Abstract
DNA methylation measured in white blood cell DNA is increasingly being used as in studies of cancer susceptibility. However, little is known about the correlation between different assays to measure global methylation and whether the source of DNA matters when examining methylation profiles in different blood cell types. Using information from 620 women, 217 and 403 women with DNA available from granulocytes (Gran), and total white blood cells (WBC), respectively, and 48 women with DNA available from four different sources (WBC, Gran, mononuclear (MN), and lymphoblastoid cell lines (LCL)), we compared DNA methylation for three repetitive elements (LINE1, Sat2, Alu) by MethyLight, luminometric methylation assay (LUMA), and [3H]-methyl acceptance assay. For four of the five assays, DNA methylation levels measured in Gran were not correlated with methylation in LBC, MN, or WBC; the exception was Sat2. DNA methylation in LCL was correlated with methylation in MN and WBC for the [3H]-methyl acceptance, LINE1, and Alu assays. Methylation in MN was correlated with methylation in WBC for the [3H]-methyl acceptance and LUMA assays. When we compared the five assays to each other by source of DNA, we observed statistically significant positive correlations ranging from 0.3-0.7 for each cell type with one exception (Sat2 and Alu in MN). . Among the 620 women stratified by DNA source, correlations among assays were highest for the three repetitive elements (range 0.39-0.64). Results from the LUMA assay were modestly correlated with LINE1 (0.18-0.20). These results suggest that both assay and source of DNA are critical components in the interpretation of global DNA methylation patterns from WBC.
Introduction
DNA methylation, an epigenetic modification, is a mark on genomic DNA created by the addition of a methyl group to the 5-carbon position of cytosine, predominantly in a 5′-CpG-3′ sequence context.Citation1 Changes in DNA methylation patterns which include global demethylation and localized hypermethylation at promoter CpG island regions of genes are some of the most common molecular alterations in tumors.Citation2,Citation3 Global demethylation has been seen in human cancer tissues, including breast,Citation4 gastric,Citation5 colonCitation6 and prostate.Citation7 Lower levels of genomic methylation in WBC DNA were also observed in head and neckCitation8 and bladderCitation9 cancer cases relative to controls. In animal models, aberrant global methylation leads to increased mutation ratesCitation10 and chromosomal instability.Citation11 Chromosomal instability is also correlated with global DNA demethylation in human colorectal cancer tissues.Citation12
Repetitive elements, which consist of interspersed repeats and tandem repeats, comprise about 45% of the human genome and contain much of the CpG methylation found in normal human postnatal somatic tissues.Citation13,Citation14 Short interspersed nucleotide elements (SINEs) and long interspersed nucleotide elements (LINEs) together account for at least 34% of the human genome.Citation15,Citation16 The Alu repeat, the most plentiful SINEs in human DNA, comprises 10% of the human genome.Citation16 LINE1, an endogenous mobile genetic element accumulated in the human genome, constitutes at least 5% of the total human genome.Citation17 Satellite 2 (Sat2) DNA sequences, located as tandem repeats in the pericentromeric and juxtacentromeric heterochromatin of several chromosomes, are composed of variants of two tandem repeats of ATT CCA TTC G followed by one or two copies of ATG.Citation18 Loss of DNA methylation in these sequences is thought to largely account for most of the global demethylation that characterizes a large percentage of human cancers.Citation19 Methylation levels of Alu, Sat2 and LINE1 repeats measured using MethyLight, a TaqMan-based real-time PCR assay,Citation20 were significantly associated with global DNA methylation, as measured by HPLC quantitation of 5-methyl cytosine (m5C) and the combined measurements of Alu and Sat2 methylation were also correlated with global DNA methylation measurements.Citation21
In contract to the MethyLight assay, which measures methylation at particular repetitive elements as a surrogate marker for estimating global methylation levels, global DNA methylation can also be measured directly. Two commonly used methods for determining genomic methylation levels are the [3H]-methyl acceptance (referred to throughout as methyl acceptance)Citation22 and the luminometric methylation assays (LUMA).Citation23 The methyl acceptance assay is based upon the ability of the bacterial CpG methyltransferase SssI to transfer radiolabeled methyl groups from S-[3H]-methyl adenosylmethionine specifically to unmethylated CpG sites in DNA. LUMA is based on digestion of genomic DNA with methylation sensitive and insensitive restriction enzymes, followed by a quantification of the resulting number of cut sites using pyrosequencing.
Measures of genomic methylation in blood cell DNA have been used as a phenotypic marker of genomic instability and potential cancer risk.Citation8,Citation9 However, there is no information about global methylation profiles of different blood cell types. In addition, although there are various methods for measuring global DNA methylation including HPLC, the methyl acceptance assay, LUMA and the MethyLight assay, few studies have directly compared these methods. Using biospecimens from the New York site of the Breast Cancer Family Registry (BCFR), we measured methylation levels using the methyl acceptance assay, LUMA and MethyLight assays for LINE1, Sat2 and Alu. We evaluated the correlation of these assays by DNA source within the same individuals (n = 48) and across individuals with the same source of DNA (n = 620) using peripheral blood DNA and a set of lymphoblastoid cell line DNA.
