Abstract
“Epigenetic plasticity” refers to the capability of mammalian cells to alter their differentiation status via chromatin remodeling-associated alterations in gene expression. While epigenetic plasticity has been best associated with lineage commitment of embryonic stem cells, recent studies have demonstrated chromatin remodeling even in terminally differentiated normal cells, and advanced-stage melanoma and breast cancer cells, in context-dependent responses to alterations in their microenvironment. In the current study, we extend this attribute of epigenetic plasticity to aggressive ovarian cancer cells, by using an integrative approach to associate cellular phenotypes with chromatin modifications (“ChIP-chip”) and mRNA and microRNA expression. While we identified numerous gene promoters possessing the well-known “bivalent mark” of H3K27me3/H3K4me2, we also report 14 distinct, lesser-known bi-, tri-, and tetravalent combinations of activating and repressive chromatin modifications, in platinum-resistant CP70 ovarian cancer cells. The vast majority (>90%) of all the histone marks studied localized to regions within 2000 bp of transcription start sites, supporting a role in gene regulation. Upon a simple alteration in the microenvironment, transition from two- to three-dimensional culture, an increase (17% to 38%) in repressive-only marked promoters was observed, concomitant with a decrease (31% to 21%) in multivalent (i.e., juxtaposed permissive and repressive histone marked) promoters. Like embryonic/tissue stem and other (non-ovarian) carcinoma cells, ovarian cancer cell epigenetic plasticity reflects an inherent transcriptional flexibility for context-responsive alterations in phenotype. It is possible that this plasticity could be therapeutically exploited for the management of this lethal gynecologic malignancy.
Introduction
In addition to the well-known phenomenon of transcriptional regulation by DNA methylation, epigenetic regulation is also largely mediated by acetylation and methylation of histone lysine residues within their N-terminal “tails” that emanate from the core nucleosome histone octamer. Five extensively modified residues include lysines 4, 9, 14, 27 and 36 of histone H3 (H3K4, H3K9, H3K14, H3K27 and H3K36 respectively).Citation1 Of these, H3K4, H3K9, H3K27 and H3K36 are mono-, di- or trimethylated by distinct histone methyltransferases and histone demethylases, while H3K9 and H3K14 are also frequently acetylated by histone acetyltransferases.Citation1 While DNA methylation is mostly repressive of gene transcription,Citation2 distinct histone modifications can facilitate a variety of biological outcomes, as set forth by the “histone code” hypothesis, which posits that specific histone alterations (i.e., the “written” code) are “read” by their cognate chromatin-interacting proteins to bring about unique (downstream) biological events, including the activation or repression of gene transcription (i.e., the “interpretation” of the code).Citation3,Citation4
Repressive histone marks frequently coordinate with (the more permanent mark of) DNA methylationCitation5 in heterochromatin, while embryonic and tissue stem cells possess numerous gene promoters concurrently having two opposing histone marks, the transcriptionally activating di-/tri-methylated H3K4 (H3K4me2/3) and the transcriptionally repressive trimethylated H3 lysine 27 (H3K27me3), marks.Citation6,Citation7 In stem cells, such promoter “bivalency” allows for transcriptional flexibility (“epigenetic plasticity”) that is subsequently restricted during differentiation-associated lineage commitment.Citation8,Citation9 In cancer cells, however, bivalency has been hypothesized to “mark” specific genes for (the more permanently repressive) DNA methylation,Citation10–Citation12 H3K27 trimethylation can also mediate gene silencing (in the absence of DNA methylation), by the commonly overexpressed oncoprotein EZH2.Citation13,Citation14
Although epigenetic plasticity is predominant in embryonic and tissue stem cells,Citation6,Citation7 recent studies have also shown that even in terminally differentiated, senescent cells, epigenetic modifications are potentially reversible to allow “reprogramming” to an altered differentiation level. One of the most dramatic examples of epigenetic reprogramming is found in studies of “induced pluripotency,” in which terminally differentiated somatic cells, including mouse fibroblasts, keratinocytes, intestinal epithelial and muscles cells were (by ectopic introduction of various transcription factors or drugs) restored to an embryonic-like pluripotent phenotype capable of producing chimeric mice and in rare cases, germline integration.Citation15,Citation16 Another example of epigenetic plasticity is “transdifferentiation” of various differentiated cells from one lineage to another.Citation17
While epigenetic plasticity is essential for normal differentiation of pluripotent stem cells, the recent “cancer stem cell” hypothesis of carcinogenesis likewise suggests a tumor cell hierarchy similar to that of normal organs (and indeed, tumors have been suggested to represent “abnormal organs”), with tumor growth, “differentiation,” and propagation instigated by a distinct subset of malignant progenitors (“cancer stem cells”).Citation18,Citation19 In support of this hypothesis, highly aggressive (possibly stem-like) cancer cells demonstrate considerable epigenetic flexibility (likely allowing adaptation to diverse microenvironments and immune evasion), including an intricate type of transdifferentiation, the epithelial-to-mesenchymal transition (EMT), a normal (microenvironment-regulated) cell detachment/dispersal and mobility process essential for embryogenesis.Citation20 Analogously, however, in cancer cells, EMT facilitates metastatic tumor cell detachment, migration, basement membrane invasion and establishment of secondary lesions,Citation20 and a recent breast cancer study revealed extensive chromatin remodeling associated with EMT induced by TGFβ.Citation21 Epigenetic plasticity of advanced stage cancer cells was also strikingly demonstrated by the differentiation of highly aggressive melanoma, breast and prostate cancer cells,Citation22 following exposure to embryonic stem cell (ESC)-derived extracellular matrices.Citation23 Similarly, malignant reversion of breast cancer and acute myelogenous leukemia cells followed antibody-mediated disruption of integrin binding, while abnormal stromal fibroblasts and loss of laminin-1-associated polarity, are now strongly associated with a number of carcinomas.Citation24,Citation25 Moreover, in support of a role for epigenetic plasticity during the process of oncogenesis, numerous ESC-bivalently marked genes (mostly tumor suppressors) were also DNA-methylated in adult colon cancer tumors,Citation6,Citation26 while numerous bivalent promoters were also identified in both normal and malignant prostate tissues (although the specific bivalently marked genes differed between malignant and normal prostate cells).Citation27 Together, these studies demonstrate that even genetically damaged cancer (or senescent, normally differentiated) cells can drastically alter their cellular phenotype, via chromatin remodeling, in response to perturbations in the microenvironment.
In this report, we examine whether aggressive, chemoresistant CP70 ovarian cancer cells also possess epigenetic plasticity (similar to other carcinomas), by assessing chromatin alterations associated with a simple change in their microenvironment, the transition from anchorage-dependent (“2D,” as cell monolayers) to anchorage-independent culture conditions (“3D,” as spherical aggregates). Similar tumor-derived spheroids are commonly seen in the ascites of ovarian cancer patients,Citation28 and a recent study of an ovarian cancer cell line part demonstrated widespread gene expression changes in spheroids, in cell culture, following chemotherapy treatment.Citation29 Consequently, we hypothesized that epigenetic plasticity, inherent to other (non-ovarian) carcinoma cells, might also facilitate adaptation to three-dimensional culturing. Using ChIP-chip analyses, we identified “epigenetic signatures” of CP70 cells, ovarian cancer stem-like cells and patient-derived tumors, while also characterizing a number of previously little-known multivalent combinations of active (H3Ac, H3K4me2) and repressive (H3K9me3, H3K27me3) histone marks. Similar to the well known bivalent domains of H3K27me3/H3K4me2/3 in stem cells, these multivalently marked promoters also conferred epigenetic plasticity to CP70 cells, in correlation with distinct changes in gene expression. Consequently, as in other carcinomas, a better understanding of chromatin plasticity in serous epithelial ovarian cancer may facilitate epigenetic strategies for ameliorating (or even reversing) the tumor-progressive and metastatic phenotypes of this disease.
Results
Transcriptionally repressive and permissive histone marks predominantly flank gene promoters and first exons in ovarian cancer cells.
