Abstract
Human embryonic stem (hES) cells and fetal mesenchymal stem cells (fMSC) offer great potential for regenerative therapy strategies. It is therefore important to characterise the properties of these cells in vitro. One major way the environment impacts on cellular physiology is through changes to epigenetic mechanisms. Genes subject to epigenetic regulation via genomic imprinting have been characterised extensively. The integrity of imprinted gene expression therefore provides a measurable index for epigenetic stability. Allelic expression of 26 imprinted genes and DNA methylation at associated differentially methylated regions (DMRs) was measured in fMSC and hES cell lines. Both cell types exhibited monoallelic expression of 13 imprinted genes, biallelic expression of six imprinted genes, and there were seven genes that differed in allelic expression between cell lines. fMSCs exhibited the differential DNA methylation patterns associated with imprinted expression. This was unexpected given that gene expression of several imprinted genes was biallelic. However, in hES cells, differential methylation was perturbed. These atypical methylation patterns did not correlate with allelic expression. Our results suggest that regardless of stem cell origin, in vitro culture affects the integrity of imprinted gene expression in human cells. We identify biallelic and variably expressed genes that may inform on overall epigenetic stability. As differential methylation did not correlate with imprinted expression changes we propose that other epigenetic effectors are adversely influenced by the in vitro environment. Since DMR integrity was maintained in fMSC but not hES cells, we postulate that specific hES cell derivation and culturing practices result in changes in methylation at DMRs.
Introduction
Maintaining cells in culture typically alters their phenotype, changing them compared to the derivative in vivo population. In order to use cultured cells as a representative biological model or in clinical therapy, we must know the extent and consequences of their in vitro adaptation. The epigenetic status of stem cells, or indeed any cells, including their genomic methylation level, chromatin accessibility and gene expression profile, may be used as phenotypic markers. Additionally, specialized gene expression pathways, ultimately controlled by epigenetic profiles such as chromatin accessibilty and DNA methylation, can reflect a cell's differentiation capacity, ability to self-renew and its susceptibility to transformation.
Genomic imprinting is an epigenetic mechanism that results in parent-of-origin monoallelic expression of certain genes. Depending on the gene, it can be stable and absolute, transient, tissue-dependent, polymorphic within a population or preferential, where both alleles are expressed albeit with one at a significantly higher level. The mechanisms controlling imprinted expression are specific to each imprinted gene cluster and include a wide range of epigenetic modifiers.Citation1 Canonically, imprinted gene clusters and lone imprinted transcripts are arranged in cis to a differentially-methylated region (DMR).Citation2 Such differential methylation is established during the process of gametogenesis of the previous generation, and is pivotal in the regulation of imprinted expression. In some cases, human and mouse DMRs have been demonstrated to be imprinting control regions (ICRs).Citation3–Citation9
A large number of imprinted genes have been analyzed extensively in vivo and characterized in detail in mouse tissues, and to some extent in humans, through a range of ontological stages.Citation10 This provides a reliable, measurable and reproducible readout of epigenetic status in a given cell line. Such information can be used to compare cell lines and delineate a normal range of epigenetic variation tolerated in normal and abnormal cell conditions. Furthermore, disrupted imprinted gene expression has been directly implicated in cell cycle defects and tumorigenesis.Citation11
In the human, reports of imprinting status in stem cells to date focus on embryonic stem (hES) cells. These studies find imprinting deregulation to be universal amongst hES cell lines in a very gene specific manner. Notably, SNRPN on human chromosome (Hsa) 15 is constitutively monoallelic, and SLC22A18 on Hsa 11 is almost always biallelic.Citation12–Citation14 The imprinted expression of some genes, however, such as H19, has been shown to be influenced by specific culture conditions and passage number, indicating that in some cases it may be possible to optimize tissue culture conditions in order to maintain appropriate imprinted expression.Citation13–Citation15
hES cells are derived from the inner cell mass of the developing blastocyst during genome-wide epigenetic remodeling.Citation16 Cell line derivation during this sensitive period may render the cells epigenetically unstable, manifesting, for example, in loss of imprinting. Although ICR methylation is already established at this developmental stage, reduced methylation due to defective reprogramming may have downstream effects on DMR maintenance. hES cells are derived from spare embryos following assisted reproductive technologies (ART), typically including superovulation. Superovulation has been shown to cause both hypo- and hypermethylation at DMRs, notably the ICRs of H19 and MEST/Mest in human and mouse oocytes.Citation17,Citation18 In addition, hES cell lines are derived at different points during blastocyst expansion. Such variation in the reprogramming stage at derivation is likely to introduce inter-line variation in methylation levels.Citation19
To be able to assess the significance and influence of ART and cell line-derivation during reprogramming on imprinting in hES cells, it is timely and useful to compare hES cells with cultured cells from other in vivo sources and cells derived later in gestation. Human first trimester fetal mesenchymal stem cells (fMSC) are derived beyond the point of genome-wide remodeling from normally conceived fetuses. fMSC are more proliferative and have greater differentiative potential than later sources of adult MSC.Citation20–Citation22 They also have a number of primitive properties similar to hES cells, but are non-tumorigenic and as such represent a promising resource for regenerative therapy.Citation23–Citation25
The allelic expression of imprinted genes was compared between fMSC and hES cells using a panel of 26 imprinted transcripts. Imprinted genes were selected from igc.otago.ac.nz/table. html and chosen to include genes in genomic imprinting clusters and genes not located within a cluster, as well as both protein coding and long non-coding imprinted transcripts. The presence of differential methylation at DMRs was also analyzed in each cell line. To evaluate the impact of the differentiation of cells on imprinted gene expression, allelic expression of a subset of genes was compared between undifferentiated fMSC and fMSC postdifferentiation to osteoblasts and adipocytes.
