Abstract
Supplementation with folic acid during pregnancy is known to reduce the risk of neural tube defects and low birth weight. It is thought that folate and other one-carbon intermediates might secure these clinical effects via DNA methylation. We examined the effects of folate on the human methylome using quantitative interrogation of 27,578 CpG loci associated with 14,496 genes at single-nucleotide resolution across 12 fetal cord blood samples. Consistent with previous studies, the majority of CpG dinucleotides located within CpG islands exhibited hypo-methylation while those outside CpG islands showed mid-high methylation. However, for the first time in human samples, unbiased analysis of methylation across samples revealed a significant correlation of methylation patterns with plasma homocysteine, LINE-1 methylation and birth weight centile. Additionally, CpG methylation significantly correlated with either birth weight or LINE-1 methylation were predominantly located in CpG islands. These data indicate that levels of folate-associated intermediates in cord blood reflect their influence and consequences for the fetal epigenome and potentially on pregnancy outcome. In these cases, their influence might be exerted during late gestation or reflect those present during the peri-conceptual period.
Introduction
DNA methylation, one of several post-replication epigenetic changes imposed upon the genome, contributes to the regulation of gene expression and maintenance of genome integrity and stability.Citation1 This epigenetic change is directed toward cytosine bases in the context of CpG dinucleotides and is considered vital in developmental processes. Aberrant DNA methylation, apparent as either hyper or hypomethylation, is associated with changes in phenotype, aging, cancer and many other diseases.Citation2–Citation5
In the context of developmental changes, experimental data, primarily derived from direct intervention studies in rodents and more recently in sheep, are instructive and reinforce a growing body of evidence highlighting the importance of B-group vitamin availability (e.g., vitamin B12 and folic acid) in the methyl donor pathway, particularly within the peri-conceptual period.Citation6–Citation12 Collectively, these studies show that manipulation of methyl donor micronutrient availability (either through supplementation or depletion) during pregnancy can lead to alterations in methylation levels in offspring and that these changes influence gene expression as well as disease- and/or health-related phenotypes. In humans, epidemiological findings reflect in several ways those seen in interventional studies. For example, the importance of nutritional status on early embryonic development and subsequently adult epigenome is apparent in offspring born during the Dutch Famine of World War II.Citation13,Citation14 However, and with these noted exceptions, there is little data on the effect of diet and more particularly methyl group availability on the human fetal epigenome.Citation5
We recently examined the relationship between folic acid supplementation and its metabolites on human cord blood methylation.Citation15 As a surrogate for genome-wide methylation, we examined methylation of LINE-1 repetitive elements by quantitative Pyrosequencing following sodium bisulphite conversion.Citation15 LINE-1 methylation was negatively correlated with plasma homocysteine levels (an inverse indicator of blood folate status and methyl group supplyCitation16) and positively correlated with fetal birth weight centile.Citation15 These findings support a link between one-carbon supply, changes in the epigenome and fetal outcome and suggest an important role for homocysteine. Plasma homocysteine is inversely correlated with serum folate in both maternal and cord bloodCitation17 and increased levels have been associated with increased risk of neural tube defects and other pregnancy outcomes.Citation18 In the context of methylation, it is the key substrate in the methylation of methionine and subsequently S-adenosyl methionine—the methyl donor in DNA methylation reactions. Indeed, some studies suggest that homocysteine is a better functional indicator of one-carbon supply for DNA methylation reactions than folate, at least in part because of the effect of B vitamins on folate-derived one-carbon supply.Citation19
A caveat associated with analyses of LINE-1 elements as a surrogate for genome-wide methylation is that changes to these repetitive elements may differ from the rest of the genome. Indeed, Yang and colleagues, who first described the advantage of analyzing these repetitive elements recognized this potential drawback.Citation20 Paradoxically, in tumor cells global hypomethylation accompanied by gene-specific hypermethylation is a nearly universal finding that is, in this case, frequently associated with gene silencing.Citation2 Although these findings reflect those apparent in a disease state, they suggest that assessment of methylation status should describe both gene-specific and genome-wide changes.
