Abstract
Chromatin is broadly compartmentalized in two defined states: euchromatin and heterochromatin. Generally, euchromatin is trimethylated on histone H3 lysine 4 (H3K4me3) while heterochromatin contains the H3K9me3 marks. The H3K9me3 modification is added by lysine methyltransferases (KMTs) such as SETDB1. Herein, we show that SETDB1 interacts with its substrate H3, but only in the absence of the euchromatic mark H3K4me3. In addition, we show that SETDB1 fails to methylate substrates containing the H3K4me3 mark. Likewise, the functionally related H3K9 KMTs G9A, GLP, and SUV39H1 also fail to bind and to methylate H3K4me3 substrates. Accordingly, we provide in vivo evidence that H3K9me2-enriched histones are devoid of H3K4me2/3 and that histones depleted of H3K4me2/3 have elevated H3K9me2/3. The correlation between the loss of interaction of these KMTs with H3K4me3 and concomitant methylation impairment leads to the postulate that, at least these four KMTs, require stable interaction with their respective substrates for optimal activity. Thus, novel substrates could be discovered via the identification of KMT interacting proteins. Indeed, we find that SETDB1 binds to and methylates a novel substrate, the inhibitor of growth protein ING2, while SUV39H1 binds to and methylates the heterochromatin protein HP1α. Thus, our observations suggest a mechanism of post-translational regulation of lysine methylation and propose a potential mechanism for the segregation of the biologically opposing marks, H3K4me3 and H3K9me3. Furthermore, the correlation between H3-KMTs interaction and substrate methylation highlights that the identification of novel KMT substrates may be facilitated by the identification of interaction partners.
Introduction
The genome is structured around an array of proteins and nucleic acids complex compacted into chromatin. Consequently, DNA-dependent processes, such as transcription, replication and repair, are generally impaired by chromatin. So, in order to facilitate genic access for the cell machinery, the chromatin template is dynamically modified by small molecules, such as acetyl and methyl moieties. Several lysine methyltranferases (KMTs) have clear links to cancer,Citation1,Citation2 suggesting a central role for methyl-lysine (Kme) in maintaining cellular homeostasis and in preventing neoplastic diseases. The SET (Su(var)3-9, Enhancer-of-zeste, Trithorax) domain is responsible for the catalytic activity of most known KMTs, which catalyze the transfer of the methyl group from the co-factor S-adenosylmethionine (SAM) to the lysine ε-amine. While the lysine side chain receives only one acetyl group, it can either be mono- (Kme1), di- (Kme2) or tri-methylated (Kme3). These incremental methylation states can lead to diverse outcomes. The relaxed state of chromatin, euchromatin, is characterized by specific histone tail marks, such as H3K4me3, while the condensed chromatin, heterochromatin, displays distinct marks as well, such as H3K9me3. Genome-wide studies of histone marksCitation3 reveal a segregation of H3K4me3 and H3K9me3. The actual mechanisms that preclude the methylation of H3K9 at H3K4me3 sites remain elusive. In human, six KMTs have a described H3K9-specific activity.Citation4 Namely, these are SUV39H1, SUV39H2, SETDB1, G9A, GLP and RIZ1. One of the KMT that harbors H3K9 specificity, SETDB1 (SET domain bifurcated 1), forms transcriptional silencing complexes with HP1α,Citation5–Citation7 KAP-1Citation7,Citation8 and mAM.Citation9 The bacterially expressed form of SETDB1 is inactiveCitation7 (personal observation O.B.), suggesting that post-translational modifications may be required for activity. Otherwise, SETDB1 may only be active in the presence of other factors such as mAM.Citation9 Furthermore, SETDB1 functions and H3K9me3 are linked to the etiology of Huntington Disease,Citation10 cancer,Citation11 and ES cell differentiation.Citation12–Citation14 Therefore, deciphering the molecular mechanisms underlying the specificity of KMTs is fundamental to understand these biological processes and possibly treat associated diseases.
Herein we describe a novel mechanism by which lysine methylation via the KMTs SETDB1, G9A and SUV39H1 is impaired by the euchromatic H3K4me3 mark. Most importantly, our results highlight that several KMTs operate via a stable interaction with their respective substrates. Thus, the discovery of novel substrates may depend on proteomic approaches to identify KMT-substrates through protein-protein interactions.
Results
SETDB1 binds histone H3 in vitro and in vivo.
In addition to the methyl-CpG binding domain (MBD) and the catalytic SET domain, SETDB1 contains several putative histone binding elements, such as a tandem Tudor domain as well as a preSET and a postSET region predicted to form a zinc finger (depicted in ). We therefore verified whether SETDB1 could bind directly to histones. Recombinant full-length SETDB1 prepared from insect cells was incubated with calf thymus histones and subjected to glutathione-S-transferase (GST)-pulldown assays. shows that SETDB1 interacts with histone H3. Moreover, H2A and H4 immunoblotting demonstrates that the interaction of SETDB1 with histones is specific to H3. The plant homeodomain (PHD) of the inhibitor of growth 2 (ING2) fused to GST (GST-ING2PHD) was used as a positive control for histone H3 binding.Citation15
To validate SETDB1 interaction with H3 in vivo, we conducted a protein-protein chromatin immunoprecipitation (ppChIP) experiment where histone H3 was immunopurified from chromatin. Endogenous SETDB1 was detected from the H3 ppChIP (). H3 and H4 were used as controls and thus were detected from the H3 ppChIP.