Results
The allelic expression of imprinted genes was used to delineate each of the cell lines (). This analysis showed that there were no striking trends in allelic expression, either within different fMSC lines (), different hES cell lines () or between the two cell types.
By analyzing results from allelic expression analysis in both fMSC and hES cells, three groups of genes were identified: (1) genes (n = 13) that were always expressed monoallelically in fMSC and hES cells (), (2) genes (n = 6) that were always expressed biallelically in fMSC and hES cells (i.e., a second allele peak present of at least a 25% proportion of the first; ) and (3) genes (n = 7) whose expression varied between cell lines (). The imprinted genes in groups (1) and (2) above we class as genes whose imprinting pattern is ‘set’ to be either monoallelic or biallelic upon expression in a cultured cell.
Allelic expression patterns between cell lines.
To detect more subtle trends in allelic expression between different genes or cell lines, we excluded genes whose expression did not vary—groups (1) and (2) above—as the proportion of biallelic to monoallelic expression for these genes was determined by the SNP informativity of the cell line. lists only genes for which allelic expression varied between cell lines—group (3) above. Of these, we found that IGF2 is expressed monoallelically in hES cells where it is informative, in lines SHEF1 and SHEF4, but it is expressed biallelically in fMSC where it is informative, in lines fBl8+3 and fLiv11+5. These data were verified by restriction digest and are shown by sequencing in Supplemental Figure S3, providing evidence that imprinted IGF2 expression may be disrupted in fMSC specifically. No other distinctions in allelic expression between fMSC and hES cells were identified.
hES cell and fMSC lines can be derived further into subgroups, according to their original laboratory: King's College London or Sheffield University for hES cells and Imperial College London for fMSC. In addition, the fMSC lines were derived from fetuses at a variety of gestational ages, from 8–12 weeks and derived either from blood, liver or bone marrow. No striking differences could be observed between any of the subgroups, although we were limited in our analysis by SNP informativity within the cell lines and cannot rule out the presence of subtle trends in the subgroups for changes in allelic expression. For ES cell lines SHEF5 and KCL-002, only two and three genes were informative, respectively, limiting the analysis of expression in these cell lines.
Alterations of cell culture conditions within cell lines.
Analysis of gene expression in the cell lines was extended to include variations in culture conditions. Allelic expression data from hES cells was initially gathered from analysis of undifferentiated, karyotypically normal lines. This data was then compared to imprinted gene expression analysis in hES cells at a high passage harboring karyotypic abnormalities: SHEF4, passage (P)104: 16% trisomy 17; SHEF1, P121: 10% trisomy 3, 90% del 2(q); and hES cells differentiated to embryoid bodies. We found no allelic expression differences between low passage and high passage/karyotypically abnormal hES cells or between undifferentiated and differentiated cells (data not shown).
The allelic expression data for imprinted genes in fMSC was generated from undifferentiated fMSC that had been carefully maintained at or below 60% confluency (“sub-confluent”). To evaluate the effect of culture conditions on imprinted gene expression, fMSC sub-cultures were fed and grown without passage for four weeks, resulting in growth in a highly confluent state, where the cells grow in layers. The proportion of monoallelic and biallelic transcripts, as a percentage of the total number of SNPs, was compared between sub-confluent fMSC and highly confluent fMSC. No expression differences were observed in any of the lines whether confluent fMSC or their undifferentiated, sub-confluent counterparts (data not shown).
Differentiation of fMSC.
To further investigate the subset of genes that lack imprinted expression in fMSC, cells were differentiated to either bone (osteoblasts) or fat (adipocytes) () and analyzed for their expression and imprinting status. Two of the imprinted genes with “set” expression patterns in culture were analyzed, KCNQ1OT1, which was universally monoallelically expressed and SLC22A18, which was universally biallelically expressed. These two genes were chosen as their SNPs were highly informative and allelic expression could be analyzed in as many of the differentiated fMSC lines as possible. In addition, allelic expression of PEG10 and the expression levels of DLK1 were analyzed, chosen due to the association of their protein products with osteogenesis and adipogenesis.Citation26,Citation27
Six fMSC lines that were informative for one or more of the SNPs in KCNQ1OT1, SLC22A18, PEG10 or DLK1 were differentiated to osteoblasts and adipocytes. Differentiation was confirmed by Von Kossa staining to identify calcium deposition as evidence of osteogenesis or Oil Red O staining to identify lipid deposition as evidence of adipogenesis (). DLK1 expression was not detectable in low passage undifferentiated fMSC and remained below detection levels following fMSC differentiation to adipocytes or osteoblasts. Therefore allelic expression could not be analyzed for this gene. Allelic expression of the remaining three genes was consistent with that observed in undifferentiated fMSC (Sup. Table S4). KCNQ1OT1 () and PEG10 () were both monoallelically expressed while SLC22A18 () was biallelically expressed in each cell line regardless of osteo- or adipogenic culture.