In the current study, we examined the relationship between the folate metabolite homocysteine, birth weight centile and methylation patterns. However, and in contrast to our previous study, we now present findings where we examine gene-specific methylation. In this case, we use high-resolution genome-wide DNA profiling that permits quantitative interrogation of 27,578 CpG loci associated with 14,496 genes at single-nucleotide resolution across 12 samples in parallel.
Results
As expected, folic acid supplementation was associated with an increased maternal serum folate concentration; though this did not achieve statistical significance as maternal values were not available on all samples (Sup. Table S1). Fetal serum folate followed a similar, but less marked relationship. Maternal and fetal plasma homocysteine were similar in the supplemented and non-supplemented groups, though numbers are too small to draw major conclusions.
Initial visual comparison of median β values revealed no gross differences in overall CpG methylation between patient samples (Sup. Fig. S1). Although interpatient variation was low, when CpG islands and non-CpG island sites were analyzed independently, average methylation levels were significantly (p < 1 × 10−16) lower in sites within CpG islands (median β value 0.06) than non-CpG islands (median β value 0.68) (). This finding is consistent with previous reports.Citation21–Citation23 The CpG sites within CpG islands were largely unmethylated (80.3% of sites with β value <0.2 and only 5.4% with β value >0.8) while those outside CpG islands were generally moderately/highly methylated (13.9% of sites with β value <0.2 and 30.9% with β value >0.8).
Hierarchical clustering.
To identify potential underlying β value-derived methylation associations across the samples, unsupervised hierarchical clustering was employed. On the basis of our exclusion criteria (see Methods section), we assessed a total of 7,259 CpG sites where β values in all samples were ≥0.2 or ≤0.8. The 6,532 autosomal and 727 X chromosomal CpG sites were analyzed separately.
Hierarchical clustering analysis of the autosomal sites generated two discrete clusters, designated A and B (). Given our previous data showing the inverse association of LINE-1 global methylation with plasma homocysteine and direct correlation with birth weight centile, we examined these characteristics along with data on maternal characteristics, folic acid supplementation and serum folate concentrations, between the two clusters (). Compared with cluster A, birth weight centile was significantly higher (p = 0.019) and plasma homocysteine was significantly lower (p = 0.038) in cluster B. Furthermore, LINE-1 methylation levels were significantly increased (p = 0.028) in cluster B. Generally median β values for patients were similar between clusters (cluster A median of medians = 0.544, cluster B median of medians = 0.545; Sup. Fig. S2). While serum folate was lower in cluster A than B, this did not achieve statistical significance. Maternal characteristics and proportion of patients taking folic acid supplementation were not different, though numbers were too small to draw firm conclusions. Separation of CpG sites into those within CpG islands and those in non-CpG islands did not result in discrete groups using hierarchical clustering. Using the 727 X chromosome CpGs, hierarchical clustering did not identify well separated clusters (results not shown).
Association of bead array data with birth weight centile, plasma homocysteine and global (LINE-1) methylation levels.
Having identified significant associations between β values and birth weight centile, plasma homocysteine concentration and LINE-1 methylation levels between the two clusters, we then examined the data to identify which CpGs were significantly correlated with each of these parameters across the 12 samples. Analysis of the 6,532 autosomal CpGs using univariate linear regression analysis identified 304 that showed significant (p < 0.05) associations with birth weight centile, 298 with plasma homocysteine and 146 with mean LINE-1 methylation (). Sixteen sites showed significant correlations with all three parameters. When we then examined the 727 X chromosome sites, an additional 17 sites demonstrated significant correlations with birth weight centile, eight with plasma homocysteine and eight with LINE-1 methylation, one of which was associated with all three parameters (Sup. Table S2 for details of all significant CpG sites).
The resultant 16 autosomal and one X chromosome CpGs with significant correlations with all three parameters are listed in . Of these 17 CpGs, all 12 sites with inverse correlations between β values and birth weight centile and LINE-1 methylation demonstrated direct correlations with plasma homocysteine (), supporting our previously observed relationship between global methylation, birth weight and plasma homocysteine. 15 Conversely, the five sites with positive correlations of β value with birth weight centile and LINE-1 methylation demonstrated inverse correlations with plasma homocysteine concentration. Examples of correlations with birth weight centile, plasma homocysteine and LINE-1 methylation are illustrated in .