In order to map the minimal region of SETDB1 necessary for H3 interaction, we have generated an extensive panel of SETDB1 truncation mutants as GST-fusion proteins. We focused on the SET domain containing carboxy terminal half of SETDB1 as the tandem Tudor domain was shown previously to fail to interact with histones.Citation16 Full-length SETDB1, but not GST alone, interacted with biotinylated H3 peptide containing unmodified amino acids 1 to 21 (). The carboxy terminal half (SETDB1(L) and SET constructs) was found to contain the minimal region necessary for H3 interaction ( and E). Thus, the carboxy terminal region of SETDB1 was dissected by removal of the preSET, postSET or both. These deletion mutants demonstrate that the preSET domain was not required for H3 binding (ΔpreSET, and E). Moreover, the preSET alone or the pre-N-SET were unable to associate with the histone H3 peptide. Interestingly, deletion of the postSET domain (ΔC-SET) abolished the histone H3 interaction and the minimal region required for this interaction was mapped to the carboxy terminal half of the SET domain (SETDB1(S) and C-SET, and E). These findings demonstrate that the carboxy terminus of SETDB1 is both necessary and sufficient for interaction with histone H3 amino terminal tail.
Altogether, these results clearly demonstrate that SETDB1 interacts with histone H3 both in vitro and in vivo by means of the SET domain.
H3K4me3 euchromatic mark impedes SETDB1 binding in vitro.
Because methylated H3K4 and H3K9 have antagonistic chromatin signaling functions, we tested whether methylated H3K4 forms bound SETDB1. While SETDB1 interaction with histone H3 was unaffected by neither H3K4me1 nor H3K4me2, H3K4me3 completely abrogated the interaction between H3 and SETDB1 (). To ascertain the quality of the peptides for use in pulldown assays, the SETDB1 pulldowns were conducted in parallel with a known H3 interactor, the PHD domain of the inhibitor of growth 4 (ING4PHD).Citation17 shows that ING4PHD interacted with neither unmodified H3 nor H3K4me1, but strongly bound to H3K4me2 and H3K4me3, as previously observedCitation17 and confirms that SETDB1 binds unmodified H3 as well as H3K4me1 and H3K4me2, but fails to interact with H3K4me3. Other KMTs with a similar topology as SETDB1, consisting of preSET, SET and postSET domains, such as SETDB2, SUV39H2, SETD2 and SETMAR also bind histone H3 (Sup. Fig. 1 and data not shown). In addition, shows that G9A interacts with H3, but fails to bind to H3K4me3. Likewise, H3K4me3 prevents SUV39H1 from binding H3 (). Equivalent amounts of peptide were used in all experiments (see Sup. Fig. 2). Therefore, our results demonstrate that these H3K9-specific KMTs bind to histone H3 only in the absence of the euchromatic mark H3K4me3.
H3K4me3 euchromatic mark hinders SETDB1 in vitro H3 methylation.
The failure of SETDB1 to associate with its histone H3 substrate when it has an euchromatic H3K4me3 mark led us to postulate that H3K4me3 could not only preclude SETDB1 from binding to H3, but also prevent SETDB1 from methylating H3K9. Thus, KMT assays were conducted with recombinant SETDB1 purified from insect cells and biotinylated H3 peptides in the presence of 3H-SAM. The peptides were then purified using streptavidin sepharose and transferred to scintillation vials for quantification of 3H incorporation and assess histone H3 methylation. shows that SETDB1 potently methylates the unmodified histone H3 peptide as well as the H3K4me1 and H3K4me2 peptides, but fails to methylate a peptide harboring H3K4me3. The H3K9ac peptide served as an unmethylatable control.Citation7 Methylation of H3K9ac and H3K4me3 by SETDB1 was respectively about 15- and 7-fold less than the unmodified H3 peptide. Equivalent amounts of each peptide were used, as confirmed by dot blot analysis (Sup. Fig. 3). The results were validated using a different approach based on the principle of scintillation proximity. The biotinylated peptides were immobilized in quadruplicate on a Flashplate, then subjected to methylation by either SETDB1 or G9A and methyltransferase activity was quantified. The latter experiment confirms that H3K4me3 impairs H3 methylation by SETDB1 (). Furthermore, G9A-dependent H3 methylation was also significantly hindered by H3K4me3. The experiment also suggests that SETDB1 and G9A mainly lead to dimethylation of H3K9 since they were both able to retain some activity on H3K9me1, but had no significant activity on neither H3K9me2 nor H3K9me3 peptides in this particular assay. The amount of each peptide used was confirmed by dot blot analysis (Sup. Fig. 4). Finally, SETDB1 KMT activity was assessed on full-length recombinant histones chemically methylated at specific sites (methyl-like analog, MLA). These KMT assays on MLAs demonstrate that SETDB1 methylates full-length histone H3, but methylation of H3K4me3 histone is severely impaired, as assessed by autoradiography of 3H incorporation and H3K9me3-specific immunoblotting (). Similar results were obtained for G9A and SUV39H1 (). Hitherto, we provide strong evidence that H3K9-specific methylation is impaired by H3K4me3 in correlation with the binding affinity of these enzymes with their substrate.
Structural basis of H3K9-specific KMT s interaction with H3.
Fine mapping of the SETDB1 H3 interaction domain indicates that the SET domain and the postSET region are essential. Moreover, deletion of the postSET in SETDB1 or single point mutation of C1279 abolishes SETDB1 KMT activity.Citation7 Similarly, deletion of the postSET region of SUV39H1 also abolishes KMT activity.Citation18 Thus, SETDB1 cysteine C1226 of the SET domain conserved active site (NHSCSPN) or postSET C1278 and C1280 were mutated, but these subtle changes failed to prevent H3 binding (data not shown). Thus, although the carboxy terminus of SETDB1 is required for H3 binding, the putative zinc coordination through the catalytic core and the postSET domain is dispensable for this interaction.