Analysis of methylation at differentially methylated regions (DMRs).
The gene-specific loss of imprinting observed could be due to changes in methylation at imprinting control region DMRs. To investigate this, methylation at known human DMRs was analyzed. DMR genomic positions and specific amplicons analyzed are listed in Supplemental Table S5. Methylation was measured using combined bisulfite and restriction analysis (COBRA) to give a ratio of methylated to unmethylated DNA (). PCR amplicons from each sample were then cloned and sequenced to show that COBRA analysis was representative of other CpGs in the DMR. In all cases, bisulfite sequencing identified both methylated and unmethylated clones and DNA strands were either entirely methylated or entirely unmethylated. This demonstrated that for each DMR, the CpGs quantitated by COBRA were representative of methylation status across the region (Sup. Fig. S4). This analysis could not determine specific alleles as no informative SNPs for the cell lines were identified within the bisulfite amplicons.
Comparison of differential methylation at imprinting control regions revealed the only major difference between fMSC and hES cells. In fMSC, differential methylation was maintained at each DMR, but in hES cells there was a locus-specific loss of differential methylation in certain lines. At the MEST DMR, PEG10 DMR, H19 Promoter and the germline DMR in this region, the H19 differentially methylated domain (H19 DMD), KvDMR and the Prader Willi/Angelman syndrome domain imprinting center (PWS/AS-IC), the COBRA ratio of methylated to unmethylated DNA was approximately 1:1 for all samples, strongly suggestive of the maintenance of differential methylation ( and G). These data were confirmed by the presence of both completely methylated and unmethylated strands when the amplicons were analyzed by sequencing Supplemental Figure S4. In fMSC lines, the DLK1 intergenic DMR (IG-DMR), PEG3 DMR, NESP Promoter and NESPAS/XL DMRs also maintained differential methylation ( and H–J). In hES cell lines, however, there was a deviation from the expected 1:1 ratio of methylation for the IG-DMR, PEG3 DMR, NESP Promoter and NESPAS/XL DMRs ( as above). This variation was cell line-specific and featured both hypo- and hypermethylation.
Correlations between DMR maintenance and allelic expression.
Where imprinted genes were expressed monoallelically, this could be correlated in most cases with normally methylated DMRs, e.g., the KvDMR maintained differential methylation at a 1:1 ratio () and KCNQ1OT1, KCNQ1 and CDKN1C were monoallelically expressed in all samples (). The IG-DMR was differentially methylated in hES line KCL-003-CF1 (), correlating with monoallelic expression of DLK1 in this line ( and Sup. Fig. S3). For a minority of samples, however, monoallelic expression was observed even when differential methylation was lost or skewed, for example, PEG3 was monoallelically expressed in the ES cells line SHEF4 despite hypermethylation at the PEG3 DMR in this cell line ( and and Sup. Fig. S1).
Conversely, where genes were expressed biallelically ( and ), the maintenance of differential methylation was still observed for the majority of samples, e.g. SLC22A18, was always biallelic and expression of SLC22A18AS was both monoallelic and biallelic, depending on the cell line, but the KvDMR was always differentially methylated ( and ), whereas hypermethylation would be expected. These data suggest that allelic histone tail modifications, known to correlate with monoallelic expression at this locus, may be configured to promote biallelic expression despite DMR maintenance.Citation28 DLK1 expression in hES cell lines SHEF2 and SHEF5 was biallelic but the IG-DMR was differentially methylated in these lines. DLK1 was also biallelically expressed in hES line SHEF4 but in this case the IG-DMR was fully methylated, which would be expected to ablate expression, rather than activating both alleles ( and and Sup. Fig. S3).
Differential methylation at the MEST DMR, which was of a 1:1 ratio in all samples (), also did not correlate with the variable allelic expression observed for both MEST isoforms. Notably, fBM10 + 4 had a ratio of 1:1 methylated/unmethylated DNA at the MEST DMR but was biallelic for both MEST Isoforms 1 and 2 (). Due to the lack of any discernible pattern, we conclude that methylation changes, although confined to hES cells, were otherwise stochastic and unrelated to imprinted expression.
Discussion
We demonstrate that biallelic expression of certain imprinted transcripts is found in human embryonic stem (hES) cells and undifferentiated fetal mesenchymal stem cells (fMSC). This biallelic expression is unrelated to maintenance of ICR differential methylation. In vitro differentiation of fMSC to adipocytes and osteoblasts does not result in a change of allelic expression of imprinted genes. Three classes of imprinted genes were revealed: those that were always monoallelically expressed (n = 13), those that were always biallelically expressed (n = 6) and those whose expression varied between cell lines (n = 7).
The 13 genes that were always expressed monoallelically we view as stable in an in vitro environment, maintaining their monoallelic status. The seven genes whose expression varied between cell lines we suggest to be intermediately sensitive to cell culture. The imprinting status of these variable genes may provide a useful index for cell line epigenetic stability.
Finally, six of the genes analyzed were always biallelic in fMSC and hES cells. We hypothesize that the allelic expression of these genes is most susceptible to disruption by in vitro culture. As their allelic expression status was the same in all the lines tested, it is likely that expression becomes biallelic upon cell line derivation.