We then examined the characteristics of the CpG loci associated with birth weight centile, homocysteine and LINE-1 methylation. As shown in , of the 304 autosomal CpG loci that showed associations with birth weight centile and 146 with LINE-1 methylation, the majority were located within CpG islands. In contrast, for those 298 associated with homocysteine, the identified loci showed an approximately equal distribution between these two regions. Of the 6,532 autosomal sites studied, there proportion of sites within CpG islands was 51.3%.
Functional analysis of identified genes.
Finally, we examined links between sites associated with each of the clustered variables, namely; birth weight centile, plasma homocysteine and LINE-1 methylation and known functional categories. As a means to condense the complex functions of multiple genes we used the GO database of terms. For each of the three variables, we identified statistical over enrichment (EASE score a modified Fisher's exact test; see Methods section) of different categories (Sup. Table S3). While any such method to summarize complex information will necessarily over-simplify, the genes correlated with birth weight centile suggest a potential relationship to lipid metabolism while those correlated to plasma homocysteine were enriched for terms related to developmental processes. Those genes correlated with LINE-1 methylation were associated with a range of processes including terms associated with the development of white blood cells. This latter association was consistent with analysis that showed enrichment of genes constituting the “hematopoietic cell lineage” KEGG pathway. These are functional categories of potential interest in future studies.
Discussion
We have previously shown that genome-wide methylation, as determined by analysis of LINE-1 sequences as a surrogate marker, is associated with plasma homocysteine concentration and birth weight centile in human cord blood.Citation15 We now show, by high-resolution genome-wide profiling, that these associations are not confined to LINE-1 sequences, but are also apparent within CpG dinucleotides associated with consensus coding sequence (CCDS) genes.
Across the samples, the majority of the 21,261 CpG sites studied demonstrated similar degrees of methylation. These findings are consistent with recent data from other groups.Citation21–Citation23 The greatest similarity in our dataset and across the samples was apparent in sites with either low or high β values. Elimination of these invariable sites from our dataset reduced the number of probes to 7,259. The substantial reduction in the proportion of total CpG sites interrogated and showing change is similar to the proportions reported by othersCitation21–Citation23 and reinforces the consensus view that intra- and inter-individual methylation patterns are stable and largely invariant.Citation24,Citation25
Several other recent reports have employed similar genomewide techniques to survey normal human and/or tumor-derived material.Citation21–Citation23 A consistent finding in these studies is that methylation within CpG islands is significantly lower than that seen in non-CpG islands. Our own findings are consistent with these reports () and suggest that methylation profiles are similar in human cord blood to those in adult tissues.
Unsupervised hierarchical clustering of the filtered autosomal probe set (n = 6,532) generated two discrete clusters in the dataset. In common with our previous report of the variables associated with genome-wide LINE-1 methylation,Citation15 we show that CpG β values of CCDS gene clusters with birth weight centile, plasma homocysteine concentrations and LINE-1 methylation levels. Within cluster B, mean birth weight centile and LINE-1 methylation were significantly higher and plasma homocysteine significantly lower than that apparent in cluster A.
In our analyses, we also considered the context of the identified loci associated with each of the variables. For this analysis, we divided CpGs into those within CpG islands and those in non CpG islands. For associations with homocysteine, identified loci are equally distributed between these regions, in keeping with the proportion of CpG islands sites in the autosomal study set (51.3%). However, for the other clustered variables, birth weight centile and mean LINE-1 methylation, identified loci are predominately located in CpG islands. These findings are at variance with the data we had derived following our initial cluster analysis. In this case, clear clustering of birth weight was only observed in combined CGI and non-CGI data (see above) and is also at variance with other recent high-resolution data generated using similar approaches. In these cases, although the number of reports are limited, where change is reported, as example, across different tissue typesCitation21,Citation22 or as a predictor of tumor subclass,Citation23 the variation was more prevalent in non CpG islands than within CpG islands. Other technological approaches used to survey the methylomeCitation26 also suggest that increase in methylation of CpG dinucleotides is found predominantly in CpG island “shores” rather than the islands themselves. We are reluctant to draw too many conclusions at this stage from our own data, but it is of interest that genes associated with observed LINE-1 methylation and the important phenotype of weight centile show changes that reside mostly in gene regulatory (CpG islands) regions.