Results
Methylation levels in different blood cell types.
reports the overall distribution (mean, median and inter-quartile range) by each cell type and by assay for the 48 subjects with DNA from multiple sources. The level of [3H]-methyl acceptance of LCL DNA was statistically significantly higher than that of each individual cell type (p < 0.0001) (). In contrast, for the LUMA assay (), global DNA methylation measured in LCL was lower than the other cell types (p < 0.0001); methylation in Gran was the highest for LUMA (p < 0.0001). The average global DNA methylation and overall distributions of LINE1, Sat2 and Alu methylation were not significantly different among the four cell types of DNA (p = 0.76, p = 0.44 and p = 0.39, respectively).
reports the within-person correlations of global methylation measures, using the methyl acceptance assay and LUMA by source of DNA. Global methylation of LCL DNA was statistically significantly associated with that from MN and WBC, with Spearman correlation coefficients of 0.47 (p = 0.0008) and 0.49 (p = 0.0004), respectively. Global methylation of MN DNA was also correlated with that of WBC, with a correlation coefficient of 0.32 (p = 0.03). Similarly, using LUMA, we observed a statistically significant correlation between MN and WBC DNAs with a Spearman correlation coefficient of 0.44 (p = 0.008).
reports the within-individual Spearman correlation coefficients by source of DNA for the 48 subjects. LINE1 methylation level in LCL DNA was correlated with that in MN and WBC DNA, with Spearman correlation coefficients of 0.31 (p = 0.049) and 0.33 (p = 0.047), respectively. Alu methylation level in LCL was also correlated with that in WBC DNA (r= 0.41; p = 0.009). Sat2 methylation in Gran, not LCL, was statistically significantly correlated with that in MN and WBC DNA.
Correlation of DNA methylation assays in blood cells.
reports the within-person correlations between markers of global DNA methylation for the methyl acceptance, LUMA and MethyLight assays by source of DNA. The levels of DPM for the methyl acceptance assay did not correlate with the methylation levels from LUMA in Gran, MN and WBC DNA, but were inversely correlated with LCL DNA (r = −0.48, p = 0.0005). The levels of DPM for the methyl acceptance assay did not correlate with the percentage of methylation from the MethyLight assays for any of the loci. LINE1 methylation was correlated with Sat2 methylation in all four DNA sources: Spearman correlation coefficients of 0.45 (p = 0.002), 0.69 (p < 0.0001), 0.51 (p = 0.0004) and 0.62 (p < 0.0001) for Gran, LCL, MN and WBC, respectively. LINE1 methylation was also correlated with Alu methylation in Gran, LCL, MN and WBC DNA: Spearman correlation coefficients were 0.70 (p < 0.0001), 0.26 (p = 0.08), 0.31 (p = 0.04), 0.33 (p = 0.04), respectively. Sat2 and Alu global DNA methylation levels were also correlated: Spearman correlation coefficients of 0.36 (p = 0.02), 0.41 (p = 0.005) and 0.31 (p = 0.049) for Gran, LCL and WBC, respectively. Sat2 and Alu levels were not correlated in MN cells. We assessed the correlations after removing 15 subjects with LINE1 levels above 100%, but the overall inferences did not materially change after the exclusion (data not shown).
reports the Spearman rank correlations between markers of global DNA methylation separately by DNA source for the individuals with only one source of DNA available (Gran n = 217 or total WBC n = 403). Consistent with the within-individual results reported in , there was no correlation between DNA methylation measured by the methyl acceptance assay and the three repetitive elements (LINE1, Alu and Sat2). The methyl acceptance assay levels were positively correlated with the LUMA levels in Gran (r = 0.24, p = 0.003). The levels of LUMA were correlated with LINE1, with Spearman correlation coefficients of 0.20 (p = 0.008) and 0.18 (p = 0.002) in Gran and WBC, respectively. Sat2 methylation was correlated with LUMA in Gran (r = 0.18; p = 0.02) but not in WBC. Global DNA methylation levels measured by the three repetitive elements were positively correlated with each other in both Gran and WBC DNAs. We also estimated Spearman correlations for the comparisons in adjusting for age, breast cancer status and smoking status. The partial correlation coefficients were not materially different after adjustments and the overall inferences were the same (data not shown). We also performed supplemental analyses examining whether combined measures of methylation in Alu and Sat2 were more highly correlated with global DNA methylation than single measures. Combined measures of Alu and Sat2 were not correlated with global DNA methylation levels measured by the methyl acceptance and LUMA assays (data not shown). The Spearman correlation coefficients for the combined methylation of Alu and Sat2 and LINE1 were 0.66 and 0.63 in Gran and WBC, respectively.
Discussion
Within-individual variability in blood DNA methylation levels by cell type.