It is possible that the variability in allelic expression status we observe is due to the common tissue-specificity of imprinted gene expression. For example, GNAS is maternally expressed in pituitary glandCitation29 and skewed towards maternal expression in the thyroid and gonads.Citation30 However, GNAS is biallelically expressed in adult lymphocytes, fetal liver, lung, skin, central nervous system, muscle and pancreas,Citation31 so our findings of biallelic expression of GNAS in fMSC and hES cells are perhaps not surprising.
To provide additional evidence that in vitro culture causes loss of imprinting, the allelic expression profiles of the population from which fMSC and hES cells were derived in vivo would be required. However, human preimplantation embryos are rarely available for such research. To derive the fMSC lines, non-homogenous cell suspensions from fetal bone marrow, blood and liver are enriched for fMSC using serial passage in vitro. Of the initial extracted tissue, fMSC represent at most 0.4% of total bone marrow mononuclear cells, less in blood and liver, making expression analysis technically demanding.Citation20 In addition, currently there are no specific cell markers that would allow enrichment of fMSC without an intermediate in vitro stage.Citation32
The only difference in allelic expression between fMSC and hES cells was the expression of IGF2. Both alleles of IGF2 were expressed in fMSC from blood and liver, but IGF2 was monoallelic in hES cells. IGF2 is known to be expressed biallelically in 10% of the white European population, at least in peripheral blood.Citation33 As there were only four informative samples available, it was not clear whether the biallelic expression we observe is characteristic of fMSC or due to polymorphic imprinting in the individuals from which the cells were derived.
Comparing allelic methylation at DMRs for selected imprinted genes revealed the first major difference between fMSC and hES cells. In fMSC, all differentially methylated regions were maintained, but in hES cells several DMRs exhibited hypo- or hypermethylation.
ART embryos, and therefore hES cells, are produced from oocytes whose growth has been accelerated using gonadotrophic ovary stimulation or “superovulation.” Superovulation causes demethylation at the Kcnq1, Snrpn and Peg3 imprinting control regions in mouse embryos, demethylation at the human MEST DMR and hypermethylation at the H19 DMD in humans and mice.Citation18,Citation34 Additionally, subfertility in couples undergoing ART may have downstream effects on cell lines derived from their embryos. Since the fetuses from which fMSC were derived were conceived without the use of ART or superovulation, these factors highlight possible explanations for why hypo- and hypermethylation of DMRs was restricted to hES cells. There are, however, several other important differences between the two cell types. The tissue culture milieu, passaging technique and derivation procedure differ widely between hES cells and fMSC. Variations in cell culture media, such as the inclusion of serum, have been shown to have profound effects on hES cell gene expression patterns, 35 while at very high passage H19 expression becomes biallelic in certain hES cell lines.Citation15
To explore the sensitivity of hES cells and fMSC to small changes in environment, we compared low passage hES cells to those at higher passage and compared subconfluent fMSC with those that were highly confluent. We did not observe any alterations in allelic expression profiles following these manipulations.
The hypo- and hypermethylation of DMRs in hES cells could not be correlated with changes in allelic expression. Similarly, the normal DMR methylation in fMSC is at odds with biallelic expression of imprinted genes in these cells. For example, differential methylation was maintained at the KvDMR, but KCNQ1OT1, KCNQ1 and CDKN1C were monoallelically expressed and SLC22A18 and PHLDA2 biallelically expressed, also observed in hES cells by Young and colleagues.Citation14 Alterations in DMR methylation thus appeared to be stochastic, and did not correlate with imprinted gene expression. We suggest that other epigenetic effectors, such as allelic histone modifications or ncRNA transcripts, are adversely affected by cell culture, resulting in biallelic imprinted gene expression in some cases and the maintenance of monoallelic expression despite deviation from differential methylation, in others.Citation28
In summary, aside from IGF2 expression, we demonstrated no overt differences between allelic expression in hES cells or fMSC. We observe no changes in allelic expression upon differentiation of fMSC to osteoblasts of adipocytes. These data indicate that the disruption to imprinted expression of certain genes is due to in vitro culture, rather than the derivation techniques used, any assisted reproductive technologies or specific cell culture conditions. Simply, loss of imprinted expression of certain imprinted genes is an inherent feature of cultured cells.
The different stabilities of DMRs in the two cell types, however, may be due to different culture strategies required or be a result of superovulation. Additionally, unlike fMSC, hES cells are derived from blastocysts during genome-wide epigenetic reprogramming, the effect of which is unknown.
fMSC are potentially an important resource for the treatment of mesodermal diseases such as osteogenesis imperfecta, muscular dystrophy and of bone trauma.Citation23,Citation36 The prevention of imprinted gene disturbance by assessing and testing culture conditions is a future challenge for the use of stem cells in therapy. Given the significance of the loss of imprinting found in tumors the data presented here represent an important characterization step on the way to the clinic.Citation11,Citation37,Citation38
Materials and Methods
Stem cell collection and culture.
Fetal mesenchymal stem cells (fMSC). Fetal tissues were collected from consenting women undergoing first-trimester termination of pregnancy (see Sup. Table S1 for details). Collection was approved by the Hammersmith and Queen Charlotte's Hospitals Research Ethics Committee and complied with national guidelines on fetal tissue collection for research. Fetal blood was obtained by ultrasoundguided fetal blood sampling and fetal liver and bone marrow dissected from the products of conception.