To define key amino acids of H3K9-specific KMTs required for H3 interaction, we conducted a multiple sequence alignment between GLP, G9A, SUV39H1 and SETDB1 and identified a region of high similarity (Sup. Fig. 5). We found that GLP D1162 is conserved in G9A and SETDB1, while D1176 is conserved between GLP, G9A and SUV39H1. Thus, we postulated that H3K4 methylation may disrupt an interaction between the positively charged lysine H3K4 and the KMTs aspartic acids. To provide mechanistic insight on the role of H3K4 on the catalysis of H3K9 methylation, D1074 and D1088 were individually substituted for alanine or tyrosine in G9A900. Also, a catalytically inactive mutant C1115A (NHLC1115DP) was generated. These mutants were tested for binding properties in biotin-pulldown assays with H3 and H3K4me3 peptides and for activity in KMT assays on recombinant H3 and H3K4me3 MLA. The binding affinity of G9A900 D mutants for H3 and H3K4me3, while lower than wild-type, seemed only mildly affected (data not shown). As expected, G9A900 C1115A retained H3 binding capability (data not shown), but had no detectable activity on H3 (). Moreover, the D1074A mutant activity on H3, while lower then wild-type G9A (see Sup. Fig. 6), seemed unaffected by the presence of H3K4me3 (). Interestingly, the double D1074A/D1088A mutant activity was significantly stronger on H3K4me3 than on unmodified H3 (). Finally, H3K4me3 did not affect the activity of G9A900 D1088Y, but H3K4me3 still impaired D1074Y activity (). These results suggest that aspartic acid residues in the SET domain play a role in differentiating between H3K4 methylation states, thus regulating H3K9 methylation.
H3K4me2/3 and H3K9me2/3 interplay in vivo.
We then addressed the biological relevance of H3K4me in regulating H3K9me in vivo. Thus, we used HP1γ, which interacts with dimethylated H3K9,Citation19 in a ChIP experiment and HP1γ-bound histones were analyzed by mass spectrometry. shows that histones enriched by HP1γ ChIP were dimethylated on H3K9 and acutely depleted of the H3K4me2 and H3K4me3 marks. In a complementary approach, we depleted genes required for H3K4me3 and analyzed histones by mass spectrometry. Knockdown of either WDR82 or RBBP5 in HeLa cells () led to a global decrease in H3K4me3 (), as expected.Citation20,Citation21 The WDR82 knockdown led to severe decrease of H3K4me2/3 and a 3–4 fold increase of both H3K9me2 and H3K9me3 (). Although the RBBP5 knockdown led to a similar decrease in H3K4me2/3 as the WDR82 knockdown, only a moderate increase in H3K9me2 was observed (). Thus, histones with H3K4me2/3 tend to lack H3K9me2/3 in vivo and reciprocally, H3K9me2-enriched histones are deprived of H3K4me2/3 in agreement with our in vitro methylation data.
KMTs interact with their substrate.
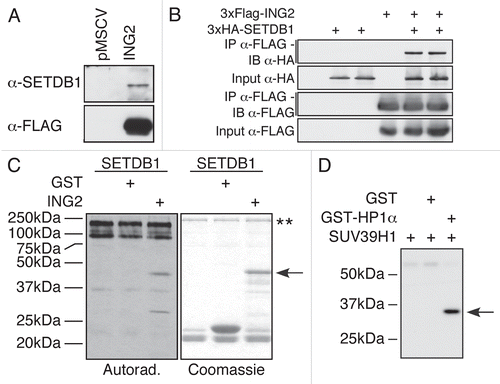
Hitherto, we have demonstrated that multiple KMTs interact with H3 for efficient lysine methylation of H3K9. This led us to hypothesize that the SET domain may interact with other substrates. Since the inhibitor of growth 2 (ING2) was recently found to associate with KMT activity,Citation22 we suspected that it may also be a target for lysine methylation. Indeed, endogenous SETDB1 can be detected from purified ING2 complex (). To confirm the specificity of the interaction and validate the identity of the KMT, we coexpressed FLAG-ING2 and HA-SETDB1 in HEK-293 cells for co-immunoprecipitation experiments. We show that two independent HA-SETDB1 clones came down only in the presence of FLAG-ING2 (), confirming that SETDB1 can associate with ING2 complex. Then, we tested whether ING2 could be methylated by SETDB1. In vitro KMT assays demonstrate that ING2 is methylated only in the presence of recombinant SETDB1 (). Thus, SETDB1 associates with and methylates ING2. To further support our hypothesis, we looked at known KMT interacting partners. The heterochromatin protein HP1α interaction with SUV39H1 is well documentedCitation23,Citation24 and HP1 proteins were recently shown to be methylated in vivo on multiple lysines.Citation25 Furthermore, HP1α was recently found to associate with a complex containing SUV39H1.Citation26 Thus, we tested whether SUV39H1 could methylate HP1α. shows that SUV39H1 potently methylates HP1α. Therefore, it appears that KMT interacting proteins are often also substrates for lysine methylation, supporting our hypothesis.
Discussion
SETDB1 is a member of a group of enzymes within the lysine methyltransferases family that possess specificity for histone H3 lysine 9 and has an important role in silencing transcription via the histone marks it deposits, H3K9me2–3. Furthermore, SETDB1 was recently shown to negatively regulate a strict subset of genes involved in embryonic stem cell differentiation.Citation12,Citation13 To date, little is known regarding the molecular mechanisms regulating SETDB1 and other KMTs to methylate histones and non-histone protein substrates.