Overall, we observed the strongest within-person correlations for LCL, MN and WBC in four of the five assays to measure global DNA methylation (methyl acceptance, LUMA, LINE1, Alu). When we compared these four assays, global DNA methylation levels measured in Gran were uncorrelated with measurements in LCL, MN and WBC. The exception was Sat2 where global DNA levels in Gran were correlated with LCL, MN and WBC. With both methods to measure direct global DNA methylation levels (methyl acceptance and LUMA assays) methylation levels were statistically significantly correlated within the same individuals for MN and WBC. WBC are divided into two categories: granulocytes and mononuclear cells, including lymphocytes and monocytes. Our results suggest that MN global DNA methylation levels are a better representation of the levels in WBC than those found in Gran.
LCL are lymphoblastoid cell lines obtained through an Epstein-Barr viral (EBV) transformation of B-lymphocyte cells. There is concern about the utility of LCL as a resource for methylation studies as it may be likely that through the transformation process to extend the replication capacity of the cells, global DNA methylation levels may decrease.Citation28 A study comparing DNA methylation profiles between WBC and EBV-transformed LCL from the same individuals found that the majority of genes have similar methylation profiles in WBC and LCL.Citation28 However, a subset of genes (∼8%) had regions that were differentially methylated in the two sources. The largest differences in methylation profile were localized in areas of low CpG density.Citation28 This is consistent with our finding that, compared to other cell types, the lowest methylation levels measured by LUMA were seen in LCL, and the levels of LUMA measured in LCL were not correlated with any of the other cell types (Gran, MN and WBC). We did not observe lower methylation levels for the three repetitive elements which contain much of the CpG methylation in LCL.
Levels of methylation of LINE1 and Alu elements measured in LCL were correlated with those in WBC and MN. These results suggest that when interested in the level of LINE1 and Alu repeat methylation, LCL DNA might serve as a valid surrogate tissue for WBC. In contrast, the levels of Sat2 methylation in Gran were statistically significantly correlated with those in MN and WBC. Sat2 DNA sequences are small tandem repeats located in the pericentromeric and juxtacentromeric regions of chromosomes. Therefore, they represent a different set of CpG sites than the LINE1 and Alu elements, and this could explain the different results found for the different elements.
The fact that there are differences in the levels of global DNA methylation for the different cell types is not unexpected. Studies have shown that the main type of Gran, neutrophils, has a distinctive gene expression profile that has been found to be significantly different from that of monocytes.Citation29 Altered gene expression and DNA methylation status of specific sequences has also been found to be correlated with neutrophil count, suggesting different cell types have alternative ways to regulate expression of their genome.Citation30 While such evidence supports the presence of differential methylation patterns in different blood cell subsets, it cannot readily explain some of our results, such as why MN DNA methylation correlates so closely with WBC methylation. We might expect that Gran, which represents a larger proportion of cells in the blood (70%), would be more correlated with WBC DNA methylation profile. However, studies using neutrophils highlight the effect that infections and inflammation have on the expression profile of these cells.Citation29,Citation31 Therefore, it is possible that epigenetic mechanisms are involved in the response of neutrophils to infections or other cellular stress, and may help some explain the lack of correlation between Gran and WBC methylation within the same individuals.
Correlation across global DNA methylation assays.
We observed substantial heterogeneity in global DNA methylation levels across assay type. For all of the assays except the methyl acceptance assay, higher values indicate more global DNA methylation. For the methyl acceptance assay, SssI methylase in the presence of [3H]-SAM specifically catalyzes the transfer of methyl groups from SAM to cytosine residues in CpG sequences, so that the ability of DNA to incorporate [3H]-methyl groups in vitro is inversely related to endogenous DNA methylation.Citation25,Citation26 Quinlivan et al. compared DNA methylation levels measured by liquid chromatography-tandem mass spectrometry and the [3H]-methyl acceptance assay and concluded that the two assays had good agreement with each other.Citation32 Despite its simplicity, the methyl acceptance assay is prone to a number of technical problems. Daily and batch variation can be considerable, primarily due to the instability of both SAM and variations in SssI methyltransferase activity. To account for this variability, we used the same batch of fresh [3H] SAM and enzyme for each set and ran the DNA from the four different sources as sets, and used another batch of fresh [3H] SAM and enzyme to measure the second larger set of samples. However, we found different results in our two sets of samples, with the larger sample size showing less consistent results probably as a result of batch variability. This problem highlights the need for assays that are easy to perform and reliable in the study of DNA methylation in large population studies. In addition, the methyl acceptance assay is also highly sensitive to DNA amount and the difficulty in dissolving genomic DNA may lead to inaccurate DNA concentration measurements.Citation20,Citation33 We quantified DNA in each reaction using PicoGreen double-strand DNA quantification reagents.