As a source of methyl groups, folic acid and other B vitamins are regarded as essential for methylation of DNA and proteins and also serve important roles in de novo purine and pyrimidine biosynthesis.Citation27 The intracellular form of folic acid, folate, is an important substrate in the methionine-folate cycle and homocysteine, as a key constituent of the cycleCitation27 is regarded an accurate inverse indicator of methyl group status.Citation16 While our data supported a relationship between folic acid supplementation and maternal serum folate, the link with plasma homocysteine in cord blood is less clear. This may be due to the balance between involvement of folate in purine/pyrimidine biosynthesis and methionine cycle, which is known to be influenced by a range of factors including vitamin B12 availability and genetic variation in key enzymes. Our study shows a significant difference in plasma homocysteine concentrations between the two clusters. In addition, the inverse relationship of this intermediary with serum folate, within each of the clusters, is also suggested (cord serum folate is higher in cluster B than A and vice versa for plasma homocysteine), though the difference in serum folate between clusters did not attain statistical significance. A similar significant inverse correlation between homocysteine and LINE-1 methylation was also apparent in our previous study.Citation15 The finding that neither study clearly identified relationship(s) with serum folate might indicate that homocysteine represents a more useful biomarker of folate utilisation in vivo. Larger studies will be necessary to test the hypothesis that the relationship with folate might reach statistical significance with increased power.
Interestingly, Wang and colleagues very recently examined genomic and LINE-1 methylation in fetal neural tissue in the aetiology of neural tube defects (NTD).Citation28 They showed lower LINE-1 methylation levels in this tissue compared to controls, and that this change was associated with significant decrease in maternal plasma vitamin B-12 relative to controls. Their study also showed a decrease in folate and increased concentrations of homocysteine relative to controls in these specimens. While not reaching statistical significance, this finding further reinforces the inverse relationship between these intermediaries and LINE-1 methylation.
Reports of maternal dietary manipulations in animal models during pregnancy, and in particular of folic acid and other B vitamins, provide compelling evidence that methyl donors affect offspring by epigenetic mechanisms.Citation6–Citation12,Citation28–Citation30 Reinforcing these animal studies are compelling data from quasi-experimental settings such as those described in the Dutch prenatal famine cohorts.Citation13,Citation14 These studies of peri-conceptual exposure to famine first identified a decrease in DNA methylation of the insulin like growth factor 2 (IGF2) differentially methylated region (DMR).Citation13 A subsequent study of multiple loci implicated in growth and metabolic disease shows that peri-conceptual exposure to famine leads to persistent changes in DNA methylation.Citation14 Their report also demonstrated that while some loci showed increased others showed diminished DNA methylation. Similar findings are also apparent in animal studies of methionine restriction.Citation10 Taken together, these observations suggest that changes may, at least in part, occur due to an adaptive response rather than a consequence of methyl donor availability alone. Our high resolution profiling of CpG loci also identified multiple loci that showed changes in methylation patterns and these were associated with birth weight centile, plasma homocysteine and LINE-1 methylation. Of the multiple loci that were associated with each of these variables a common set of 17 CpG sites showed significant correlations with all three variables, that is birth weight centile, plasma homocysteine concentration and LINE-1 methylation level.