We observed that SETDB1 binds to its substrate histone H3 via its catalytic domain. The SET domain of SETDB1 has the particularity of being split in two halves by a so-called bifurcation region of unknown function. The minimal region of SETDB1 both necessary and sufficient for H3 binding comprises the half of the SET domain located to the carboxyterminal side of the bifurcation and the postSET region. We have found that other KMTs with the preSET-SET-postSET topology found in SETDB1, such as G9A, GLP, SETDB2, SUV39H1, SUV39H2, SETD2 and SETMAR, also have the capability to interact with histone H3. Thus, this broad aptitude of several KMTs to interact with their substrates suggests a potential enzymatic mechanism for regulating lysine methylation.
Interestingly, the euchromatic mark H3K4me3 blocks SETDB1 binding to H3. This observation applies as well to G9A and SUV39H1, which fail to bind H3K4me3. Because H3K4me3 impairs the physical association of these KMTs with H3, we hypothesized that this particular histone mark also hinders substrate methylation. Indeed, we found that the KMT activity of the H3K9 methyltransferases SETDB1 and G9A on histone peptides was impaired by H3K4me3. Interestingly, di-methylation of H3 peptides at K4 has no effect on SETDB1 activity, in agreement with previous reports,Citation7 but H3K4me3 significantly impairs H3 methylation by SETDB1. To further demonstrate that H3K4me3 impairs H3 methylation by SETDB1, we used methyl-like analog recombinant histones. This complementary approach also shows that the H3K4me3 mark impairs H3 methylation by SETDB1 and G9A, but also that of SUV39H1.
To dissect the molecular mechanism behind the binding of H3K9 methyltransferases and histone H3, several point mutations of SETDB1 were generated. Neither the mutation of SETDB1 catalytic center C1226 nor the postSET mutations at C1278 and C1280 could prevent physical interaction between SETDB1 and H3. Based on fission yeast H3K9 methyltransferase Clr4 crystal structure,Citation27 the postSET cysteines coordinate a zinc ion with the catalytic site and is necessary for enzymatic activity. Our results establishing that SETDB1 mutations that abrogate the methyltransferase activity and retain H3-binding capability, suggest that the catalytic activity is uncoupled from the substrate interaction. Since bacterially expressed G9A is enzymatically active, we used G9A as our prototype KMT to test the effect of various mutations. The recent publications regarding trimethyl-lysine interacting domainsCitation15,Citation17 highlight the role of tyrosine and tryptophane amino acids in the affinity for trimethylammonium groups. We thus modified G9A SET domain based on a multiple alignment of several KMTs in an attempt to accommodate the H3K4me3. Our data on the methyltransferase activity of the D1074 and D1088 mutants suggest that the interaction between G9A aspartic acids and H3 lysine 4 side chain can be adjusted to accommodate the trimethylammonium group on K4 thereby allowing G9A to methylate H3 while K4 is trimethylated. Moreover, the G9A D1074 and D1088 mutants highlight the importance of H3K4 interaction with the methyltransferase for optimal activity. While preparing this manuscript, the structure of GLP in complex with histone H3 tail was published (PDB Id 3HNA)Citation28 and provided clues that histone H3 lysine 4 is indeed buried in an acidic fold. However, we provide evidence that these conserved aspartic acids are involved in a mechanism where H3K4me3 impedes H3K9 methylation by preventing protein-protein interaction between H3K9-specific KMTs and H3.
To address the biological relevance of our in vitro observations, we globally disrupted H3K4 methylation by targeting either the SETD1A/B complex subunit WDR82 or the SETD1A/B and MLL complexes subunit RBBP5.Citation20,Citation29 As expected, this led to a dramatic depletion of H3K4me2/3. Importantly, histones lacking the H3K4me2/3 marks were being di- and tri-methylated on H3K9. Another approach we used was to enrich H3K9me2 histones using HP1γ. These histones had the particularity of lacking the H3K4me2/3 marks. Thus, in agreement with the antagonistic biological functions of H3K4 and H3K9 methylation, we conclude that H3K4me2/3 and H3K9me2/3 are reciprocally regulated in cis.
Cross-talk between histone tail modifications occurs either in trans, as exemplified by the requirement of H2B ubiquitination (K123ub) for methylation of H3K4,Citation30,Citation31 or in cis, as shown for phosphorylation of H3S10 and methylation of H3K9.Citation32 Recent work conducted on stem cells showed that the promoter of differentiation genes can be marked by opposing marks (bivalent marks) such that H3K4me3 and H3K27me3 marks can occupy the same promoter.Citation33 Likewise, H3K4me3 and H3K9me3 can also be found together at promoters in stem cells.Citation12 Such unorthodox promoters are poised and believed to neither be active nor silenced, but in a ready state. It remains unclear whether bivalent marks such as H3K4me3 and H3K9me3 or H3K27me3 reside on the same histone tail molecule or can be found within the same nucleosome or are merely found in broad chromatin regions on distinct nucleosomes. Nevertheless, here we show that several H3K9 methyltransferases fail to efficiently methylate H3K4me3 substrates, supposedly due to impairment of enzyme-substrate interaction. Therefore suggesting that some bivalent marks, such as H3K4me3 and H3K9me3, found at the promoter of differentiation genes in stem cells, may be found on distinct nucleosomes or seperate histones within the octamer.