Fetal mesenchymal stem cells were selected by plastic adherence and cultured as described previously.Citation20 Briefly, cells were cultured in Dulbecco's modified Eagle's media (DMEM®) (Sigma-Aldrich®, Steinheim, Germany), supplemented with 10% fetal bovine serum (FBS) (Gibco®/Invitrogen™, Paisley, UK), 2 mM L-glutamine, 500 U/ml penicillin and 50 µg/ml streptomycin (Gibco/Invitrogen) in 100 mm polystyrene cell culture dishes (Corning, Hemel Hempstead, UK). Adherent cells were passaged before reaching confluency using 0.25% Trypsin-EDTA (Gibco/Invitrogen).
fMSC differentiation. fMSCs were grown to 70% confluency and differentiation initiated by incubation with DMEM plus 10% FBS supplemented with 10 µM dexamethasone, 5 µg/ml insulin (Sigma) and 60 µM indomethacin (Sigma-Aldrich) (adipogenesis) or with 10 µM dexamethasone, 0.2 mM ascorbic acid and 10 mM b-glycerol phosphate (Sigma-Aldrich®) (osteogenesis) for four weeks as previously described.Citation39 Cells were stained with fresh Oil Red O solution or silver nitrate Von Kossa stain (Sigma-Aldrich) to assess lipid deposition or mineralization, respectively.Citation40,Citation41 Confluent fMSC cultures were produced in 100 mm dishes. Cells were fed twice weekly with DMEM plus 10% FBS as above and maintained without passaging for a total of four weeks.
Human embryonic stem (hES) cells. Sheffield hES cell lines (Sup. Table S1) were cultured in T25 flasks with 5 ml Embryonic Stem Cell Media containing 80% Knockout™ Dulbecco's modified Eagle's medium (Gibco/Invitrogen), 20% Knockout™ Serum Replacement (Gibco), 1 mM L-glutamine, 0.1 mM β-mercaptoethenol (Sigma-Aldrich), 1% non-essential amino acids, supplemented with 20 ng/ml FGF-4. Cells were grown on a sub-confluent layer of Swiss-strain mouse embryonic fibroblasts (MEFs), except for hES cell line SHEF7, which was grown on human gonadal interstitial cell feeders. Colonies were dissociated for passaging using trypsin with 3 mm glass beads. King's College London hES cell lines (Sup. Table S1) were grown in 60 mm dishes (Nunc™, UK) with ES media and MEFs (as above) supplemented with either FGF-2 (KCL-001 and KCL-002) or LIF (KCL-003-CF1). Colonies were dissociated for passaging manually with a glass pipette.
Fetal tissues. Fetal tissues (from liver, brain, heart, skin, intestine, limb and placenta) from first trimester termination of pregnancy and placental trios from term placenta with parental peripheral bloods, were from the Moore Fetal Tissue and Moore Placental trio cohorts respectively.Citation42 Collection was approved by the Hammersmith, Queen Charlotte's and Chelsea and Acton Hospitals Research Ethics Committee.
Imprinting analysis.
Expression of imprinted transcripts in hES and fMSC. Expression of selected imprinted genes (see Sup. Tables S2 and S3) was measured semi-quantitatively in fMSC and hES cell pools using reverse transcriptase PCR (RT-PCR). Following TURBO DNase™ (Ambion®/Applied Biosystems™, Warrington, UK) treatment, 1 µg total RNA was treated with DNaseI (Promega), primed with random hexamers and reverse-transcribed with Murine-Maloney Leukemia virus (MMLV) reverse transcriptase (RT; Promega), in parallel with samples omitting the RT enzyme. cDNA synthesis and the absence of contaminating genomic DNA was checked by amplification of both positive- and negative-RT samples using β-ACTIN primers in a 32 cycle RT-PCR reaction. Non-quantitative expression profiles, using increments of five cycles up to a total of 50, defined cycle number ranges corresponding to the log-linear phase of the RT-PCR (Sup. Table S2). The templates created omitting the RT enzyme were included in the RT-PCR in each case, amplified to the maximum cycle number, again to control for amplification of any genomic DNA present. Where possible, primers were designed to cross intron-exon boundaries and were isoform specific where required (MEST Isoforms 1 and 2, GNAS, GNAS EXON 1A and GNAS XL and PLAGL1, which has a non-imprinted isoform). Primer sequences listed in Supplemental Table S6. Pooled samples of fetal tissues derived in vivo, from the Moore fetal tissue cohort,Citation42 were used as positive expression controls.
Genotyping and RT PCR. Genomic DNA extraction from snap frozen cell pellets was performed using standard phenol/chloroform extraction. Exonic single nucleotide polymorphisms (SNPs) were chosen based on their validation from the UCSC Genome Browser dbSNP build 129 (genome.ucsc.edu/cgi-bin/hgGateway) and expressed genes genotyped with respect to each SNP as indicated in –, Supplemental Tables S2 and S6. Total RNA from each cell line was extracted, reverse transcribed in parallel with samples omitting RT and checked using β-ACTIN primers as before. RT-PCR was carried out on individual cDNA samples from each cell line, cycle numbers are shown in Supplemental Table S2. Genotypes and allelic expression were determined using a combination of sequencing (Applied Biosystems, CA) and restriction fragment length polymorphism (RFLP) analysis (RFLP enzymes (New England Biolabs) listed in Sup. Table S2). Sequencing traces for each polymorphism in each gene for exemplar informative samples are shown in Supplemental Figures S1–S3.