LUMA is a global measure of methylation using distinctive enzymatic restriction cleavage with the isoschizomers Hpall and MspI. Cleavage of the CCG G sequence by both enzymes is dependent on the methylation of the internal cytosine. Therefore, LUMA reflects the overall contribution of a specific set of m5C including both CpG island and non-CpG island regions. In addition, LUMA requires good quality DNA, as even partially degraded samples will bias the measurements. Dependence on enzymatic activity variations is also a possible difficulty with the assay; however, we found our samples to be completely digested after 4 h. Validation experiments of the LUMA with m5C measurements have not been published to date. Although both the methyl acceptance assay and LUMA measure global DNA methylation levels across the entire genome, the lack of association between two assays might be because the methyl acceptance assay measures methylation in CpG sequences across the genome, while LUMA only measures methylation in the CCG G sequence, which accounts for only 8% of the genomic CpG sites.Citation34 Thus, studies are needed to establish it as an appropriate indicator of global methylation levels.
Methylation levels of LINE1, Alu and Sat2 by MethyLight were more strongly correlated with each other. Correlations were consistently found in the overall study population as well as the within-individual samples. Correlations between methylation of Alu and LINE1 elements have been reported previously.Citation35 However, percentage methylation of the repetitive elements measured by the MethyLight assay was not correlated with the methyl acceptance assay. The lack of correlation of the MethyLight and methyl acceptance data was not unexpected since these different techniques provide information on DNA methylation at different levels, spanning from genome-wide methylation content to methylation of a set of specific CpG sites in specific regions of the repetitive elements. However, LINE1 methylation did correlate with LUMA in the larger sample set, which is consistent with other data.Citation36
Limitations related to the MethyLight assay may also explain some of the variability in the overall correlations. MethyLight is a high-throughput assay to make bulk estimates of methylation in repetitive elementsCitation21 where bisulfite-treated DNA is used directly as a template in a standard PCR reaction. One major limitation of MethyLight is that bisulfite treatment is prone to reaction artifacts, which may affect its reliability. The extent of this reaction cannot be easily measured and incomplete conversion of cytosine to uracil is common because of incomplete denaturation of the DNA template or partial renaturation during bisulfite treatment.Citation20,Citation33 DNA degradation due to partial acid-catalyzed depurination and long incubations can result in extensive damage to template sequences, which subsequently may be difficult to amplify.Citation37 To account for this limitation, we bisulfite treated DNA from different cell types as sets and used the same incubation times for within-person comparison in the 48 sets. However, we did not track bisulfite batch and cannot determine if it influenced the correlations between persons in the larger study. Another limitation is that all the sites within the primer and probe binding regions must be methylated for detection. These limitations may partially explain why there are very modest to no correlations between the MethyLight and the other assays used in our study. It is unclear why the methylation levels of the three loci were different and why some of the percentages of methylation levels in LINE1 were above 100 percent. We have recently measured LINE1 methylation in several commercial samples of methylated DNA and found that they are around 85% methylated. It is well known that populations are polymorphic for the presence of LINE1 elements and the human genome contains more than 105 truncated LINE1 elements. Data on methylation of these three repetitive elements in WBC DNA by MethyLight assay are limited;s however, our data are consistent with the studies using DNA from tumor and normal tissue.Citation21 Despite its limitations, the MethyLight method has several important advantages, including its quantitative format and the high-throughput nature of the assay with minimal DNA requirements.
DNA methylation measurements in epidemiologic studies.
Although the repertoire of methylation screening methodologies has expanded widely and different approaches have been used to estimate genomic methylation, no single technique fulfills all criteria for generating unambiguous data on methylation.Citation20 In the present study, we found good correlations between methylation in three loci. This is consistent with the fact that DNA methylation typically occurs in CpG dinucleotides and up to 80% of CpG dinucleotides occur in repetitive sequences. It is known that more than 90% of all m5C lies within retrotransposon sequences, including satellite repeats, SINE and LINE-1.Citation38 We also found that LUMA was correlated with Sat2 methylation in Gran and LINE1 methylation in Gran and WBC, suggesting that measuring particular repetitive elements may serve as a surrogate marker for estimating global DNA methylation levels.
The measurement of biomarkers in blood specimens has become an integral component of many epidemiologic studies. There is widespread interest in exploring the associations between WBC genomic m5C content and clinical outcomes, as well as environmental, lifestyle and dietary exposures.Citation26,Citation27,Citation39,Citation40 Studies suggest that global DNA methylation levels decrease with time and thus a role for both endogenous and exogenous factors in changing global DNA methylation levels has been supported.Citation39 Although there are various methods for studying DNA methylation on a genome-wide scale,Citation41 most epidemiologic studies conducted in large populations have focused on the [3H]-methyl acceptance,Citation26,Citation40 LUMACitation42 and the MethyLightCitation21 assays.
Whole blood and blood fractions comprise a major portion of biospecimen collections for epidemiologic studies. In the present study, we found that the methylation level of Gran DNA was not correlated with that of LCL, MN and WBC while WBC and MN DNA methylation levels were correlated for assays that measure this content directly. These results suggest that cell type matters in methylation studies. Although the effect of method collection, time from blood draw to processing and freezing, and storage conditions may have affected the integrity of m5C in different sources of DNA, we used the same method to extract DNA from the different cell types. In addition, the same protocols of blood collection, processing, storage and DNA extraction were applied for all the specimens in this study.