Our observations on the correlation between KMT binding to histone H3 and lysine methylation led us to postulate that nonhistone protein substrates of methyltransferase could be identified through a protein-protein interaction based approach. Since the inhibitor of growth ING2 was recently found to associate with H3K9 methyltransferase activity and that ING2 is a subunit of the mSIN3A/HDAC1 complex,Citation15,Citation34 which also associates with SETDB1,Citation35 we looked for a potential interaction between ING2 and SETDB1. We found that not only SETDB1 could interact with ING2, but it could also methylate this novel substrate. To further validate our hypothesis, we looked at known protein-protein interactions involving other KMTs. SUV39H1 interacts with HP1α to propagate heterochromatin. Thus, we tested whether SUV39H1 could methylate HP1α. In agreement with the recent finding that HP1α is methylated on multiple lysines in vivo,Citation25 we found that SUV39H1 could methylate HP1α. Furthermore, recent publications find that NSD1 interacts with NFκB subunit RelA and promotes methylation events on RelA in vivo,Citation36 while G9A binds and methylates the ATPase reptin.Citation37 Together, our results and others, make a strong case of the correlation between lysine methylation and physical interaction between KMT and their substrates; therefore providing a novel approach for the identification of novel lysine methylation substrates.
Because SETDB1 efficiently methylates H3K9 only in the absence of H3K4me3 ( and B), we postulate that H3K4me3 level may modulate SETDB1 and other H3K9 methyltransferases intra-nuclear targeting and promoter specific activity. It will be critical to explore the biological relevance of H3K4me3 on H3 methylation by H3K9-specific methyltransferases in vivo by modulating H3K4me3 level by ectopic expression and knockdown of H3K4-specific lysine demethylases. Importantly, the structural determination of SETDB1 bound to its substrate will be critical to understand the role of the bifurcation region and further delineate the catalytic mechanism, which appears to require more than just SETDB1.
Materials and Methods
Plasmids, cDNA, peptides and antibodies.
The cDNA and expression plasmids for the KMTs were obtained from the Gozani lab. SETDB1 cDNA was inserted in pcDNA3. The FLAG-tagged human ING2 cDNA was described elsewhere.Citation38 HP1α cDNA was a gift from Dr. Monika Lachner. Histone peptides were synthesized at Yale University's W.M. Keck Foundation Biotechnology Resource Laboratory. SETDB1 truncations were generated by PCR using Pfu turbo (Stratagene) polymerase and were inserted in pGEX6-P-1 (Pharmacia). G9A SET domain (G9A900—amino acids 900–1210) was inserted in pET28c (Novagene). The HRP-conjugated α-GST (ab3416), α-H2A (ab18255), α-H3 (ab1791 and ab21054), α-H3K4me3 (ab8580), α-H4 (ab7311) and α-SETDB1 (ab12317) antibodies were bought from Abcam.
Recombinant proteins purification.
Briefly, BL21 DE3 (Stratagene) were transformed with appropriate pGEX or pET28 plasmids and purified as described previously,Citation17,Citation38,Citation39 or using α-FLAG M2-agarose (Sigma). GST-tagged full-length SETDB1 was expressed from pDEST vector in Sf9 insect cells.
KMT assay.
GST-purified proteins (1 µg) were incubated with recombinant KMT (2 µg) with 3H-S-adenosine-methionine (3H-SAM) in KMT reaction buffer (50 mM Tris-Cl pH8.0, 10% glycerol, 20 mM KCl, 5 mM MgCl2). The reactions were incubated for 1 h at 37°C. Then, the samples separated by SDS-PAGE, transferred to PVDF membrane, which were then dried and sprayed with En3Hance (Perkin Elmer) prior to autoradiography. Recombinant histone H3 and chemically modified H3K4me3, H3K9me1 and H3K9me2 by methyl-like analog (MLA) were obtained from Active Motif.
Flashplate KMT assay.
The carboxy-terminally biotinylated histone peptides (330 pmol in 50 µL ddH2O) were allowed to bind to the streptavidin-coated Flashplate (Perkin Elmer) overnight at 4°C. The peptides were then removed by suction and the Flashplate rinsed four times with PBS (0.05% Tween-20) and twice with ddH2O. The KMT reactions were then carried out by adding to each well 1 µg KMT and 0.5 µL 3H-SAM in 50 µL KMT reaction buffer and incubated at 37°C for 2 h. The lysine methylation was measured by 3H scintillation counting on a Perkin Elmer Wallac MicroBeta.
Calf thymus histones pulldown.
Essentially as conducted previously.Citation15 Briefly, histones (20 µg) were incubated with GST proteins (1 µg) in binding buffer (1 M NaCl, 1% NP-40, 50 mM Tris-Cl pH7.5) prior to GST-pulldown.
Biotin-pulldown.
Biotinylated peptides (1 µg) were incubated with GST-KMT or 6xHisFLAG-KMT (1 µg) and pulldowns carried out with streptavidin-sepharose (GE Healthcare) as described.Citation17
Protein-protein ChIP.
Briefly, ∼1.5E07 U2OS cells were crosslinked with formaldehyde (1% final) and quenched with glycine (125 mM final). Cell pellets were washed in PBS and resuspended in a hypotonic buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl and protease inhibitors), dounce homogenized and nuclei pelleted at 11,000 g. Nuclei were sonicated in lysis buffer (50 mM Tris-Cl pH 8.0, 10 mM EDTA, 1% SDS) and centrifuged at 16,000 g. The supernatant was diluted five fold in IP buffer (10 mM Tris-Cl pH 7.5, 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% Triton, 0.1% Sodium Deoxycholate and protease inhibitors). Then IPs were performed with Protein A Dynabeads alone or coupled to 1 µg of H3 antibody (Abcam ab1791), incubated overnight at 4°C with shaking. Next, samples were washed three times with IP buffer and once in TE buffer (10 mM Tris-Cl pH 8.0, 10 mM EDTA). Beads were resuspended in Laemmli loading buffer and boiled for 45 min.