To assess the presence of multivalent (juxtaposed permissive and repressive, suggestive of epigenetic plasticity) histone modifications in ovarian cancer cells, two transcriptionally permissive chromatin marks, acetylated H3 (H3Ac) and H3K4me2 and two repressive marks, H3K9me3 and H3K27me3, were examined in CP70 ovarian cancer cells cultured under anchorage-dependent (2D) or -independent (3D) conditions. By chromatin immunoprecipitaton (ChIP), using antibodies against these four specific histone modifications, followed by hybridization to 244K tiled CpG island microarrays (Agilent G4492A Human CpG island array; “ChIP-on-chip”), in conjunction with our previously developed “peaksPicking” program for identifying the most statistically confident hybridization fluorescent signals,Citation30 we identified a total of 5,053 epigenetically marked genes under the two conditions. All four H3 marks were detected equally in the ChIP-chip experiments and the regions containing these marks (i.e., the hybridized microarray fluorescence signal peaks) were quite similar in size (median value of ∼300 bp), with a few long tails stretching to 5,000 bp (). Each mark was easily identified even within the more stringent top 5% and top 2% of the peaksPicking results, although multivalent marks were somewhat less frequently observed in the top 5% than in the top 10% of hybridized loci (Suppl. Fig. S1).
Chromosomal alignment revealed these histone marks to preferentially locate in gene promoters, rather than intragenic regions (). More precisely, comprehensive location analysis using the human genome (HG17) browser (genome.ucsc.edu) further revealed these histone marks to be largely proximal (−2,000 bp to +2,000 bp, ) to transcription start sites (TSSs). Subsequently, utilizing our peaksPicking program, we further studied the top 10% of peaks from CP70 cells grown under 2D (2734 total ChIP-hybridized genes) or 3D (2,424 total genes) conditions (). Both peaksPicking and Integrated Genome Browser (www.igb.org) analyses revealed multiple (often opposing) histone marks to occupy any given promoter region.
In embryonic and various tissue stem cells, numerous gene promoter regions coordinately possess the transcriptionally permissive histone marks H3K4me2/3 and the transcriptionally repressive H3K27me3 mark, to create a “bivalent” epigenetic signature and remain “poised” for further, differentiation-associated activation or repression.Citation6 ChIP-chip analyses revealed these four H3 modifications to occur in 15 distinct combinations, allowing us to now extend this phenomenon of “multivalent” epigenomic domains to ovarian cancer cells, including the well-characterized H3K4me2-H3K27me3 mark, in addition to other little-reported combinations of active (H3Ac, H3K4me2) and repressive (H3K9me3, H3K27me3) chromatin modifications present in distinct bivalent, trivalent and tetravalent associations. Those previously uncharacterized promoter combinations include the bivalent combinations H3K4me2/H3K9me3, H3K4me2/H3K27me3, H3Ac/H3K27me3 or H3Ac/H3K9me3, the trivalent combinations H3K42me2/H3K9me3/H3K27me3, H3Ac/H3K4me2/-H3K27me3, H3Ac/H3K9me3/H3K27me3, H3Ac/H3K4me3/H3K9me3 and an interesting tetravalent combination, H3Ac/H3K4me2/H3K9me3/H3K27me3 ().
Epigenetic flexibility contributes to ovarian cancer cell adaptation to a change in microenvironment (anchorage-dependent to -independent culture).
We next investigated the epigenetic consequences of the altered cellular architecture and behavior induced by 3D cell culture. While similar numbers of monovalent marks were found in 2D and 3D cells (1879 vs. 1895), the combinations of H3 modifications among these genes were quite varied. While H3Ac was the most common overall (42% in 2D cells and 24% in 3D cells), H3K9me3 was more abundant in 3D cells, equal to H3Ac at 24% (). Although H3K4me2 was relatively similar between 2D and 3D cells (12 vs. 16%), a larger number of permissive marks (H3Ac and H3K4me2) were found in 2D culture, while 3D cells had more repressive histone marks overall (H3K9me3, 24 vs. 7% and H3K27me3, 12 vs. 7%, 3D vs. 2D, respectively, ). The H3Ac permissive mark, however, remained the most abundant univalent mark in 2D cells, although its combination with other histone marks was fairly infrequent, with its greatest colocalization occurring in the tetravalent mark H3Ac-H3K4me2/-H3K9me3/H3K27ME3me3 (). Also in 2D cells, the well-known bivalent H3K4me2/H3K27me3 combination (a stem cell histone mark associated with “transcriptional readiness”)Citation26,Citation31 was more prevalent in 3D than in 2D cells (6.8 vs. 1%, respectively), as was the previously little-studied trivalent mark H3K4me2/H3K9me3/H3K27me3 (3D cells, 9.7% vs. 2D cells, 0.3%, ). We also observed two other previously uncharacterized combinations, including trivalent H3Ac/H3K4me2/H3K9me3 domains (present only in 3D cells) and a tetravalent combination, H3Ac/H3K4me3/H3K9me3/-H3K27me3 (present only in 2D cells, ); these newly characterized mutlivalent marks suggest an alternative means to transcriptional flexibility. Together, these results suggest that this commonplace change in CP70 cell microenvironment (i.e., transition from 2D monolayers to 3D spheres) induces extensive chromatin remodeling that facilitates phenotypic adaptation.
Several multivalent epigenetic marks in 2D cells become monovalent upon 3D culturing.
We next globally assessed the similarity (or divergence) of the total changes in the four abovementioned histone H3 marks upon 2D to 3D transition. Of all the array-hybridized genes possessing any of the four H3 marks in 2D (2,734 total genes) and 3D (2,319 genes, ) cells (using the top 10% of ChIPhybridized genes from our peaksPicking program), we found only 1,149 (42%) genes to overlap (thus possessing at least one of the four marks in both cellular states) (). The epigenetic signatures of these 1,149 genes were then evaluated for changes induced by 3D spheroid culture conditions. Interestingly, we found that among these common genes, a large number underwent epigenetic reprogramming through gain or loss of epigenetic marks. While a majority of monovalently marked genes in 2D cells transitioned to alternative monovalent, repressive marks in 3D ( and Sup. Table S2), many other 2D-actively marked genes reshaped their promoter regions to possess various alternative mono- or multivalent histone marks in 3D ( and and Sup. Table S2). Significantly, 52% of the 2D genes possessing the H3Ac permissive mark retained an active mark in 3D culture, either as monovalent H3Ac (18%), H3K4me2 (19%) or both (H3Ac/H3K4me2, 15%) (Sup. Table S2), suggesting that these genes remain active upon the change in micro-environment. Overall, however, the total number of active marks decreased in 3D (). Similarly, several genes possessing permissive H3K4me2 in 2D remained permissive in 3D culture, by either retaining H3K4me2 or changing H3K4me2 to H3Ac (12 and 32%) and numerous 2D H3K9me3-repressed genes reverted to active chromatin by replacing H3K9me3 with H3Ac (49%, Sup. Table S2). The other repressive mark, H3K27me3, remained unchanged in 39% of genes (Sup. Table S2), thus representing the mark least likely to convert to an active H3Ac mark, suggesting that its increased stability may serve an important role in maintaining gene repression.
Because 2D monolayer-cultured cells possessed a greater proportion of multivalently marked genes than 3D cells (), we examined the fate of these potentially “plastic” genes upon 3D culturing. This multivalency was generally lost upon CP70 cell transition from 2D to 3D culture (855 genes in 2D vs. 275 in 3D, a 68% decrease, data not shown), with 50% of the multivalent promoters becoming monovalent, primarily due to loss of the active H3 marks and retention of the repressive H3K27me3 modification (). This resolution to monovalency suggests an overall loss of chromatin plasticity in 3D culture, due to the change in cellular architecture, with 3D conditions directing epigenetic fate toward more strict gene expression or repression. The multivalent domains that changed most significantly from 2D to 3D included the well-studied bivalent mark H3K4me2/H3K27me3 (6 vs. 1%, ), the trivalent mark, H3K4me2/H3K9me3/H3K27me3 (10 vs. 0.3%, ) and the above-mentioned tetravalent mark (2 vs. 0.1%, ). Similar to the H3K27me3 monovalently marked genes ( and bottom pie chart), bi- and tri-valent domains containing H3K27me3 were also more likely to retain repressive marks, suggesting continued epigenetic repression by (the more stable) H3K27me3 (Sup. Table S2). Tetravalent domains were much more common in 2D (55 genes) than in 3D (3 genes) cells (data not shown). However, while tetravalency was greatly decreased upon 3D culturing, a substantial fraction (45%) of 2D-tetravalently marked genes remained bi- or trivalent (, top bar, Sup. Table S2).
Epigenetic remodeling correlates with transcriptional changes and pathway dysregulation, following the 2D-to-3D transition.