Further, for the comparisons within individuals, these associations cannot be explained by other factors that are fixed within an individual such as cancer status, smoking, age or diet. For example, even though dietary patterns can influence methylation,Citation43,Citation44 for within-individual comparisons, diet, smoking status, cancer status and age cannot explain any of the correlations across cell type. For the larger study where we had only a single source of DNA, it is possible that cancer status, age, diet, and smoking affected DNA methylation levels. Our samples were not collected after fasting, but some studies suggest that short-term folate restriction is not sufficient to modify methylation content in leukocyte DNA.Citation45–Citation47 However, we were able to adjust for these individual-level factors in the larger study but this adjustment did not affect any of the inferences about the correlations across assay type.
In summary, our study highlights the importance of using the same source of blood DNA when measuring global methylation. We also found differences across assays in global DNA methylation levels suggesting that using different techniques to measure changes in m5C might affect interpretation of data across molecular epidemiologic studies. Because results from both the MethyLight and methyl acceptance assays have been correlated with endpoint phenotypes, multiple methods may be needed when studying the associations between environmental exposures and global DNA methylation profiles.
Materials and Methods
Study participants.
The study population was selected from families participating in the New York site of the Breast Cancer Family Registry (BCFR).Citation24 The New York site of the BCFR recruited high risk breast and/or ovarian cancer families from cancer clinics within the Metropolitan New York area. Families were eligible to participate if the family had at least one of the following: (1) a female relative with breast or ovarian cancer diagnosed before age 45 years, (2) a female relative with both breast and ovarian cancer regardless of age at diagnosis, (3) two or more relatives with breast or ovarian cancer diagnosed after age 45 years, (4) a male with breast cancer diagnosed at any age or (5) a family member with a known BRCA mutation. All consenting family members were asked to complete an epidemiologic questionnaire which collected information on demographics, ethnicity, smoking, alcohol consumption, reproductive history, hormone use, weight, height, physical activity and dietary intake. We also completed an extensive family history pedigree for each family. We collected blood from participants at the time of recruitment to permit the isolation of plasma and white blood cell fractions. Specifically, we collected a 30 ml blood sample; granulocyte and mononuclear cell fractions were collected by centrifugation over Ficoll. The study was approved by Columbia University's Institutional Review Board; written informed consent was obtained from all subjects and strict quality controls and safeguards were used to protect confidentiality.
At the beginning of the study we stored DNA from multiple sources. Specifically, we used samples from 48 subjects (20 breast cancer cases and 28 unaffected sisters) from whom four different sources white blood cells (WBC), granulocytes (Gran), mononuclear (MN) and lymphoblastoid cell lines (LCL) of DNA were available to investigate the within-person correlations of methylation markers. In addition, we also used samples from 620 women, 217 with Gran (91 cases and 126 unaffected sisters) and 403 with total WBC (175 cases and 228 unaffected sisters) who had a single source of DNA to investigate the correlation between the global DNA methylation markers.
DNA extraction and bisulfite treatment.
We extracted genomic DNA from all cells by the salting out procedure. Cells were lysed with SDS in a nuclei lysis buffer and treated with RNase A (final 133 µg/mL) and RNase T1 (final 20 units/mL) to remove RNA. Proteins were coprecipitated with NaCl (330 µL of saturated NaCl added per 1 mL solution) by centrifugation. Genomic DNA was recovered from the supernatant by precipitation with 100% ethanol, washed in 70% ethanol and dissolved in the Tris-EDTA buffer.
For the MethLight assay, aliquots of DNA (500 ng) were bisulfite-treated with the EZ DNA methylation kit (Zymo Research, Orange, CA) to convert unmethylated cytosines to uracils while leaving methylated cytosines unmodified. The DNA was resuspended in 20 µL of distilled water and stored at −20°C until used.
Methylight assay.
After sodium bisulfite conversion, MethyLight was carried out using the following forward and reverse primers and probes (LINE1-M1, Alu-M2 and Sat2-M1 referred to throughout the manuscript as LINE1, Alu and Sat2 respectively) as described.Citation21 PCR was performed in a 10 ul reaction volume with 0.3 uM forward and reverse PCR primers, 0.1 uL probe, 3.5 uM MgCl2, using the following PCR program: 95°C for 10 min, then 55 cycles of 95°C for 15 s followed by 60°C for 1 min. Standard curves for the Alu C4 repeat control reaction were generated from 1:25 serial dilutions of bisulfite-converted, CpGenome universal methylated and unmethylated DNAs. A pooled sample of DNA from five controls was used as a quality control and analyzed with each batch of test samples. Assays were run on an ABI Prism 7900 Sequence Detection System (Perkin-Elmer, Foster City, CA). Intra- and interassay coefficients of variation (CVs) were 1.2% and 1.9%, respectively.
Methylation calculations.
