M, Mikhalkevich N, et al. Chromosome 5q deletion and epigenetic suppression of the gene encoding alpha-catenin (CTNNA1) in myeloid cell transformation. Nat Med 2007; 13:78 - 83
- Iwai M, Kiyoi H, Ozeki K, Kinoshita T, Emi N, Ohno R, et al. Expression and methylation status of the FHIT gene in acute myeloid leukemia and myelodysplastic syndrome. Leukemia 2005; 19:1367 - 1375
- Zheng S, Ma X, Zhang L, Gunn L, Smith MT, Wiemels JL, et al. Hypermethylation of the 5′ CpG island of the FHIT gene is associated with hyperdiploid and translocation-negative subtypes of pediatric leukemia. Cancer Res 2004; 64:2000 - 2006
- Lenz G, Hutter G, Hiddemann W, Dreyling M. Promoter methylation and expression of DNA repair genes hMLH1 and MGMT in acute myeloid leukemia. Ann Hematol 2004; 83:628 - 633
- Horton TM, Thompson PA, Berg SL, Adamson PC, Ingle AM, Dolan ME, et al. Phase I pharmacokinetic and pharmacodynamic study of temozolomide in pediatric patients with refractory or recurrent leukemia: a Children's Oncology Group Study. J Clin Oncol 2007; 25:4922 - 4928
- Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Methylation of p15INK4B is common, is associated with deletion of genes on chromosome arm 7q and predicts a poor prognosis in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 2003; 17:1813 - 1819
- Wong IH, Ng MH, Huang DP, Lee JC. Aberrant p15 promoter methylation in adult and childhood acute leukemias of nearly all morphologic subtypes: potential prognostic implications. Blood 2000; 95:1942 - 1949
- Chen CY, Tsay W, Tang JL, Shen HL, Lin SW, Huang SY, et al. SOCS1 methylation in patients with newly diagnosed acute myeloid leukemia. Genes Chromosomes Cancer 2003; 37:300 - 305
- Wu SJ, Yao M, Chou WC, Tang JL, Chen CY, Ko BS, et al. Clinical implications of SOCS1 methylation in myelodysplastic syndrome. Br J Haematol 2006; 135:317 - 323