Overall, the presence of the little-known bivalent H3H3K4me2/H3K9me3 and tetravalent H3Ac/H3K4me2/H3K9me3/H3K27me3 marks in ovarian cancer cells suggested possible epigenetic and phenotypic plasticity of the 2D cultures. Consequently, we performed transcriptome analysis of 2D- and 3D-cultured CP70 cells, to examine possible correlations of gene expression with changes in histone marks (as assessed by our PeaksPicking analysis of the ChIP-chip results). Affymetrix gene expression arrays for CP70 2D and 3D cells were compared to normal ovarian surface epithelial cells (nOSE). As shown in and B, 2D cell genes possessing the activating marks H3Ac and H3K4me2 were transcriptionally upregulated, while those having the repressive histone marks H3K9me3, H3K27me3 and H3K9me3/H3K27me3 mark correlated with transcriptional downregulation, as predicted by the histone code hypothesis.Citation3 Similarly in 2D cells, the bivalent marks H3K4me3/H3K9me3 and H3K4me2/H3K27me3 associated with transcriptional downregulation, suggesting dominance of the repressive (H3K9me3 and H3K27me3) marks in bivalent domains (, left part). By contrast, in 3D cells, the H3K4me2/H3K9me3 and H3K4me2/H3K27me3 bivalent domains associated with transcriptional upregulation (, right part), suggesting a reversal of the dominant mark upon cellular transition to substrate independence, possibly to facilitate transcriptional activity allowing adaptation to this specific (3D) microenvironment.
To identify specific gene families or biological pathways, regulated by the aforementioned epigenetic patterns, the gene expression microarray datasets were subjected to Gene Set Enrichment Analysis (GSEA, www.broad.mit.edu/gsea). Hierarchical clustering demonstrated distinct segregation of the 2D and 3D gene expression patterns, (the 15 most upregulated and 15 most downregulated genes between CP70 2D and 3D cells, and D). Members of the inhibitor of DNA binding (Id) family of genes (Id1, Id2, Id3) were most significantly downregulated in 3D cells, while matrix metalloprotease MMP genes were also downregulated (). Gene Ontology (GO, www.geneontology.org/) and GSEA determinations revealed that among the multivalently marked genes, genes for chromatin remodeling, development and cellular differentiation were enriched (Sup. Table S3). Those results validated the ChIP-chip data, which showed numerous up- or downregulated genes to associate, respectively, with the H3Ac/H3K4me2 activating (data not shown) or H3K9me3/H3K27me3 repressive () marks. Opposing bivalently marked genes were not preferentially up- or down-regulated, although the H3K27me3 mark generally associated with downregulation. Of the 20,700 annotated genes on the Affymetrix microarray, 5,514 (27%) showed significant fold-changes (p < 0.05). The significant expression changes in chromatin remodeling genes is supportive of a high degree of phenotypic plasticity.Citation32
To further identify biological processes associated with altered histone marks in 3D culture, we analyzed four gene sets, with each having the permissive H3Ac mark in 2D and various histone marks in 3D, using the integrated database/software suite MetaCore.Citation33 This pathway analysis tool uses each gene in the set as a “node,” generating networks between the nodes, which are further broken down into smaller subnetworks centered on receptors and transcription factors.Citation33 These four sets included: (A) 14 2D genes retaining the activating H3Ac mark in 3D; (B) eight genes with H3Ac replaced by the permissive H3K4me2 mark; (C) eight 2D genes having H3Ac replaced by the repressive mark H3K9me3 in 3D and (D) 13 2D genes with H3Ac replaced by the binary permissive mark H3Ac/H3K4me2 in 3D. Other gene sets were excluded from the analysis due to: (1) their gene number was less than five; or (2) failure to infer significant subnetworks. The results are shown in Supplemental Figure S2 and Table S4. For Group A genes, a 50-node network derived from the five nodes could be parsed into sub-networks involved in positive regulation of cell cycle and organelle organization (Sup. Fig. S2A). Group B genes, however (4 nodes of a total of 50), formed a network negatively regulating cell cycle (Sup. Fig. S2B), while the network resulting from Group C genes (again 4 nodes of a total of 50) that positively regulated biological processes including stress/stimulus responses and immune response (Sup. Fig. S2C and Sup. Table S4). Finally, the Group D (changing from H3Ac to H3Ac/H3K4me2) network included positive regulation of apoptosis (Sup. Table S4 and Sup. Fig. S2D). While reconciling these pathway predictions with our 2D/3D culture is difficult, without further experimental validation (e.g., pathway reporters, pharmacologic inhibitors, etc.,), the analyses are consistent with the phenotypes of our 3D spheroids, including increased differentiation, decreased epigenetic plasticity and slower proliferation (see above and previous section).
H3K27 trimethylation regulates microRNA expression in CP70 cells.
As another cancer-associated epigenetic phenomenon, microRNA (miRNA)-mediated regulation of tumor suppressors and oncogenes, is strongly implicated in ovarian tumor progression,Citation34–Citation41 we evaluated a possible role for miRNA expression in microenvironment-associated epigenetic or genetic reprogramming by custom microarray analysesCitation42 of 2D and 3D CP70 cell cultures. Only minor miRNA changes were noted between the 2D and 3D cultures, suggesting that miRNA profiles do not change significantly during this transition (). However, the repressive H3K27me3 mark strongly associated with downregulated gene expression ( and both in 2D and 3D cells), and genes initially marked with H3K27me3 tended to retain the mark, thus reinforcing our earlier studies showing the stability of this repressive modification ( and Sup. Table S2). Consequently, we wished to examine the role of H3K27me3 in repression of miRNA expression, utilizing a previously established CP70 cell subline (CP70-H3K27R) expressing a dominant-negative histone H3 having lysine 27 replaced by (the similarly basic residue) arginine.Citation43
Western blot analysis of CP70-H3K27R cells revealed, as we demonstrated previously,Citation43 a decrease of the H3K27me3 mark, in addition to an increase of the activating H3Ac mark, as compared to the parental CP70 cell line cultured under 2D or 3D conditions (Sup. Fig. S3). Further comparison of CP70-H3K27R cells under both 2D- and 3D-culture conditions revealed a dramatic difference in miRNA profiles (, left and right insets). Loss of H3K27me3 methylation resulted in distinct up- or downregulated miRNA gene families, demonstrating a strong role for this mark in regulating miRNA expression. The largest cluster of significantly overexpressed miRNA genes in CP70-H3K27R cells was the miR-200 family (, right inset), well known suppressors of the metastasis-associated epithelial-to-mesenchymal transition.Citation44–Citation47 These results also suggest that the repressive histone mark H3K27me3 may play a role in ovarian cancer metastasis, as the H3K27-trimethylating enzyme, EZH2, is overexpressed in highly aggressive (and metastatic) breast and prostate cancers.Citation48–Citation50 Thus, both miRNA-mediated epigenetic regulation, as well as the reciprocal epigenetic-induced changes in miRNA expression, highlights an additional complexity in ovarian cancer epigenomics.
Ovarian tumor, xenografts and ovarian cancer stem cells extensively possess the H3K27me3 repressive mark.
To further investigate the in vivo occurrence of the four H3 marks, and their possible roles in tumor initiation and progression, we performed ChIP-chip analyses on DNA from ovarian cancer stem-like cells and bulk tumor cells. Based on the increasingly accepted “cancer stem cell hypothesis,” a distinct tumor subpopulation of malignant progenitors is directly responsible for tumor growth, drug resistance and metastasis.Citation18,Citation19 Based on recently described attributes of tumor progenitors,Citation51 we isolated ovarian cancer-initiating cells (OCICs) from human tumors,Citation52 and compared OCIC histone patterns to CP70-H3K27R, bulk tumor cells, CP70-3D and CP70-2D cells (Sup. Fig. S4). In contrast to CP70-2D cells (but similar to CP70-3D cells), OCICs, CP70-H3K27R and bulk tumor cells possessed substantially fewer H3Ac-marked genes, with similar levels of the other monovalent and multivalent marks (Sup. Fig. S4).