Universal methylated DNA served as a methylated reference and the Alu-based control reaction (Alu-C4) was used as a control reaction to measure the levels of input DNA to normalize the signal for each methylation reaction. The MethyLight data specific for methylated repetitive elements were expressed as percent of methylated reference (PMR) values.
PMR = 100% * 2 exp − [Delta Ct (target gene in sample − control gene in sample) − Delta Ct (100% methylated target in reference sample − control gene in reference sample)].
Each MethyLight reaction was performed in duplicate and the PMR values represent the mean.
[3H]-methyl acceptance assay.
The [3H]-methyl acceptance assay was carried out as described by Balaghi and WagnerCitation25 and Pilsner et al.Citation26 The DNA was incubated with [3H]S-adenosylmethionine in the presence of the SssI prokaryotic methylase enzyme, which indiscriminately methylates all unmethylated CpG sequences. Therefore, the ability of DNA to incorporate [3H] methyl groups in vitro is inversely related to endogenous DNA methylation. Briefly, 200 ng of DNA was incubated with 3 U of SssI methylase (New England Biolabs); 3.8 µmol/L (1.1 µCi) [3H]-labeled S-adenosylmethionine (Perklin-Elmer); and EDTA, DTT and Tris-HCL (pH 8.2) in a 30 µL mixture and incubated for 1 h at 37°C. The reaction was terminated on ice and 15 µL of the reaction mixture applied onto Whatman DE81 filter paper. The filter was washed on a vacuum filtration apparatus three times with 5 mL of 0.5 mol/L sodium phosphate buffer (pH 8.0), follow by 2 mL each of 70% and 100% ethanol. Dried filters were each placed in a vial with 5 mL of scintillation fluid (Scintisafe, Fisher) and analyzed by a Packard scintillation counter. Each DNA sample was processed in duplicate, and each processing run included samples for background (reaction mixture with all components except SssI enzyme) and universal methylated and unmethylated DNA as positive and negative controls, respectively. For the methyl acceptance assay, the quality of the DNA sample and the reproducibility with which DNA concentration can be measured are major factors in determining inter- and intra-assay variability. A pooled sample of DNA from five controls was used as a quality control and analyzed with each batch of test samples. Intra- and inter-assay CV were 2.5% and 5.9%, respectively. To quantify the amount of double-strand DNA in each reaction, an aliquot of DNA was used to determine DNA concentration using PicoGreen double-strand DNA quantification reagent (Molecular Probes). All disintegrations per minute (DPM) values were expressed per µg DNA as quantified by PicoGreen. The laboratory investigator who performed the assays was blinded to epidemiologic data.
Luminometric methylation assay (LUMA).
The assay was carried out as described previously.Citation27 Genomic DNA (200 ng) was cleaved with HpaII + EcoRI or MspI + EcoRI in two separate 10 µl reactions containing 33 mM Tris-acetate, 10 mM Mg-acetate, 66 mM K-acetate pH 7.9, 0.1 mg/ml BSA and five units of each restriction enzyme. The reactions were set up in a 96-well format and incubated at 37°C for 4 h. Then 10 µl annealing buffer (20 mM Tris-acetate, 2 mM Mg-acetate pH 7.6) was added to the cleavage reactions, and samples were placed in a PyroMark Q24 system with the following dispensation order: GTG TCA CAG TGT. For calculations, the peak heights of dispensations 9 through 12 were used, with a cut off for a peak height of 3. Dispensations 1 through 4 were used to assess the quality of the digested DNA. A cut off value of 3 was used for the peak heights corresponding to these dispensations, and values higher than 3 were indicative of a sample with low quality DNA that could not be used in this assay. Percentage of DNA methylation was expressed as [1 − (HpaII + EcoRI ∑G/∑T)/(MspI + EcoRI ∑G/∑T)]*100. The inter-assay CV was 1.5%.
Statistical methods.
We used Spearman Rank correlation coefficients to estimate the within-individual correlations by source of DNA and by assay (–). For each of the four cell types (Gran, LCL, MN and WBC), we estimated pairwise Spearman Rank Correlation Coefficients and 95% corresponding confidence limits (95% CI). These analyses were done separately for each of the five global DNA methylation assays. We then compared each of the five assays to each other using Spearman Rank correlation coefficients and separately stratifying by cell type. For these analyses, all individual-specific information (e.g., cancer type, smoking history, age) was the same for each individual so we did not further adjust for these factors. For the larger study where we only had a single source of DNA we compared the five assays to each other separately by Gran and WBC. For these analyses, we examined both overall Spearman Rank correlation coefficients as well as partial Spearman correlation coefficients after adjusting for age, cancer status and smoking history. As a supplemental analysis we also explored whether combined measures of Alu and Sat2 were correlated with global DNA methylation levels measured by the methyl acceptance and LUMA and LINE1 assays. All analyses were performed with SAS software 9.2 (SAS Institute, Cary, NC).