Analysis of DMR methylation.
Combined bisulfite and restriction analysis (COBRA). DNA from each sample was treated with sodium bisulfite and purified using the EZ DNA Methylation-Gold Kit™ (Zymo, CA), before amplification using primers specific for each DMR (primer sequences listed in Sup. Table S7). DNA was tested for bisulfite conversion by PCR with bisulfite-specific PWS/AS-IC DMR primers in each case. Hotstar Taq polymerase (Qiagen, West Sussex, UK) was used for 45 PCR cycles. Amplicons were digested using either Taqα1 (-TCGA-), Tai1 (-ACGT-), MboI (-GATC-) or BstUI (-CGCG-) restriction enzymes (New England Biolabs) depending on the location of recognition sites in each region. Digested products were resolved on 3–4% agarose gels and stained with ethidium bromide.
Bisulfite sequencing. Three microliters of the COBRA PCR was not digested but analyzed by bisulfite sequencing instead. Amplicons were ligated into PGEM-T® Vector (Promega) as per the manufacturers instructions. JM109 competent cells, of 108 cfu/µg efficiency (Promega) were transformed and blue/white selection used to pick colonies. Forty transformed colonies were selected per cell line for each DMR and all were amplified using M13 primers for 30 cycles (Promega). Colony PCRs were sequenced, allowing calculation of bisulfite conversion rate and methylation profile of each CpG in the amplified region. Only strands with a C to T conversion efficiency over 95% were included in the analysis.
Figures and Tables
Figure 1 Interline variation in allelic expression status of imprinted genes in fMSC and hES cells. Gray shaded bars = monoallelically expressed genes, black shaded bars = biallelically expressed genes, with the number of informative genes analyzed plotted against the cell line the gene expression was analyzed in (A) fMSC cell lines. Each fMSC line displays both mono- and biallelic expression of some imprinted genes. Adult human fibroblast cells are used as a control, and show a similar pattern of both mono- and biallelic expression. (B) hES cell lines: Similarly to fMSC, hES cell lines express imprinted genes monoallelically in the case of some genes, but biallelically for others. Graphs outline the overall profile of allelic expression of the imprinted genes analyzed for each cell line. Both fMSC and hES cell lines expressed some genes biallelically and others monoallelically. There were two exceptions, hES line KCL-002 and SHEF5 only expressed genes monoallelically and biallelically respectively. Similarly, in fMSC, fMSC line fLiv11 + 5 expressed more genes biallelically than monoallelically. This difference can be attributed to the expression profile of the genes informative in these lines, rather than an inherent feature of that line.
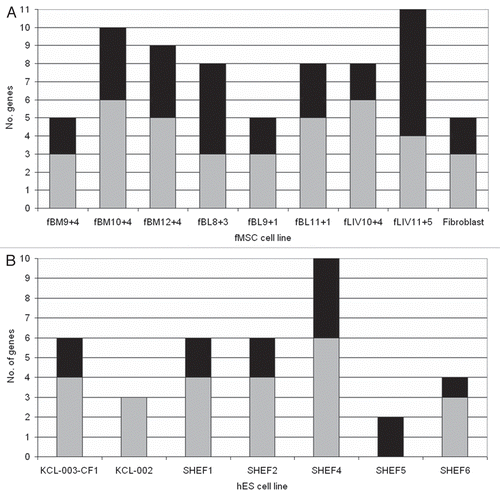
Figure 2 Cell staining following fMSC differentiation and post-differentiation allelic expression analysis. (A) Light microscopy of live fMSC in culture following incubation with either osteogenic cell culture medium or adipogenic cell culture medium. Images were taken at the end of the four week differentiation. All lines showed evidence of calcium deposition during osteogenic culture and of lipid deposition during adipogenic culture. Von Kossa staining (silver nitrate) was used to stain calcium deposits, seen above as dark brown or black staining and Oil Red O to stain lipid droplets red. Sequence chromatograms for RT-PCR of (B) KCNQ1OT1, (C) PEG10 and (D) SLC22A18 are shown for each cell line, corresponding with the differentiated images in (A). KCNQ1OT1 and PEG10 were monoallelic before differentiation, and following differentiation to bone and fat in each cell line. SLC22A18 was biallelically expressed in undifferentiated fMSC and following differentiation to bone and fat. To view color, see the online publication.
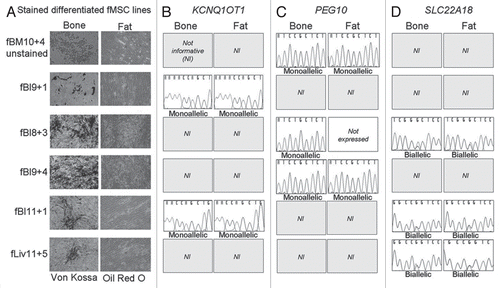
Figure 3 COBRA analysis of DNA methylation in fMSC and hES cells. COBRA analysis of the DMRs associated with the imprinted genes investigated in this study, listed in genome order from Hsa 7 (MEST DMR; A) to Hsa 20 (NESP Promoter; J). Digested bisulfite PCRs were visualized on ethidium bromide stained agarose and the intensity of the stain quantitated using ImageMaster 1D Prime software V.2.01 (bisulfite sequences shown in Fig. S4). Black, methylated; gray, unmethylated. The y-axis represents the ratio of the methylated and unmethylated bands. A 1:1 ratio of methylated to unmethylated DNA is represented by equal lengths of black and gray bars, so the boundary between the bars will lie at 1 on the y-axis. Hypermethylation is represented by the bar boundary being between 1 and 2, and hypomethylation by the bar boundary being between 0 and 1. For the MEST DMR (A), PEG10 DMR (B) H19 Promoter (C) and DMD (D), KvDMR (E) and the PWS-IC (G), ratios of methylated to unmethylated alleles were approximately 1:1 for each sample. For the DLK1 IG-DMR (F), PEG3 DMR (H) and GNAS region DMRs (I and J) there was variation from the expected 1:1 ratio of methylation, with both hyper and hypomethylation observed, but only in hES cell lines, fMSC lines were normally methylated.