Functional characterization revealed enrichment of particular genes/pathways associated with each of these variables. The enriched terms for lipid metabolism in the birth weight centile subset supports the established link between lipid metabolism, metabolic syndrome and pregnancy outcomes.Citation31,Citation32 In terms of the link between homocysteine and enrichment of terms for developmental process, impairments in folate-mediated one-carbon metabolism are associated with several common diseases and developmental anomalies including intestinal cancers, vascular disease, cognitive decline and neural tube defects, all of which are affected by disruption to normal cell cycle control.Citation33 While these pathways may offer potential opportunities for further research, validation would be essential before these could be investigated with confidence.
The associations with global methylation (using LINE-1 as surrogate), described in our previous study,Citation15 and of plasma homocysteine concentration and birth weight centile with gene-specific methylation apparent in this current investigation are of particular note. In this context, most reports suggest that the peri-conceptual period of particular importance in the establishment of methylation patterns.Citation5,Citation13,Citation14,Citation34 Our measurements of plasma homocysteine and of serum folate represent levels are at term, and while we could speculate that these might be similar to the peri-conceptual levels this has not be verified in our study. We are therefore reluctant to draw firm conclusions regarding the likely influence of these intermediaries in the periconceptual period on gene methylation. However, several recent reports suggest that, for particular genes, the late gestational period might also be important.Citation14,Citation35,Citation36 Clearly if will be useful for future studies to address this issue.
In conclusion, genome-wide interrogation of CpG dinucleotides identifies gene-specific CpG methylation patterns associated with plasma homocysteine concentration, LINE-1 methylation level and birth weight centile. For two of these variables, LINE-1 methylation and birth weight centiles, the majority of identified CpG dinucleotides were within CpG islands. While the small numbers and multiple testing increase the risk of false positive results, our initial data highlight the importance of changes within these regulatory regions that, while requiring further investigation, may uncover genes important in fetal development. These findings also suggest that homocysteine might be a useful biomarker of one carbon utilization and methyl group supply in DNA methylation processes.
Methods
Patients.
Cord blood samples were obtained from twelve women attending the Maternity Unit of the University Hospital of North Staffordshire between 2007–2008. These samples formed part of our original cohortCitation15 and were selected to give a range of LINE-1 methylation values. The study was approved by the Local Research Ethics Committee and all women provided written informed consent. Demographic data on maternal age at delivery, parity, body mass index, fetal gender, anti-epileptic drug usage and folate supplementation during pregnancy were collected prospectively by the study research midwife and these were verified against the mother's clinical record. Given the known anti-folate and teratogenic effects of carbamazepine,Citation37 women on this medication were excluded from the study. Blood samples were collected immediately after delivery from the umbilical cord into sample bottles containing ethylene diamine tetraacetic acid (for methylation analysis), heparin (for plasma homocysteine measurement) and without anti-coagulant (for serum folate measurement). The demographic and biochemical characteristics of the 12 subjects are shown in .
Measurement of blood folate parameters.
Serum folate was measured using a competitive immunoassay-based assay employing direct chemiluminescent technology with an acridinium ester label on Siemens Advia Centaur equipment and reagents (Siemens Medical Solutions Diagnostics) as per manufacturer's instructions. Plasma homocysteine was measured using High Performance Liquid Chromatography (HPLC) as described by Martin et al.Citation38 Mean LINE-1 methylation levels were calculated as described previously.Citation15
Illumina bead array analysis.
DNA was extracted from cord blood samples collected into potassium EDTA using standard phenol/chloroform procedures. DNA concentrations were quantified using the nanodrop 8000 spectrophotometer (Thermo Fisher Scientific Inc.) before being normalized to 25 ng/µl. 500 ng of total DNA was used in the EZ gold bisulphite conversion reaction (Zymoresearch, Orange, CA) as per the manufacturer's instructions but with elution in 12 µl elution buffer. 4 µl of bisulphite DNA was used as template in the Infinium Methylation 27K (Illumina, San Diego, CA) and was processed as per the manufacturer's instruction. This array examines the methylation status of 27,578 CpG sites across 14,496 genes. Data was collected using the Illumina Bead Array (Illumina) reader and analyzed with GenomeStudio V2009.1 methylation module 1.1.1 (Illumina). This assigns a “β value” which is a quantitative measure of methylation for each CpG site and ranges from 0 (no methylation) to 1.0 (100% methylation of both alleles).