Abbreviations
| BCFR | = | Breast Cancer Family Registry |
| DPM | = | disintegration per minute |
| Gran | = | granulocytes |
| HPLC | = | high performance liquid chromatography |
| LCL | = | lymphoblastoid cell lines |
| LINE1 | = | long interspersed nucleotide element-1 |
| LUMA | = | luminometric methylation assay |
| m5C | = | 5-methylcytosine |
| MN | = | mononuclear |
| SAM | = | S-adenosylmethionine |
| Sat2 | = | satellite 2 |
| SINEs | = | short interspersed nucleotide elements |
| WBC | = | white blood cell |
Figures and Tables
Figure 1 Box plot of within-person DNA methylation levels using the methyl acceptance, MethyLight and LUMA assays by source of DNA.
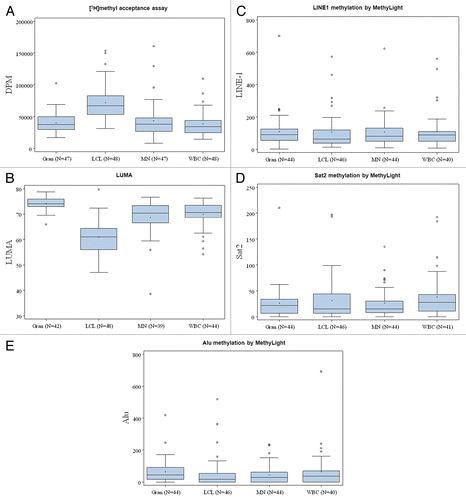
Table 1 Within-person Spearman correlation coefficients for global DNA methylation measured using the [3H]methyl acceptance assay and LUMA by source of DNA (n = 48)
Table 2 Within-person correlation of global methylation measured with the MethyLight assay by source of DNA and assay (N = 48)
Table 3 Within-person spearman correlation coefficients for markers of global methylation measured with the [3H]methyl acceptance, LUMA and MethyLight assays by source of DNA (N = 48)
Table 4 Spearman correlation coefficients for markers of global methylation measured with the [3H] methyl acceptance, LUMA and MethyLight assays by source of DNA
Acknowledgements
This work was supported by an award from the Breast Cancer Research Foundation and NIH grants U01 CA69398, P30 CA13696 and P30 ES009089. This work was also supported by the National Cancer Institute, National Institutes of Health under RFA # CA-06-503 and through cooperative agreements with members of the Breast Cancer Family Registry (CFR) and Principal Investigators. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the CFR, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government or the CFR.
References
- Wolffe AP, Matzke MA. Epigenetics: Regulation through repression. Science 1999; 286:46 - 48
- Laird PW, Jaenisch R. DNA methylation and cancer. Hum Mol Genet 1994; 3:1487 - 1495
- Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet 2001; 10:687 - 692
- Esteller M, Fraga MF, Guo M, Garcia-Foncillas J, Hedenfalk I, Godwin AK, et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet 2001; 10:3001 - 3007
- Suzuki K, Suzuki I, Leodolter A, Suzuki K, Suzuki I, Leodolter A, et al. Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell 2006; 9:199 - 207
- Goelz SE, Vogelstein B, Hamilton S, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 1985; 228:187 - 190
- Florl AR, Steinhoff C, Muller M, Seifert HH, Hader C, Engers R, et al. Coordinate hypermethylation at specific genes in prostate carcinoma precedes LINE-1 hypomethylation. Br J Cancer 2004; 91:985 - 994
- Ting HD, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, et al. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 2007; 16:108 - 114
- Moore LE, Pfeiffer RM, Poscablo C, Real FX, Kogevinas M, Silverman D, et al. Genomic DNA hypomethylation as a biomarker for bladder cancer susceptibility in the Spanish Bladder Cancer Study: a case-control study. Lancet Oncol 2008; 9:359 - 366
- Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature 1998; 395:89 - 93
- Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300:455
- Rodriguez J, Frigola J, Vendrell E, Risques RA, Fraga MF, Morales C, et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res 2006; 66:8462 - 9468
- Initial sequencing and analysis of the human genome. Nature 2001; 409:860 - 921
- Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet 2003; 19:68 - 72
- Deininger PL, Moran JV, Batzer MA, Kazazian HH. Mobile elements and mammalian genome evolution. Curr Opin Genet Dev 2003; 13:651 - 658
- Weiner AM. SINEs and LINEs: the art of biting the hand that feeds you. Curr Opin Cell Biol 2002; 14:343 - 350
- Kazazian HH Jr. Mobile elements: Drivers of genome evolution. Science 2004; 303:1626 - 1632
- Jeanpierre M. Human satellites 2 and 3. Ann Genet 1994; 37:163 - 171
- Melanie E. DNA methylation in cancer: too much, but also too little. Oncogene 2002; 21:5400 - 5413
- Christina D, Per G. DNA methylation analysis techniques. Biogerontology 2003; 4:233 - 250
- Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, et al. Analysis of repetitive element DNA methylation by MethyLight. Nucl Acids Res 2005; 33:6823 - 6836