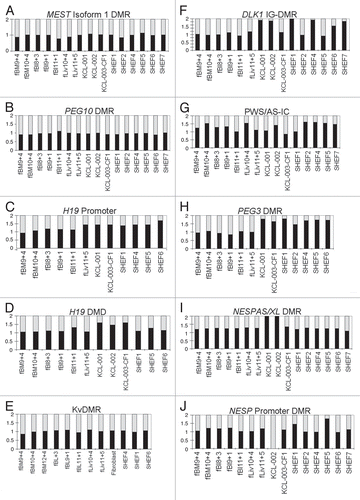
Table 1 Imprinted genes that were monoallelically expressed in all cell lines
Table 2 Imprinted genes that were biallelically expressed in all cell lines
Table 3 Genes whose allelic expression varied according to cell line
Table 4 Comparison of allelic expression of variable genes
Additional material
Download Zip (6.5 MB)Acknowledgements
This work was supported by the Medical Research Council (Ph.D. Studentship 10-10-2005 to J.M.F., G0801438 to G.E.M.), the March of Dimes 6-FY06-1266 to D.M., Wellbeing of Women (PG642/03), the Wellcome Trust (084358/B/07/Z) and Sport Aiding Medical Research for Kids (07/ICH/05) to G.E.M. H.D.M. would like to acknowledge the MRC and Sheffield Hospitals Charitable Trust.
The authors would like to thank Zhenling Luo at King's College London and Katherine Amps from Sheffield University for their careful culture and harvesting of the human embryonic stem cells. Thanks also to Rebecca Oakey, Ruth McCole and William Puszyk for critical reading of the manuscript.
References
- Edwards CA, Ferguson-Smith AC. Mechanisms regulating imprinted genes in clusters. Curr Opin Cell Biol 2007; 19:281 - 289
- Arnaud P. Genomic imprinting in germ cells: imprints are under control. Reproduction 2010; 140:411 - 423
- Thorvaldsen JL, Duran KL, Bartolomei MS. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev 1998; 12:3693 - 3702
- Yang T, Adamson TE, Resnick JL, Leff S, Wevrick R, Francke U, et al. A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat Genet 1998; 19:25 - 31
- Buiting K, Lich C, Cottrell S, Barnicoat A, Horsthemke B. A 5 kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum Genet 1999; 105:665 - 666
- Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet 2002; 32:426 - 431
- Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, et al. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet 2003; 35:97 - 102
- Arima T, Yamasaki K, John RM, Kato K, Sakumi K, Nakabeppu Y, et al. The human HYMAI/PLAGL1 differentially methylated region acts as an imprint control region in mice. Genomics 2006; 88:650 - 658
- Williamson CM, Turner MD, Ball ST, Nottingham WT, Glenister P, Fray M, et al. Identification of an imprinting control region affecting the expression of all transcripts in the Gnas cluster. Nat Genet 2006; 38:350 - 355
- Morison IM, Ramsay JP, Spencer HG. A census of mammalian imprinting. Trends Genet 2005; 21:457 - 465
- Hernandez L, Kozlov S, Piras G, Stewart CL. Paternal and maternal genomes confer opposite effects on proliferation, cell cycle length, senescence and tumor formation. Proc Natl Acad Sci USA 2003; 100:13344 - 13349
- Sun BW, Yang AC, Feng Y, Sun YJ, Zhu Y, Zhang Y, et al. Temporal and parental-specific expression of imprinted genes in a newly derived Chinese human embryonic stem cell line and embryoid bodies. Hum Mol Genet 2006; 15:65 - 75
- Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, Andrews PW, Beighton G, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol 2007; 25:803 - 816
- Kim KP, Thurston A, Mummery C, Ward-van OD, Priddle H, Allegrucci C. Gene-specific vulnerability to imprinting variability in human embryonic stem cell lines. Genome Res 2007; 17:1731 - 1742
- Rugg-Gunn PJ, Ferguson-Smith AC, Pedersen RA. Epigenetic status of human embryonic stem cells. Nat Genet 2005; 37:585 - 587
- Fulka H, Mrazek M, Tepla O, Fulka J Jr. DNA methylation pattern in human zygotes and developing embryos. Reproduction 2004; 128:703 - 708
- Shi W, Haaf T. Aberrant methylation patterns at the two-cell stage as an indicator of early developmental failure. Mol Reprod Dev 2002; 63:329 - 334
- Sato A, Otsu E, Negishi H, Utsunomiya T, Arima T. Aberrant DNA methylation of imprinted loci in super-ovulated oocytes. Hum Reprod 2007; 22:26 - 35
- Abeyta MJ, Clark AT, Rodriguez RT, Bodnar MS, Pera RA, Firpo MT. Unique gene expression signatures of independently-derived human embryonic stem cell lines. Hum Mol Genet 2004; 13:601 - 608
- Campagnoli C, Roberts IA, Kumar S, Bennett PR, Bellantuono I, Fisk NM. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver and bone marrow. Blood 2001; 98:2396 - 2402