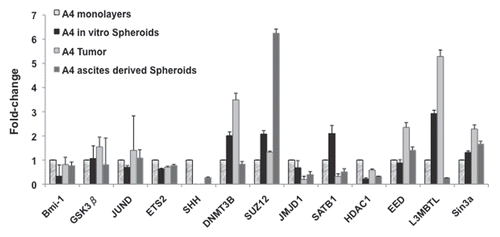
In another study of in vivo microenviromental effects on ovarian cancer cells, we compared OCIC and bulk tumor cell ChIPchip results to those of an ascites-derived ovarian tumor-initiating clonal cell population,Citation51 designated “A4.” Both OCICs and A4 cells represent tumor progenitors capable of serially recapitulating their original ovarian tumors in nude mice.Citation51,Citation52 H3K27me3 ChIP-chip assays of A4 cells identified 730 genes possessing this repressive mark, including inhibitor of DNA-binding (Id) family of genes (GSEA analysis, data not shown) and the Polycomb (PcG) and Trithorax (TTX) gene families, chromatin-modifiers, developmental regulators (Sup. Table S5 and ). Furthermore, target genes of specific histone modifications were differentially expressed in tumors vs. spheroids formed in animal ascites, again suggestive of the influence of the in vivo microenvironment. Specifically, A4-derived tumors overexpressed the PcG genes EED and L3MBTL and the DNA methyltransferase gene DNMT3B, with downregulation of the histone H3K9me3 demethylase (thus gene-activating) gene JMJD1A, as compared to expression levels in steady state, monolayer A4 cells (). In A4-derived spheroids, however, expression of JMJD1A and the PcG gene L3MBTL was decreased, while the PcG gene SUZ12 was increased >6-fold (). The variable expression of chromatin-remodeling genes, with diverse combinations of histone marks, is highly suggestive of phenotypic plasticity, similar to differentiation of embryonic stem cells.Citation32
Similarly, several H3K9me3-associated genes (13%) were also found dysregulated in spheroids. However, genes associated with the H3K9me3 mark alone (6%) exhibited only limited transcriptional flexibility, while tetravalency correlated with highly plastic gene expression (38%, data not shown). These profiling studies demonstrate the complex influence of expression patterns of key cancer cell genes on higher order chromatin remodeling, in response to cell-extrinsic signals emanating from in vitro vs. in vivo microenvironments.
Discussion
The phenomenon known as “epigenetic plasticity,” (i.e., a flexibility of chromatin modifications) is a phenotype of uncommitted cells that is progressively lost during lineage commitment, due largely to cues from the local stroma, extracellular matrix, autocrine/paracrine loops and ligand/receptor interactions on juxtaposed cells.Citation53 In tissue and embryonic stem cells (ESCs), epigenetic plasticity is associated with “bivalent” gene promoters concurrently possessing a transcriptionally repressive histone mark, trimethylated histone H3 lysine 27 (H3K27me3) and a permissive mark, di-/tri-methylated histone H3 lysine 4 (H3K4me2/3).Citation6,Citation7,Citation42 Moreover, several ESC-bivalently marked genes are also DNA-methylated in colon tumors,Citation26 supporting the long-held analogy of cancer as a reversion to an embryoniclike state.Citation54 Consequently, to assess epigenetic plasticity in ovarian cancer, we utilized the cisplatin-resistant, aggressive cell line CP70 to globally examine (using “ChIP-chip” analyses) four well-known histone marks, the activating marks, H3K4me2 and acetylated histone H3 (H3Ac) and the repressive marks, H3K27me3 and trimethylated histone H3 lysine 9 (H3K9me3). The vast majority of these marks localized to gene promoters and first exons (), while also conforming to the “histone code” hypothesis,Citation3 with activating marks correlating with gene upregulation and repressive marks correlating with gene downregulation (). We next examined the effect of the local microenvironment on CP70 epigenetic plasticity in response to two cellular conditions commonly present in ovarian malignancies, three-dimensional multicellular aggregates (spheroids) and two-dimensional monolayers, typically present in patients' ascites and peritoneal mesothelia, respectively.Citation28,Citation55 Those two disparate microenvironments correlated with numerous altered histone modifications and gene expression levels ( and Sup. Table S3), predicting significant dysregulation of various biological processes (Sup. Fig. S2 and Sup. Table S4). Moreover, our ChIP-chip assessments identified several previously uncharacterized multivalent histone marks, facilitating a “transcription-ready” chromatin state of various proto-oncogenes and stemness and chromatin-remodeling genes (Sup. Fig. S5). These assessments also highlight a functional role for the bivalent H3K4me2/H3K9me3 mark, for the first time, in aggressive cancer cells, in addition to the largely unknown binary H3K9me3/H3K27me3, trivalent H3K4me2/H3K9me3/H3K27me3 and tetravalent H3Ac/H3K4me2/H3K9me3/H3K27me3 marks.
While epigenetic plasticity is well associated with normal stem cell lineage commitment, even terminally differentiated cells may be epigenetically “reprogrammed” to a stem-like chromatin state.Citation16,Citation17 Specifically, this “induced pluripotency” occurs in the absence of changes in DNA sequence, and is associated with widespread remodeling of chromatin;Citation16,Citation56 interestingly, one agent that greatly facilitated induced pluripotency was the histone deacetylase inhibitor valproic acid.Citation57 A somewhat analogous phenomenon, “transdifferentiation,” is the process whereby a terminally differentiated cell transposes to a disparate lineage;Citation17 examples of transdifferentiation include the conversion of mammary adipocytes into secretory epithelial cells during pregnancy and lactation (with the subsequent reversion to adipocytic tissue following weaning) and the development-associated, recriprocal conversion between hepatocytes and pancreatic exocrine cells.Citation58,Citation59 Similarly, we also observed differentiation-altering, microenvironment-associated plasticity in CP70 cells, demonstrating reduced epigenetic plasticity upon their transition from 2D to 3D culture. Those results also showed H3K27me3 as a stable repressive mark, in accord with prior studies demonstrating (the H3K27me3-associated) Polycomb proteins to mediate repression in stem/cancer cells and to “pre-mark” specific genes for subsequent DNA methylation.Citation8,Citation12,Citation14,Citation31,Citation60 Additionally, CP70-H3K27R cells (largely incapable of H3K27 trimethylation)Citation43 demonstrated significantly altered miRNA expression patterns ().
Similar to differentiated somatic cells, cancer cells can also retain considerable epigenetic plasticity that is influenced by external cues. The increasingly accepted “cancer stem cell” hypothesis regards a tumor as an abnormal organ, possessing a hierarchy of progeny cells derived from multipotent tumor progenitors;Citation18,Citation19 our group identified such progenitors in human ovarian cancer patients' ascitesCitation51 and primary tumors.Citation52 Because advanced-stage carcinoma cells exhibit considerable phenotypic plasticity (allowing for adaptation to diverse microenvironments), this plasticity could possibly be exploited against those cells to circumvent their malignant potential. For example, melanoma cells, when plated atop ESC-derived extracellular matrices, remarkably, differentiate into sphere-forming melanocytes, while undifferentiated melanoma cells grow only as monolayers.Citation22,Citation23 Analogously, the melanocyte-containing spheres can be “de-differentiated” into two-dimensional melanoma cells when plated atop melanoma cell-derived extracellular matrices.Citation23 Epigenetic plasticity has also been shown by similar reversion of aggressive breast cancer cells,Citation22 and indeed, many inhibitors of epigenetic repressive enzymes (e.g., DNA methyltransferase and histone deacetylase inhibitors) are well established differentiating agents.Citation4,Citation61 Thus, even advanced-stage cancer cells, despite considerable genetic damage, retain considerable epigenetic plasticity in response to their specific microenvironments, which can either facilitate or hinder malignant progression.
In extension to prior studies of ESC, melanoma, and breast cancer cells, we now demonstrate epigenetic plasticity in monolayers of ovarian cancer cells, with frequent promoter co-localization of transcriptionally opposing bi-, tri- and tetra-valent histone marks. This epigenetic plasticity, permissive for transcriptional changes, might reflect the inherent phenotypic flexibility of the normal ovarian surface epithelium (nOSE), whose mesenchymal properties facilitate postovulatory wound repair.Citation62 Such flexibility was lost or restructured in the 3D state, which had less multivalency and more repressive monovalent marks (concomitant with a loss of permissive marks, and Sup. Table S2). While other in vitro cultured multiaggregate spheroids were enriched in stem cells (i.e., less differentiated), this property would appear to be tissue-specific (including neural and mammary tissues),Citation63,Citation64 as other sphere-forming cells actually demonstrate reduced differentiation capacity (i.e., reduced plasticity/stemness).Citation65,Citation66 It is conceivable that our in vitro spheroids resemble those in the abovementioned melanoma differentiation studies. However, our in vivo ascites-derived spheroids also overexpressed various Polycomb proteins (), which facilitate tumor cell self-renewal and associate with poor differentiation and metastatic phenotypes,Citation49,Citation50,Citation67 consistent with the in vivo properties of these ovarian tumor-derived structures.Citation28,Citation55
While the cell-of-origin of epithelial ovarian cancer remains unclear, it appears that (in contrast to other carcinomas) early stage disease is characterized by a “mesenchymal-to-epithelial transition.”Citation62,Citation68,Citation69 During malignancy progression, tumor-exfoliated spheroids (possibly analogous to our currently used culture system) transcoelomically migrate or traverse through ascites fluid (while also avoiding immunodetection), followed by attachment to the collagen-rich peritoneal mesothelial monolayer, forming multiple, invasive foci (a process known as “peritoneal seeding”).Citation28,Citation55 Those mesothelium-attached cells must exhibit a large degree of phenotypic adaptability, including integrinand cadherin-associated submesothelial anchoring, secondary tumor outgrowth, invasiveness and the establishment of matrixrich, channel-forming networks expressing vascular markers (“vasculogenic mimicry”).Citation28,Citation70,Citation71 These late-stage events show a high degree of adaptability unlikely to be mediated by genetic mutation alone,Citation72 and in contrast to early stage malignancy, these advanced disease phenotypes associate with typical EMT,Citation68 largely regulated by epigenetic permutations.Citation39,Citation62,Citation69 Moreover, another study demonstrated tumorigenic capacity of ascites-derived ovarian cancer cells to be highly dependent on their local microenvironmental “niche.”Citation73 Thus, the increased epigenetic plasticity of monolayered (vs. sphere-forming) ovarian cancer cells might facilitate the necessary phenotypic diversity that allows for adaptation to the harsh conditions encountered during ovarian cancer metastasis. Although we did not examine reversion of the 3D state to the 2D state, those previously well established in vivo findings, in combination with our current study, would allow us to predict a re-establishment of epigenetic and phenotypic plasticity under monolayer conditions.