- Kenneth PN, Curt B, David GS. Methyl group acceptance assay for the determination of global DNA methylation Levels. Methods Mol Biol 2008; 507:35 - 41
- Karimi M, Johansson S, Stach D, Martin C, Dan G, Martin S, et al. LUMA (LUminometric Methylation Assay)—A high throughput method to the analysis of genomic DNA methylation. Exper Cell Res 2006; 312:1989 - 1995
- John EM, Hopper JL, Beck JC, Knight JA, Neuhausen SL, Senie RT, et al. The Breast Cancer Family Registry: an infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res 2004; 6:375 - 389
- Balaghi M, Wagner C. DNA Methylation in Folate Deficiency: Use of CpG Methylase. Biochem Biophys Res Commun 1993; 30:1184 - 1190
- Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, et al. Genomic methylation of peripheral blood leukocyte DNA: influences of arsenic and folate in Bangladeshi adults. Am J Clin Nutr 2007; 86:1179 - 1186
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change in DNA methylation over time with familial clustering. JAMA 2008; 299:2877 - 2883
- Brennan EP, Ehrich M, Brazil DP, Crean JK, Murphy M, Sadlier DM, et al. Comparative analysis of DNA methylation profiles in peripheral blood leukocytes versus lymphoblastoid cell lines. Epigenetics 2009; 4:159 - 164
- Subrahmanyam YVBK, Yamaga S, Prashar Y, Lee HH, Hoe NP, Kluger Y, et al. RNA expression patterns change dramatically in human neutrophils exposed to bacteria. Blood 2001; 97:2457 - 2468
- Sharma P, Sahni NS, Tibshirani R, Skaane P, Uradal P, Berghagen H, et al. Early detection of breast cancer based on gene-expression patterns in peripheral blood cells. Breast Cancer Res 2005; 7:634 - 644
- Zhang X, Kluger Y, Nakayama Y, Poddar R, Whitnery C, DeTora A, et al. Gene expression in mature neutrophils: early responses to inflammatory stimuli. J Leukoc Biol 2004; 75:358 - 372
- Quinlivan EP, Gregory JF III. DNA methylation determination by liquid chromatography-tandem mass spectrometry using novel biosynthetic [U-15N]deoxycytidine and [U-15N]methyldeoxycytidine internal standards. Nucl Acids Res 2008; 36:119
- Oakeley EJ. DNA methylation analysis: a review of current methodologies. Pharmacol Ther 1999; 84:389 - 400
- Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA, Jeddeloh JA, et al. Comprehensive high-throughput arrays for relative methylation (CHARM). Cancer Res 2008; 18:780 - 790
- Cho NY, Kim BH, Choi M, Yoo EJ, Moon KC, Cho YM, et al. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol 2007; 211:269 - 277
- Romermann D, Hasemeier B, Metzig K, Gohring G, Schlegelberger B, Langer F, et al. Global increase in DNA methylation in patients with myelodysplastic syndrome. Leukemia 2008; 22:1954 - 1956
- Raizis AM, Schmitt F, Jost JP. A bisulfite method of 5-Methylcytosine mapping that minimizes template degradation. Anal Biochem 1995; 226:161 - 166
- Yang AS, Estecio MRH, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucl Acids Res 2004; 32:38
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 2005; 102:10604 - 10609
- Terry MB, Ferris JS, Pilsner R, et al. Genomic DNA methylation among women in a multiethnic New York City birth cohort. Cancer Epidemiol Biomarkers Prev 2008; 17:2306 - 2310
- Shen L, Waterland RA. Methods of DNA methylation analysis. Curr Opin Clin Nutr Metab Care 2007; 10:576 - 581
- Karimi M, Johansson S, Stach D, Martin C, Dan G, Martin S, et al. LUMA (LUminometric Methylation Assay)—A high throughput method to the analysis of genomic DNA methylation. Exper Cell Res 2006; 312:1989 - 1995
- Johanning GL, Heimburger DC, Piyathilake CJ. DNA methylation and diet in cancer. J Nutr 2002; 132:3814 - 3818
- Jacob RA, Gretz DM, Taylor PC, James J, Pogribny IP, Miller BJ, et al. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA Mmthylation in postmenopausal women. J Nutr 1998; 128:1204 - 1212
- Axume J, Smith SS, Pogribny IP, Moriarty DJ, Caudill MA. Global leukocyte DNA methylation is similar in African American and Caucasian women under conditions of controlled folate intake. Epigenetics 2007; 2:66 - 68
- Jacob RA, Pianalto DS, Henning SM, Zhang J, Swendseid M. In vivo methylation capacity is not impaired in healthy men during short-term dietary folate and methyl group restriction. J Nutr 2009; 125:1495 - 1502
- Axume J, Smith SS, Pogribny IP, Moriarty DJ, Caudill MA. The MTHFR 677TT genotype and folate intake interact to lower global leukocyte DNA methylation in young Mexican American women. Nutr Res 2007; 27:1365 - 1371