- Guillot PV, O'Donoghue K, Kurata H, Fisk NM. Fetal stem cells: betwixt and between. Semin Reprod Med 2006; 24:340 - 347
- Guillot PV, Gotherstrom C, Chan J, Kurata H, Fisk NM. Human first-trimester fetal MSC express pluripotency markers and grow faster and have longer telomeres than adult MSC. Stem Cells 2007; 25:646 - 654
- Chan J, O'Donoghue K, Gavina M, Torrente Y, Kennea N, Mehmet H, et al. Galectin-1 induces skeletal muscle differentiation in human fetal mesenchymal stem cells and increases muscle regeneration. Stem Cells 2006; 24:1879 - 1891
- Zhang ZY, Teoh SH, Chong MS, Schantz JT, Fisk NM, Choolani MA, et al. Superior osteogenic capacity for bone tissue engineering of fetal compared to perinatal and adult mesenchymal stem cells. Stem Cells 2008; 27:126 - 137
- Kennea NL, Waddington SN, Chan J, O'Donoghue K, Yeung D, Taylor DL, et al. Differentiation of human fetal mesenchymal stem cells into cells with an oligodendrocyte phenotype. Cell Cycle 2009; 8:1069 - 1079
- Abdallah BM, Jensen CH, Gutierrez G, Leslie RG, Jensen TG, Kassem M. Regulation of human skeletal stem cells differentiation by Dlk1/Pref-1. J Bone Miner Res 2004; 19:841 - 852
- Hishida T, Naito K, Osada S, Nishizuka M, Imagawa M. peg10, an imprinted gene, plays a crucial role in adipocyte differentiation. FEBS Lett 2007; 581:4272 - 4278
- Monk D, Arnaud P, Apostolidou S, Hills FA, Kelsey G, Stanier P, et al. Limited evolutionary conservation of imprinting in the human placenta. Proc Natl Acad Sci USA 2006; 103:6623 - 6628
- Hayward BE, Barlier A, Korbonits M, Grossman AB, Jacquet P, Enjalbert A, et al. Imprinting of the G(s) alpha gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest 2001; 107:31 - 36
- Mantovani G, Ballare E, Giammona E, Beck-Peccoz P, Spada A. The gsalpha gene: predominant maternal origin of transcription in human thyroid gland and gonads. J Clin Endocrinol Metab 2002; 87:4736 - 4740
- Zheng H, Radeva G, McCann JA, Hendy GN, Goodyer CG. Galphas transcripts are biallelically expressed in the human kidney cortex: implications for pseudohypoparathyroidism type 1b. J Clin Endocrinol Metab 2001; 86:4627 - 4629
- Dominici M, Le BK, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006; 8:315 - 317
- Sakatani T, Wei M, Katoh M, Okita C, Wada D, Mitsuya K, et al. Epigenetic heterogeneity at imprinted loci in normal populations. Biochem Biophys Res Commun 2001; 283:1124 - 1130
- Market-Velker BA, Zhang L, Magri LS, Bonvissuto AC, Mann MR. Dual effects of superovulation: loss of maternal and paternal imprinted methylation in a dose-dependent manner. Hum Mol Genet 2010; 19:36 - 51
- Allegrucci C, Wu YZ, Thurston A, Denning CN, Priddle H, Mummery CL, et al. Restriction landmark genome scanning identifies culture-induced DNA methylation instability in the human embryonic stem cell epigenome. Hum Mol Genet 2007; 16:1253 - 1268
- Le Blanc K, Gotherstrom C, Ringden O, Hassan M, McMahon R, Horwitz E, et al. Fetal mesenchymal stem-cell engraftment in bone after in utero transplantation in a patient with severe osteogenesis imperfecta. Transplantation 2005;; 79:1607 - 1614
- Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science 2003; 299:1753 - 1755
- Holm TM, Jackson-Grusby L, Brambrink T, Yamada Y, Rideout WM 3rd, Jaenisch R. Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell 2005; 8:275 - 285
- Guillot PV, De BC, Dell'Accio F, Kurata H, Polak J, Fisk NM. Comparative osteogenic transcription profiling of various fetal and adult mesenchymal stem cell sources. Differentiation 2008; 76:946 - 957
- Sheehan D, Hrapchak B. Theory and Practise of Histotechnology 2. Battelle Press 1980; 226 - 227
- Collins KA, Geisinger KR, Wagner PH, Blackburn KS, Washburn LK, Block SM. The cytologic evaluation of lipid-laden alveolar macrophages as an indicator of aspiration pneumonia in young children. Arch Pathol Lab Med 1995; 119:229 - 231
- Apostolidou S, bu-Amero S, O'Donoghue K, Frost J, Olafsdottir O, Chavele KM, et al. Elevated placental expression of the imprinted PHLDA2 gene is associated with low birth weight. J Mol Med 2007; 85:379 - 387