In summary, our integrated genomic, epigenomic and miRNA assessment reveals a complex network of epigeneticmediated regulation of gene and miRNA expression which is largely influenced by the cellular microenvironment.Citation74 Similar to previous studies of breast cancer and melanoma, our findings of multivalent histone marks extend the attribute of epigenetic plasticity to ovarian cancer. As this plasticity is strongly influenced by the cancer cell microenvironment, characterization of the signal cascades responsible for relaying external cues to the nuclear epigenetic machinery could provide new strategies for combating this devastating malignancy.
Materials and Methods
Cell culture, cell lines and tumor samples.
Cisplatin-resistant CP70 ovarian cancer cells (a kind gift from Dr. Robert Brown, Imperial College London and Institute of Cancer Research, UK) were used to generate the primary chromatin immunoprecipitation DNA microarray hybridization (ChIP-chip) data. To correlate the epigenetic marks associated with genes differentially expressed in the various ovarian cancer and ovarian cancerderived cell types, we subjected the CP70 cells to 2D or 3D culture conditions. To generate 3D cultures, CP70 cells were grown in a liquid basal medium (RPMI without any serum) in lowadherence dishes at 37oC for 3–4 days and were harvested by centrifugation.
All cell lines used are listed in Supplemental Table S1. A4 is a cell line previously established from a single isolated cell derived from a multi-layered spheroid present in the ascites of a serous ovarian epithelial cancer patient,Citation51 and was maintained in MEM(E), 5% FBS and 1% nonessential amino acids (each from Gibco Invitrogen, Carlsbad, CA, USA). Furthermore, we examined the epigenetic status of ovarian cancer stem-like cells (CSCs),Citation51 ovarian cancer-initiating cells (OCICs),Citation52 and ovarian side-population (SP) cells,Citation75 for the presence of multivalantly marked gene promoters. Human ovarian tissue and tumor samples were obtained by informed consent, and all studies were approved by the Institutional Review Boards of the National Centre for Cell Science, Indiana University and the Ohio State University. Normal ovarian surface epithelial (nOSE) cells were acquired from patients during surgery for benign gynecological disease, as previously described,Citation76 with cytokeratin and vimentin staining to validate their dual epithelial/mesenchymal character.Citation62
Chromatin immunoprecipitation and DNA microarray analysis (ChIP-chip).
All antibodies used for ChIP were from Millipore (Billerica, MA, USA): acetylated histone H3 (catalog #06-599), H3K4me2 (#07-030), H3K9me3 (#07-441) and H3K27me3 (#07-449) and chromatin immunoprecipitation to microarray hybridization carried out as we described previously.Citation77 Independent immunoprecipitations were performed for each analysis. Approximately 108 cells CP70 cells were used for each experiment. Cells were chemically crosslinked by the addition of one-tenth volume of fresh 11% formaldehyde solution for 15 min at room temperature, rinsed twice with ice-cold 1X PBS and harvested using a silicon scraper. The harvested cells were then resuspended in lysis buffer and sonicated to solubilize and shear crosslinked DNA to a size range of ∼500–1,000 bp. Samples were kept on ice at all times. The resulting whole cell extract was incubated overnight at 4°C with 80 ml Protein A-agarose magnetic beads that had been pre-incubated with approximately 10 µg of the appropriate antibody. Bound complexes were further eluted from the beads in TE buffer after sequential washing with low salt, high salt and LiCl buffers and crosslinks then removed by adding NaCl to the eluted DNA and heating overnight at 65°C. Whole cell extract DNA (nonimmunoprecipitated, reserved from the sonication step as a positive control) was also treated for crosslink reversal and the ChIPed and whole cell extract DNA then treated with RNaseA and proteinase K, followed by purification using DNeasy columns (QIAGEN, Valencia, CA, USA). The purified DNA was then blunted, ligated to linkers and amplified using a two-stage PCR protocol. Amplified DNA was labeled using Bioprime random primer labeling kits (Invitrogen). ChIP'ed DNA was labeled with Cy5 fluorophore, while whole cell extract DNA was labeled with Cy3. Cy3- and Cy5-labeled DNA (5 µg of each) was mixed and hybridized to 244K Agilent G4492A Human CpG island microarrays (Santa Clara, CA, USA), using Agilent hybridization chambers for 40 hours at 40°C. After hybridization, arrays were washed and scanned, using an Axon GenePix® 4,200. A scanner and GenePix® (version 6.0) software (Molecular Devices, Sunnyvale, CA, USA) used to automatically align the data, while abnormal features were identified manually, resulting in the final hybridization intensity results. The binding regions on each promoter were identified using the peaksPicking program developed in our previous study.Citation30 Briefly, this program employs a probe-based enrichment threshold to define peaks (binding regions) for each promoter array, determining three enrichment threshold levels (top 1%, top 5% and top 10%), followed by a permutation-based statistical method to control the false discovery rate for peaks identified at each enrichment threshold.Citation30 The identified binding regions are then mapped to the UCSC HG17 RefGene human genome database to locate the corresponding target genes.Citation30 In the current study, the percentage of number of genes was based on the total 16,264 annotated genes on the Agilent G4492A array. Histone mark localizations were determined using the UCSC HG17 RefGene reference database, with gene promoter regions defined as -2 kb to +2 kb of the TSS of any specific gene.
RNA preparation, microarray hybridization, data analysis and validation.
Total RNA was prepared using the QIAGEN RNeasy MiNi Kit. A DNase I digestion step was included to eliminate DNA contamination. cRNA was generated, labeled and hybridized to the Affymetrix Human Genome U133 Plus 2.0 Arrays by the Center for Medical Genomics at Indiana University School of Medicine (http://cmg.iupui.edu/), as we have described previously.Citation78 The hybridized Human Genome U133A 2.0 Array was scanned and analyzed using the Affymetrix Microarray Analysis Suite (MAS) version 5.0. The average density of hybridization signals from four independent samples was used for data analysis and genes with signal density less than 300 pixels were omitted from the data analysis.
RNA isolation, reverse-transcription PCR (RT-PCR) and real-time quantitative PCR (q-PCR ).
Total RNA was extracted from cultured cells, tumors or ascites using TRIzol reagent (Invitrogen). The primary tissue samples were initially harvested and placed in liquid nitrogen before lysis and homogenization in TRIzol, whenever necessary. The concentration and purity of RNA was measured by A260/A280 measurements using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). For quantitative PCR (qPCR) analysis, 2 µg total RNA was reverse transcribed using random hexamer primers and the SuperScript® First-Strand Synthesis System (Invitrogen) to produce cDNA, which was then used as the qPCR template, performed in a Mini Opticon real-time thermal cycler (Bio-Rad, Hercules, CA, USA), using 20 µl reactions containing buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 10 pM of each primer and 0.2 units of recombinant Taq DNA polymerase, as we have previously described.Citation43,Citation78 Amplifications were performed in triplicate. Q-PCR standard curves were produced in triplicate and Opticon Monitor software was used to calculate Ct (threshold cycle) relative gene expression values, using the well established 2exp(-ΔΔCt) method,Citation79 with normalization to expression levels of the same genes in primary cultured nOSE cells. In addition to SYBR green fluorescent detection, the amplified products were also resolved on 1.8% agarose gels and visualized by staining with ethidium bromide. Densitometric analysis was done using Genetools 3.6 (Syngene, Cambridge, England). Gene expression levels were normalized to the expression of the housekeeping gene βactin.
Statistical analysis.