Dot blot.
Each biotinylated peptide was analyzed by spotting 1 µg onto a nitrocellulose membrane in quadruplate and left to dry overnight at 4°C. The membranes were then blocked with 5% milk in Tris-buffered saline before analysis using HRP-streptavidin (Millipore).
siRNA knockdowns.
HeLa cells were transfected with Dharmacon SMARTpools siCTRL (D-001206), siWDR82 (M-016629) or siRBBP5 (M-012008) using Lipofectamine RNAiMAX (Invitrogen).
Chromatin immunoprecipitation with HP1γ for mass spectrometry.
FLAG-HP1γ was expressed in HEK293 cells by calcium phosphate precipitation. The cells were harvested 36 h post-transfection and nuclei were prepared by hypotonic lysis in TMSD buffer (10 mM Tris-HCl pH 7.8, 1.5 mM MgCl2, 0.25 M sucrose and 0.5 mM PMSF) on ice. Nuclei were pelleted and then resuspended in micrococcal nuclease (MNase) digestion buffer (10 mM Tris-HCl pH 7.8, 10 mM NaCl, 3 mM MgCl2, 3 mM CaCl2 and 0.5 mM PMSF) and the chromatin was digested to mononucleosomes with micrococcal nuclease (Sigma) at 37°C for 15 min. The MNase digestion was terminated by the addition of 20 mM EGTA, the nuclear debris were removed by centrifugation and the HP1γ-bound nucleosomes were immunoprecipitated overnight with anti-FLAG M2 agarose (Sigma). The next day, the immunoprecipitates were washed with wash buffer (10 mM Tris-HCl pH 7.8, 75 mM NaCl, 3 mM MgCl2, 0.5 mM PMSF and 5% glycerol) three times and the proteins were eluted overnight using competitive FLAG peptide at 4°C for 12 h.
Histone preparation for mass spectrometry.
Bulk histones (≥50 µg) from HeLa cell pellets were isolated, derivatized, trypsinized and prepared for mass spectrometry as previously described.Citation40,Citation41 Purified histones derived from HP1. immunoprecipitations (≥5 µg) were prepared in a similar, although scaleddown manner.
LC-CAD-MS/MS and data analysis.
Histone peptides were separated with an Agilent 1200 series HPLC and mass spectrometry was performed on a LTQ-Orbitrap mass spectrometer (ThermoFisher Scientific, San Jose, CA). Spectra were analyzed and quantified as previously described.Citation41
Abbreviations
| H3K4me3 | = | trimethyl histone H3 lysine 4 |
| H3K9me3 | = | trimethyl histone H3 lysine 9 |
| KMT | = | lysine methyltransferase |
| SET | = | Su(var)3-9, enhancer-of-zeste, trithorax |
| SETDB1 | = | SET domain bifurcated 1 |
| PHD | = | plant homeodomain |
| ING2 | = | inhibitor of growth 2 |
| SAM | = | S-adenosylmethionine |
| ppChIP | = | protein-protein chromatin immunoprecipitation |
Figures and Tables
Figure 1 SETDB1 binds histone H3 in vitro and in vivo. (A) A graphical depiction of SETDB1 functional domains. Of note, the SET domain (orange) is interrupted by a so-called bifurcation region (green) of unknown function. (B) GST-pulldown experiment. Full-length recombinant GST-SETDB1 was incubated with calf thymus histones. GST alone and GST-ING2PHD were used as negative and positive controls, respectively. Following SDS-PA GE separation and transfer, the membranes were probed with α-H3, α-H4 or α-H2A antibodies. Input lane is 5%. (C) α-H3 ppChIP. Chromatin was immunoprecipitated with α-H3 and analyzed by immunoblotting using α-H3, α-H4 and α-SETDB1. Input level for α-H3 and α-H4 is 1 and 10% for α-SETDB1. (D) Mapping of SETDB1 minimal region necessary and sufficient for H3-binding. Biotinylated histone H3 peptides were incubated with recombinant SETDB1 and streptavidin-sepharose beads used to pulldown the peptides and interacting proteins. Following SDS-PAGE separation and transfer, the membranes were probed with an HRP-conjugated α-GST antibody. (E) Representation of the truncation mutants used in (D) and a summary table of their respective affinity for H3 binding.
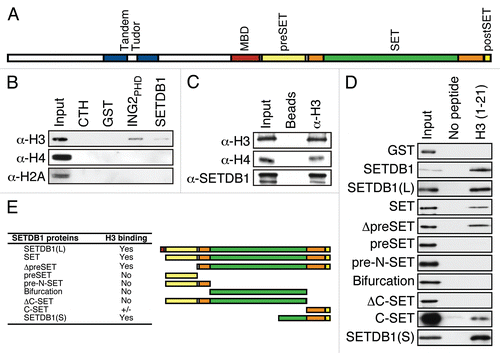
Figure 2 H3K4me3 euchromatic mark impedes SETDB1 binding in vitro. (A) Pulldowns using biotinylated H3 peptides with the indicated modifications as in . (B) A similar experiment as in (A), but ING4PHD was used in parallel as a control for H3K4me3 binding. (C) G9A and SUV39H1 were used in a biotin-pulldown experiment.