Affymetrix (Santa Clara, CA, USA) Human Genome U133 Plus 2.0 Arrays, consisting of 54,614 probe sets, were used for measuring basal level gene expression in CP70 cells. Three biological and three control samples were assessed, with the resulting Affymetrix image files (*.CEL files) analyzed simultaneously and normalized using the RMA algorithm provided in the Affymetrix Expression Console software, as we have described previously.Citation78 Probe sets were annotated using RefSeq and full-length GenBank Transcripts (ncbi.nlm.nih.gov), resulting in 20,554 annotated genes. A log2 scale was used for calculating the intensity of each Affymetrix gene probe for each sample and Welch's test used to compute p values (with p < 0.05 used as a cutoff threshold). Box plots were performed using R2.9.2 (www.r-project.org/).
Cluster analysis, gene set enrichment analysis (GSEA ) and network/sub-network predictions.
Hierarchical clustering and GSEA analyses were performed as we have described previously.Citation80,Citation81 Briefly, clustering of the miRNA and mRNA microarray profiles was performed using Cluster 3.0 (J. van Huissteden, Vrije Universiteit, Amsterdam, The Netherlands, www.falw.vu/∼huik/cluster.htm). The profiles were first pre-filtered by the fraction present and then input into the program, in which they were further log-transformed. Expression values were normalized by array median centering and gene median centering and average linkage clustering was performed by using a correlation (centered) similarity metric. Clustering results were viewed using TreeView version 1.60 (Michael Eisen, Stanford University, rana.lbl.gov/EisenSoftware.htm). GSEA is a method for evaluating specific gene sets based on prior knowledge, about biochemical pathways or coexpression in previous experiments. GSEA determines whether the specific genes in a set tend to occur randomly or toward the top (or bottom) of a sorted gene list, based on correlation of the gene set calculated by an enrichment score at particular significance level.Citation82 Specific biological pathways were defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa Laboratories, Japan, www.genome.jp/kegg/).
For additional pathway analysis, gene datasets, corresponding to various 2D-to-3D histone mark alterations and gene expression changes, were analyzed using a trial version of the integrated software suite MetaCore™ (GeneGo, St. Joseph, MI, USA www.genego.com).Citation33 This pathway analysis tool employs a vast, manually curated database of interactions between biological objects and a variety of tools for functionally analyzing high-throughput data. Networks and sub-networks enriched for the input genes are generated by a database and depicted by the MapEditor™ module of the software suite. Sub-networks are determined using a p value based on overrepresented pathways. Only significant (p < 0.05) maps were considered and subjected to Gene Ontology (www.geneontology.org) terms and processes. For a key to the various symbols and interactions between objects, please visit the MetaCore™ Quick Reference legend at www.genego.com/pdf/MC_legend.pdf.
Microarray analysis of miRNA in CP70 cells.
A custom microarrayCitation33 was used to determine miRNA expression, using two replicates for each cell line, as we have described previously.Citation83 Clustering of miRNA expression data was performed using CLUSTER,Citation85 with filtering to remove inconsistencies between replicates. For clustering, we first log-transformed the data and median-centered the array and genes, followed by average linkage clustering. Clustering results were visualized by TreeView (http://rana.lbl.gov/EisenSoftware.htm). The expanded images flanking the heat map highlight the most significantly changed genes (p > 0.02, top 98%). The top 98% of significantly changed miRNA included 16 downregulated miRNAs in K27R cells and 24 upregulated miRNAs in K27R cells compared to CP70. No significant differences were found between replicated samples or between 2D and 3D for each cell line (p > 0.95) graphs in “miRNA microarray raw data + fold-change.xls”).
Conclusions
Epigenetic plasticity is well known to facilitate phenotypic adaptation, in response to external signals, during normal mammalian development, and also likely promotes the abnormal differentiation that occurs in tumor initiation and progression. Similarly, it has now been demonstrated that even advanced-stage breast cancer and malignant melanoma cells possess considerable epigenetic plasticity that is responsive to external differentiation cues,Citation22,Citation23 while various inhibitors of epigenetic repression are well established differentiating agents.Citation61 In the current study, we extend this attribute of microenvironmentresponsive epigenetic plasticity to ovarian cancer and cancer stem-like cells, as mediated by well-known and previously little-characterized multivalent histone marks, including a possible in vitro and in vivo functional role for the largely unknown bivalent mark, H3K4me2/H3K9me3. Additionally, we have now identified bivalent H3K4me2/H3K9me3 and H3K9me3/H3K27me3, trivalent H3K4me2/H3K9me3/H3K27me3 and tetravalent H3Ac/H3K4me2/-H3K9me3/H3K27me3 domains in cancer cells, further illustrating the influence of higher order chromatin regulation on phenotype adaptability. Upon a commonplace alteration in cellular microenvironment, transition from anchorage dependence to anchorage independence, we utilized an integrative approach to associate phenotype with chromatin remodeling and transcriptome alterations, suggesting that the necessary phenotypic adaptations are mediated by epigenetic responses to reorganization of cellular architecture. A greater understanding of the mechanism(s) responsible for these microenvironment-influenced chromatin alterations will likely lead to improved epigenetic strategies to reduce tumor cell adaptation and possibly, even diminish or revert the malignant characteristics of these cells.
Abbreviations
| 5-aza-dC | = | 5-aza-2′-deoxycytidine |
| Bmi-1 | = | B-lymphoma MMLV insertion region-1 |
| ChIP | = | chromatin immunoprecipitation |
| CpGI | = | CpG island |
| CSC | = | cancer stem cell |
| DNMT | = | DNA methyltransferase |
| EMT | = | epithelial-to-mesenchymal transition |
| ESC | = | embryonic stem cell |
| EZH2 | = | enhancer of zeste homolog-2 |
| FACS | = | fluorescence-activated cell sorting |
| H3Ac | = | acetylated histone H3 |
| H3K4me2 | = | dimethylated histone H3 lysine 4 |
| H3K9me3 | = | trimethylated histone H3 lysine 9 |
| H3K27me3 | = | trimethylated histone H3 lysine 27 |
| miRNA | = | microRNA |
| nOSE | = | normal ovarian surface epithelium |
| OCIC | = | ovarian cancer-initiating cell |
| Oct4 | = | octamer-4 |
| OSE | = | ovarian surface epithelium |
| PRC | = | polycomb repressive complex |
| SCF-1 | = | stem cell factor-1 |
| TSG | = | tumor suppressor gene |
Figures and Tables
Figure 1 Genome-wide identification of genes associated with specific histone modifications in drug-resistant ovarian cancer cells. (A) Relative peak intensity for each mark. Target genes were identified using a peaksPicking program developed by Jin et al. (Genome Research 2006). Percentages of genes were calculated based on the total number of genes identified in the top 10% of genes from the Agilent array. A plot of each binding site based on the size of occupancy into a particular bin with an increment of 100 bp for each bin. (B) Histone H3 marks in Intragenic vs. promoter CpG islands (CpGIs). Number of individual peaks for each of the four H3 modifications examined (based on CpGIs featured on the array). The top 10% of the total hybridized microarray features (using peaksPicking software, see text for details) were used for the analysis. (C) Genes with H3 marks in proximity to transcriptional start sites (TSSs). Histone mark occupancies were predominately located in regions corresponding to −2 kb to 2 kb relative to the TSS.
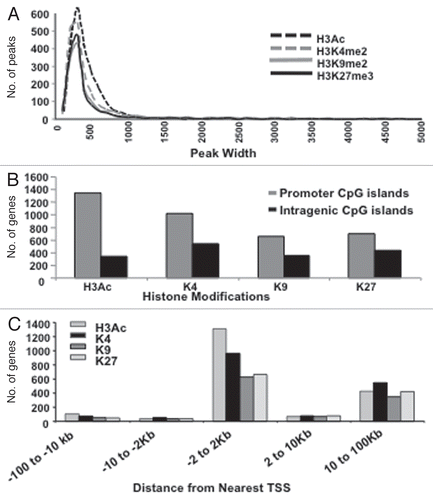
Figure 2 Histone marks and their various combinations, are redistributed or erased upon transition from 2D to 3D culture. PeaksPicking detection of monovalent, bivalent, trivalent and tetravalent H3 modifications in 2D and 3D cells. (A) Monovalent marks. All genes (the top 10%, as determined by the peaksPicking algorithm) were analyzed for the presence of individual histone marks. The proportion and type of such monovalently marked genes in 2D cells were compared to those in 3D cells. (B) Multivalent marks. All genes found by peaksPicking were analyzed for the presence of multivalent histone marks. The proportion and type of such multivalent genes in 2D cells were compared to those in 3D cells. (C) Distinctiveness and overlap of histone-marked genes between the 2D and 3D cellular states. Of the 2,734 genes detected by peaksPicking (top 10%) from 2D cells and 2,424 genes detected in 3D cells, 1149 were found to possess any of the four histone marks between the 2D and 3D.