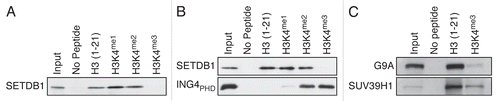
Figure 3 H3K4me3 euchromatic mark hinders SETDB1 in vitro H3 methylation. (A) The indicated histone peptides were incubated with SETDB1 and 3H-SA M, then pulled-down with streptavidin beads in 300 mM NaCl buffer and washed three times. The beads were then transferred to liquid scintillation vials for quantification of incorporated methyl groups. (B) SETDB1 and G9A were tested for KMT activity on the indicated biotin-peptides using a Flashplate. The values are in (%) with the activity on H3 (1–21) set at 100%. The error bars represent the S.E.M. of quadruplicates from a representative experiment. (C) Methyltransferase reactions using either SETDB1, G9A or SUV39H1 were conducted on full-length recombinant histone H3 with the indicated MLA modifications. The samples were analyzed by autoradiography (3H-SA M) and western blotting using α-H3K9me3. Either α-H3 or α-GST antibodies were used as controls for substrate and enzyme input in each reaction. (D) KMT activity of wild-type G9A900 and mutants C1115A, D1074A, D1074A/D1088A, D1074Y and D1088Y.
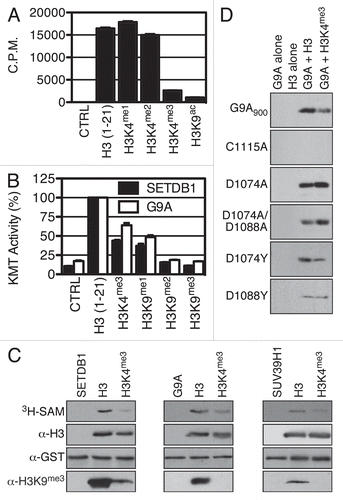
Figure 4 H3K4me2/3 and H3K9me2/3 interplay in vivo. (A) HP1γ was immuno-precipitated from cross-linked chromatin and bound histone analyzed by MS. (B) Table of the raw values obtained for (A). (C) HeLa cells were transfected with either siCTRL, siWDR82 or siRBBP5, the RNA isolated and the level of WDR82 and RBBP5 transcripts analyzed by qPCR. (D) Immunoblot analysis of H3K4me3 in cells from (C). (E) Heatmap of quantitative mass spectrometric analysis of total histones from cells in (C). (F) Raw values used to generate (E) heatmap.
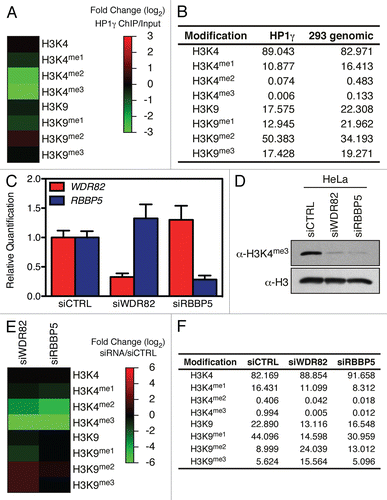
Figure 5 KMTs interact with their substrate. SETDB1 interacts with ING2. (A) FLAG-purified ING2 complex from a stably expressing cell line was probed for the presence of SETDB1. The SETDB1 antibody detected the presence of the endogenous enzyme only in the ING2 complex, but not in the control lane. (B) HEK-293T cells were transfected with an empty vector or 2 independent clones of HA-SETDB1 in the absence or presence of FLAG-ING2. The cells were lysed and proteins immunoprecipitated using FLAG-M2 agarose. The samples were separated by SDS-PAGE, transferred to PVDF and analysed with either α-HA or α-FLAG M2-HRP antibodies. (C) ING2 is methylated in vitro by SETDB1. Recombinant SETDB1 purified from insect cells was incubated alone or with either GST or GST-ING2 in the presence of 3H-SA M. Autoradiography of the experiment (left) and Coomassie staining of the gel (right). The arrow indicates GST-ING2. The two stars (**) indicate SETDB1 and its auto-methylated forms. (D) HP1α is methylated in vitro by SUV39H1. Autoradiography of an enzymatic reaction using bacterially expressed recombinant SUV39H1 and HP1α. In the first lane, only the KMT is present. In the second lane, GST was used as a negative control for SUV39H1 methylation. In the last lane, both SUV39H1 and HP1α were incubated. The arrow indicates HP1α.
Additional material
Download Zip (6.5 MB)Acknowledgements
O.B. is a recipient of a Canadian Institutes of Health Research (CIHR) fellowship and received research support through a grant from Stanford University Cancer Centre's Cancer Biology Program. D.J.B. is funded by the NIH (PA-05-015). S.R. is funded by the CIHR (MOP-67070). O.G. is funded by the NIH (R01 GM079641).