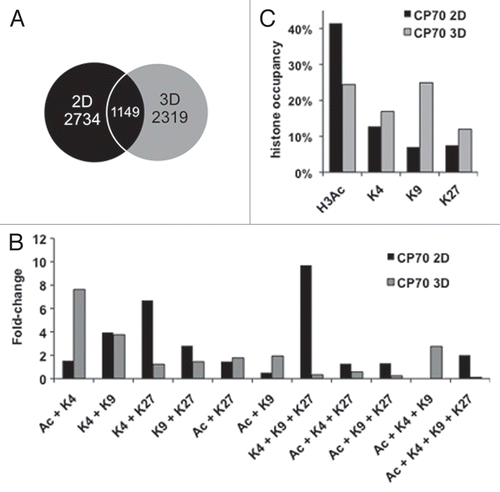
Figure 3 Multivalently histone-marked promoters, abundant in 2D cells, resolve largely to repressive monovalency under 3D culture conditions. (A) Both active and repressive monovalent promoters in 2D cells become mostly monovalent and repressive in 3D cells. All genes containing a monovalent mark in 2D cells were analyzed for the presence of histone marks in 3D cells. Inner pie is modifications less than 5% abundance. (B) Multivalent marks in 2D cells become predominantly monovalent in 3D cells. 2D cells contained 31% multivalent marks, which decreased to 21% multivalent marks in 3D cells. The 2D gene possessing the Ac + K4 + K9 mark was not marked in 3D cells. Black lines delineate monovalent marks from multivalent marks. All possible histone mark combinations are represented.
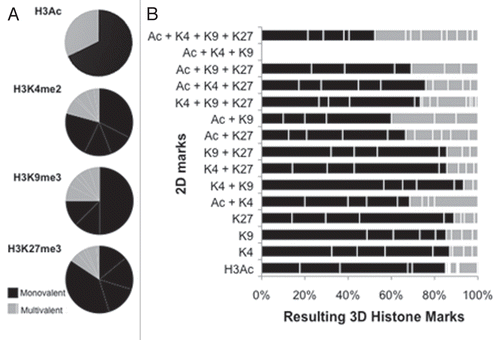
Figure 4 “Histone code”-based correlation of histone modification changes with changes in gene expression. (A) Box-plot analysis of gene expression and chromatin marks in 2D (left upper part) and 3D (right upper part) CP 70 cells. Gene expression arrays were performed on 2D and 3D CP70 cells and compared to those of nOSE cells, with changes in gene expression then correlated with specific chromatin marks associated with each gene. In accord with the histone code hypothesis, promoter-localized H3Ac and H3K4me2 correlated with gene upregulation, while H3K9me3 and H3K27me3 correlated with gene downregulation; genes having any two methylated H3 marks were mostly repressed. (B) DNA-binding, MMP and Wnt family genes are regulated by cellular architecture (2D vs. 3D). The most significant up-and-downregulated genes are shown.
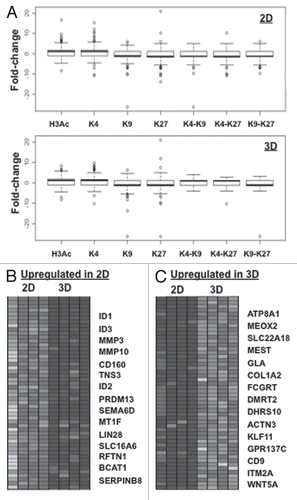
Figure 5 miRNAs are targeted during 2D to 3D transition. (A) miRNA microarray heat map. Top 15 upregulated and downregulated miRNA genes in CP 70-K27R cells, as compared to the parental CP 70 cells. A custom microarrayCitation84 was used to determine miRNA expression, using two replicates for each cell line. Clustering of miRNA expression data was performed using CLUSTER,Citation85 with filtering to remove inconsistencies between replicates. For clustering, we first log-transformed the data and median-centered the array and genes, followed by average linkage clustering. The expanded images flanking the heat map highlight the most significantly changed genes (p > 0.02, top 98%). No significant difference between replicated samples or between 2D and 3D for each cell line (p > 0.95).
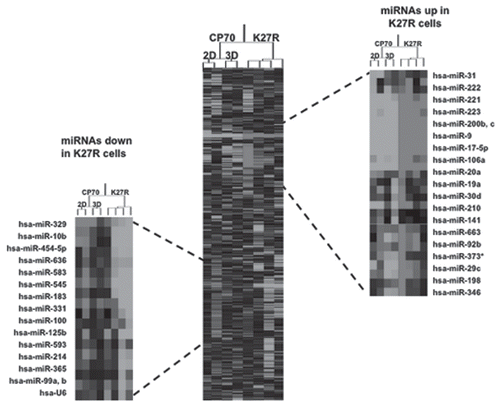
Figure 6 Flexibility of gene expression in xenografts. RT-qPCR results indicating flexibility of gene expression of monolayer CP 70 cultured cells vs. a primary sub-cutaneous tumor (A4 s.c) vs. spheroids in peritoneal metastases (A4 Sph-vivo). Q-PCR standard curves were produced in triplicate and Ct values used to determine relative expression analysis. The measured Ct value was further used to estimate the transcripts levels. Beta-actin was used as a reference housekeeping gene, with expression levels normalized to those of the same genes as expressed in normal ovarian surface epithelial (nOSE) cells.
Additional material
Download Zip (1.8 MB)Acknowledgements
The authors thank Henry Paik for bioinformatics assistance. This work was funded by National Cancer Institute awards CA113001 (to T.H.-M.H.) and CA085289 (to K.P.N.) and an NCCS Institutional Grant (to S.A.B.). S.A.B. received a Short-Term Overseas Fellowship from the Department of Biotechnology, Govt. of India. N.S. and N.K. receive a Senior Research Fellowship from the Council of Scientific & Industrial Research, India. N.B. received a postdoctoral Bridge Fellowship from Indiana University. The authors also wish to thank Andrea Caperell-Grant (Indiana University Department of Obstetrics and Gynecology) for establishment of normal ovarian surface epithelial (nOSE) cell primary cultures used in the study. The microarray studies were performed at the Center for Medical Genomics, Indiana University School of Medicine (Dr. H.J. Edenberg, Director), supported in part by the Indiana Genomics Initiative of Indiana University, INGEN, which is supported in part by the Lilly Endowment, Inc. All analyzed data sets can be obtained from www.broad.mit.edu/seq_platform/chip/.