References
- Spannhoff A, Sippl W, Jung M. Cancer treatment of the future: inhibitors of histone methyltransferases. Int J Biochem Cell Biol 2009; 41:12 - 14
- Schneider R, Bannister AJ, Kouzarides T. Unsafe SETs: histone lysine methyltransferases and cancer. Trends Biochem Sci 2002; 27:396 - 402
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823 - 37
- Krauss V. Glimpses of evolution: heterochromatic histone H3K9 methyltransferases left its marks behind. Genetica 2008; 133:93 - 106
- Loyola A, Tagami H, Bonaldi T, Roche D, Quivy JP, Imhof A, et al. The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep 2009; 10:769 - 775
- Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell 2007; 28:823 - 837
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 2002; 16:919 - 932
- O'Geen H, Squazzo SL, Iyengar S, Blahnik K, Rinn JL, Chang HY, et al. Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS Genet 2007; 3:89
- Wang H, An W, Cao R, Xia L, Erdjument-Bromage H, Chatton B, et al. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol Cell 2003; 12:475 - 487
- Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, et al. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc Natl Acad Sci USA 2006; 103:19176 - 19181
- Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem 2006; 281:19489 - 19500
- Bilodeau S, Kagey MH, Frampton GM, Rahl PB, Young RA. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev 2009; 23:2484 - 2489
- Yuan P, Han J, Guo G, Orlov YL, Huss M, Loh YH, et al. Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev 2009; 23:2507 - 2520
- Lohmann F, Loureiro J, Su H, Fang Q, Lei H, Lewis T, et al. KMT1E mediated H3K9 methylation is required for the maintenance of embryonic stem cells by repressing trophectoderm differentiation. Stem Cells 2009; 28:201 - 212
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006; 442:96 - 99
- Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep 2006; 7:397 - 403
- Hung T, Binda O, Champagne KS, Kuo AJ, Johnson K, Chang HY, et al. ING4 mediates crosstalk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol Cell 2009; 33:248 - 256
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000; 406:593 - 599
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001; 410:116 - 120
- Wu M, Wang PF, Lee JS, Martin-Brown S, Florens L, Washburn M, et al. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol 2008; 28:7337 - 7344
- Steward MM, Lee JS, O'Donovan A, Wyatt M, Bernstein BE, Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol 2006; 13:852 - 854
- Goeman F, Otto K, Kyrylenko S, Schmidt O, Baniahmad A. ING2 recruits histone methyltransferase activity with methylation site specificity distinct from histone H3 lysines 4 and 9. BBA-Mol Cell Res 2008; 1783:1673 - 1680
- Yamamoto K, Sonoda M. Self-interaction of heterochromatin protein 1 is required for direct binding to histone methyltransferase, SUV39H1. Biochem Biophys Res Commun 2003; 301:287 - 292
- Aagaard L, Laible G, Selenko P, Schmid M, Dorn R, Schotta G, et al. Functional mammalian homologues of the Drosophila PEV-modifier Su(var)3-9 encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO J 1999; 18:1923 - 1938
- LeRoy G, Weston JT, Zee BM, Young NL, Plazas-Mayorca MD, Garcia BA. Heterochromatin protein 1 is extensively decorated with histone code-like posttranslational modifications. Mol Cell Proteomics 2009; 8:2432 - 2442
- Fritsch L, Robin P, Mathieu JRR, Souidi M, Hinaux H, Rougeulle C, et al. A subset of the histone H3 lysine 9 methyltransferases Suv39 h1, G9a, GLP and SETDB1 participate in a multimeric complex. Mol Cell 2010; 37:46 - 56
- Min J, Zhang X, Cheng X, Grewal SIS, Xu RM. Structure of the SET domain histone lysine methyltransferase Clr4. Nat Struct Mol Biol 2002; 9:828 - 832
- Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, et al. Structural biology of human H3K9 methyltransferases. PLoS One 2010; 5:8570
- Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol 2008; 20:341 - 348
- Wood A, Krogan NJ, Dover J, Schneider J, Heidt J, Boateng MA, et al. Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol Cell 2003; 11:267 - 274
- Dover J, Schneider J, Tawiah-Boateng MA, Wood A, Dean K, Johnston M, et al. Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J Biol Chem 2002; 277:28368 - 28371
- Duan Q, Chen H, Costa M, Dai W. Phosphorylation of H3S10 blocks the access of H3K9 by specific antibodies and histone methyltransferase. Implication in regulating chromatin dynamics and epigenetic inheritance during mitosis. J Biol Chem 2008; 283:33585 - 33590
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315 - 326
- Doyon Y, Cayrou C, Ullah M, Landry AJ, Côté V, Selleck W, et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell 2006; 21:51 - 64
- Yang L, Mei Q, Zielinska-Kwiatkowska A, Matsui Y, Blackburn ML, Benedetti D, et al. An ERG (ets-related gene)-associated histone methyltransferase interacts with histone deacetylases 1/2 and transcription corepressors mSin3A/B. Biochem J 2003; 369:651 - 657
- Lu T, Jackson MW, Wang B, Yang M, Chance MR, Miyagi M, et al. Regulation of NFκB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc Natl Acad Sci USA 2010; 107:46
- Lee JS, Kim Y, Kim IS, Kim B, Choi HJ, Lee JM, et al. Negative regulation of hypoxic responses via induced Reptin methylation. Mol Cell 2010; 39:71 - 85
- Binda O, Nassif C, Branton PE. SIRT1 negatively regulates HDAC1-dependent transcriptional repression by the RBP1 family of proteins. Oncogene 2008; 27:3384 - 3392
- Binda O, Roy JS, Branton PE. RBP1 family proteins exhibit SUMOylation-dependent transcriptional repression and induce cell growth inhibition reminiscent of senescence. Mol Cell Biol 2006; 26:1917 - 1931
- Garcia BA, Mollah S, Ueberheide BM, Busby SA, Muratore TL, Shabanowitz J, et al. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat Protoc 2007; 2:933 - 938
- Plazas-Mayorca MD, Zee BM, Young NL, Fingerman IM, LeRoy G, Briggs SD, et al. One-pot shotgun quantitative mass spectrometry characterization of histones. J Proteome Res 2009; 8:5367 - 5374