References
- Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet 2009; 43:559 - 599
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008; 9:465 - 476
- Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074 - 1080
- Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358:1148 - 1159
- Rice JC, Allis CD. Code of silence. Nature 2001; 414:258 - 261
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315 - 326
- Weishaupt H, Sigvardsson M, Attema JL. Epigenetic chromatin states uniquely define the developmental plasticity of murine hematopoietic stem cells. Blood 2010; 115:247 - 256
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 2006; 20:1123 - 1136
- Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007; 447:425 - 432
- Rodriguez J, Munoz M, Vives L, Frangou CG, Groudine M, Peinado MA. Bivalent domains enforce transcriptional memory of DNA methylated genes in cancer cells. Proc Natl Acad Sci USA 2008; 105:19809 - 19814
- McGarvey KM, Van Neste L, Cope L, Ohm JE, Herman JG, Van Criekinge W, et al. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res 2008; 68:5753 - 5759
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 2007; 39:232 - 236
- Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 2008; 40:741 - 750
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 2006; 6:846 - 856
- Wernig M, Lengner CJ, Hanna J, Lodato MA, Steine E, Foreman R, et al. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat Biotechnol 2008; 26:916 - 924
- Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development 2009; 136:509 - 523
- Graf T, Enver T. Forcing cells to change lineages. Nature 2009; 462:587 - 594
- Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med 2007; 58:267 - 284
- Jordan CT. Cancer stem cells: controversial or just misunderstood?. Cell Stem Cell 2009; 4:203 - 205
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009; 9:265 - 273
- Papageorgis P, Lambert AW, Ozturk S, Gao F, Pan H, Manne U, et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res 2010; 70:968 - 978
- Postovit LM, Margaryan NV, Seftor EA, Kirschmann DA, Lipavsky A, Wheaton WW, et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc Natl Acad Sci USA 2008; 105:4329 - 4334
- Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer 2007; 7:246 - 255
- Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004; 432:332 - 337
- Bissell MJ, Labarge MA. Context, tissue plasticity and cancer: are tumor stem cells also regulated by the microenvironment?. Cancer Cell 2005; 7:17 - 23
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007; 39:237 - 242
- Ke XS, Qu Y, Rostad K, Li WC, Lin B, Halvorsen OJ, et al. Genome-wide profiling of histone h3 lysine 4 and lysine 27 trimethylation reveals an epigenetic signature in prostate carcinogenesis. PLoS One 2009; 4:4687
- Shield K, Ackland ML, Ahmed N, Rice GE. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol Oncol 2009; 113:143 - 148
- L'Esperance S, Bachvarova M, Tetu B, Mes-Masson AM, Bachvarov D. Global gene expression analysis of early response to chemotherapy treatment in ovarian cancer spheroids. BMC Genomics 2008; 9:99
- Jin VX, Rabinovich A, Squazzo SL, Green R, Farnham PJ. A computational genomics approach to identify cis-regulatory modules from chromatin immunoprecipitation microarray data—a case study using E2F1. Genome Res 2006; 16:1585 - 1595
- Balch C, Nephew KP, Huang TH, Bapat SA. Epigenetic “bivalently marked” process of cancer stem cell-driven tumorigenesis. Bioessays 2007; 29:842 - 845
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature 2007; 447:433 - 440
- Ekins S, Nikolsky Y, Bugrim A, Kirillov E, Nikolskaya T. Pathway mapping tools for analysis of high content data. Methods Mol Biol 2007; 356:319 - 350
- Tsuda N, Kawano K, Efferson CL, Ioannides CG. Synthetic microRNA and double-stranded RNA targeting the 3′-untranslated region of HER-2/neu mRNA inhibit HER-2 protein expression in ovarian cancer cells. Int J Oncol 2005; 27:1299 - 1306
- Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA 2006; 103:9136 - 9141
- Shell S, Park SM, Radjabi AR, Schickel R, Kistner EO, Jewell DA, et al. Let-7 expression defines two differentiation stages of cancer. Proc Natl Acad Sci USA 2007; 104:11400 - 11405
- Giannakakis A, Coukos G, Hatzigeorgiou A, Sandaltzopoulos R, Zhang L. miRNA genetic alterations in human cancers. Expert Opin Biol Ther 2007; 7:1375 - 1386
- Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res 2007; 67:8433 - 8438
- Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, et al. MicroRNA signatures in human ovarian cancer. Cancer Res 2007; 67:8699 - 8707
- Gillis AJ, Stoop HJ, Hersmus R, Oosterhuis JW, Sun Y, Chen C, et al. High-throughput microRNAome analysis in human germ cell tumours. J Pathol 2007; 213:319 - 328
- Corney DC, Nikitin AY. MicroRNA and ovarian cancer. Histol Histopathol 2008; 23:1161 - 1169
- Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, et al. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 2007; 1:299 - 312
- Abbosh PH, Montgomery JS, Starkey JA, Novotny M, Zuhowski EG, Egorin MJ, et al. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res 2006; 66:5582 - 5591
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10:593 - 601
- Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 2008; 283:14910 - 14914
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704 - 715
- Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009; 15:195 - 206
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006; 441:349 - 353
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 2003; 100:11606 - 11611
- Bryant RJ, Cross NA, Eaton CL, Hamdy FC, Cunliffe VT. EZH2 promotes proliferation and invasiveness of prostate cancer cells. Prostate 2007; 67:547 - 556
- Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res 2005; 65:3025 - 3029
- Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008; 68:4311 - 4320
- Schuebel KE, Chen W, Cope L, Glockner SC, Suzuki H, Yi JM, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet 2007; 3:1709 - 1723
- Dreesen O, Brivanlou AH. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev 2007; 3:7 - 17
- Moss NM, Barbolina MV, Liu Y, Sun L, Munshi HG, Stack MS. Ovarian cancer cell detachment and multicellular aggregate formation are regulated by membrane type 1 matrix metalloproteinase: a potential role in I.p. metastatic dissemination. Cancer Res 2009; 69:7121 - 7129
- Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007; 1:55 - 70
- Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 2008; 26:1269 - 1275
- Shen CN, Slack JM, Tosh D. Molecular basis of transdifferentiation of pancreas to liver. Nat Cell Biol 2000; 2:879 - 887
- Morroni M, Giordano A, Zingaretti MC, Boiani R, De Matteis R, Kahn BB, et al. Reversible transdifferentiation of secretory epithelial cells into adipocytes in the mammary gland. Proc Natl Acad Sci USA 2004; 101:16801 - 16806
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, et al. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 2008; 30:755 - 766
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 692
- Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology and pathology. Endocr Rev 2001; 22:255 - 288
- Liao MJ, Zhang CC, Zhou B, Zimonjic DB, Mani SA, Kaba M, et al. Enrichment of a population of mammary gland cells that form mammospheres and have in vivo repopulating activity. Cancer Res 2007; 67:8131 - 8138
- Golmohammadi MG, Blackmore DG, Large B, Azari H, Esfandiary E, Paxinos G, et al. Comparative analysis of the frequency and distribution of stem and progenitor cells in the adult mouse brain. Stem Cells 2008; 26:979 - 987
- Cicero SA, Johnson D, Reyntjens S, Frase S, Connell S, Chow LM, et al. Cells previously identified as retinal stem cells are pigmented ciliary epithelial cells. Proc Natl Acad Sci USA 2009; 106:6685 - 6690
- Li L, Fukunaga-Kalabis M, Yu H, Xu X, Kong J, Lee JT, et al. Human dermal stem cells differentiate into functional epidermal melanocytes. J Cell Sci 2010; 123:853 - 860
- Suva ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res 2009; 69:9211 - 9218
- Ahmed N, Thompson EW, Quinn MA. Epithelial-mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: an exception to the norm. J Cell Physiol 2007; 213:581 - 588
- Bendoraite A, Knouf EC, Garg KS, Parkin RK, Kroh EM, O'Briant KC, et al. Regulation of miR-200 family microRNAs and ZEB transcription factors in ovarian cancer: evidence supporting a mesothelial-to-epithelial transition. Gynecol Oncol 2010; 116:117 - 125
- Kusumbe AP, Mali AM, Bapat SA. CD133-expressing stem cells associated with ovarian metastases establish an endothelial hierarchy and contribute to tumor vasculature. Stem Cells 2009; 27:498 - 508
- Sood AK, Seftor EA, Fletcher MS, Gardner LM, Heidger PM, Buller RE, et al. Molecular determinants of ovarian cancer plasticity. Am J Pathol 2001; 158:1279 - 1288
- Hudson LG, Zeineldin R, Stack MS. Phenotypic plasticity of neoplastic ovarian epithelium: unique cadherin profiles in tumor progression. Clin Exp Metastasis 2008; 25:643 - 655
- Katz E, Skorecki K, Tzukerman M. Niche-dependent tumorigenic capacity of malignant ovarian ascitesderived cancer cell subpopulations. Clin Cancer Res 2009; 15:70 - 80
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008; 134:521 - 533
- Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci USA 2006; 103:11154 - 11159
- Ahluwalia A, Hurteau JA, Bigsby RM, Nephew KP. DNA methylation in ovarian cancer. II. Expression of DNA methyltransferases in ovarian cancer cell lines and normal ovarian epithelial cells. Gynecol Oncol 2001; 82:299 - 304
- Qin H, Chan MW, Liyanarachchi S, Balch C, Potter D, Souriraj IJ, et al. An integrative ChIP-chip and gene expression profiling to model SMAD regulatory modules. BMC Syst Biol 2009; 3:73
- Fan M, Yan PS, Hartman-Frey C, Chen L, Paik H, Oyer SL, et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res 2006; 66:11954 - 11966
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402 - 408
- Li M, Balch C, Montgomery JS, Jeong M, Chung JH, Yan P, et al. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med Genomics 2009; 2:34
- Nam S, Li M, Choi K, Balch C, Kim S, Nephew KP. MicroRNA and mRNA integrated analysis (MMIA): a web tool for examining biological functions of microRNA expression. Nucleic Acids Res 2009; 37:356 - 362
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102:15545 - 15550
- Xin F, Li M, Balch C, Thomson M, Fan M, Liu Y, et al. Computational analysis of microRNA profiles and their target genes suggests significant involvement in breast cancer antiestrogen resistance. Bioinformatics 2009; 25:430 - 434
- Thomson JM, Parker J, Perou CM, Hammond SM. A custom microarray platform for analysis of microRNA gene expression. Nat Methods 2004; 1:47 - 53
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 1998; 95:14863 - 14868