Of the 27,578 sites, all sites where one or more of the 12 samples demonstrated either detection p values of >0.05 (internal quality control) or null (missing) β values, were excluded from the analysis (n = 6,221). We took the decision to remove those probes that failed in any one of the 12 samples rather than excluding probe values for individual patient samples as we deemed this a more robust way to identify probes that varied between patients in a limited number of samples. For individual samples the number of probes with a p value greater than 0.05 ranged from 1,153 (4.2% of probes) to 5,971 (21.7%). While this is a large proportion of the total number of probes, many (1,755) overlapped with invariant sites (see below). We also eliminated those probes with known single nucleotide polymorphisms, deletions or microsatellites within the detection probe as these were predicted to affect the β values obtained (SNP information obtained from dbSNPv126). This left a core set of 21,261 CpG sites. To reduce the number of non-variable sites from subsequent analyses, those where β values in all samples were ≥0.8 or ≤0.2, an approach similar to that described by Byun and associatesCitation21 were excluded. This resulted in a final data set of 7,259 CpGs, 6,532 representing 5,318 autosomal genes and 727 CpGs representing 473 genes on the X chromosome.
Bead Array β value data was validated in three cord blood samples using bisulphite sequencing as previously describedCitation15 of eight selected candidates demonstrating a range of mean β values. As expected, the correlation between β values and proportion of methylated clones by bisulphite sequencing was highly significant (Sup. Fig. S3; r = 0.79, p < 0.001).
Statistical analysis.
Unsupervised hierarchical clustering analysis was performed using the Cluster 3.0 program (version 1.47) by applying complete linkage clustering with a Euclidean distance metric, visualized by Java Treeview version 1.1.4r3. Differences in plasma homocysteine, birth weight centile and mean LINE-1 methylation between the two clusters were assessed by the Mann-Whitney U test using the STATA statistical software package (College Station, TX; version 8). Associations between β values and plasma homocysteine, birth weight centile and mean LINE-1 methylation were examined using univariate linear regression analysis. Birth weight was expressed as a birth weight centile calculated on a customized growth chart.Citation39 Potential enrichment of identified genes of interest for particular gene ontogeny (GO) terms, biochemical pathways (Kyoto Encyclopedia of Genes and Genomes; KEGG pathways), disease association (Online Mendelian Inheritance in Man; OMIM disease identifiers) and tissue expression (GNF U133A quartile) analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID)Citation40 software package (http://david.abcc.ncifcrf.gov/, version 6.0) with a EASE score cut-off of 0.05.
Figures and Tables
Figure 1 Density function plots showing the distribution of methylation as reported by infinium array β values. Distribution of β values for the 21,261 CpG sites, separated by (A) those in CpG islands (n = 16,866) and (B) those outside these islands (n = 4,395) for each of the 12 samples.
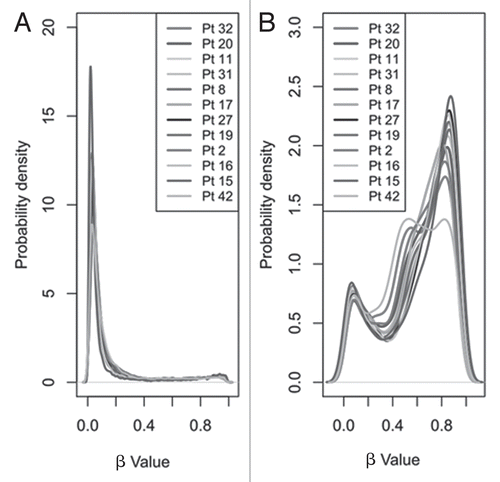
Figure 2 Hierarchical clustering using BeadArray data across the 12 samples. (A) β values for all 6,532 autosomal CpG sites where values in all samples were ≥0.8 and ≤0.2. (B) Hierarchical clustering results showing two discrete clusters. Sample numbers are shown with birth weight centile in parentheses. (C) Examples of genes showing significant correlations with birth weight centile across the 12 samples.
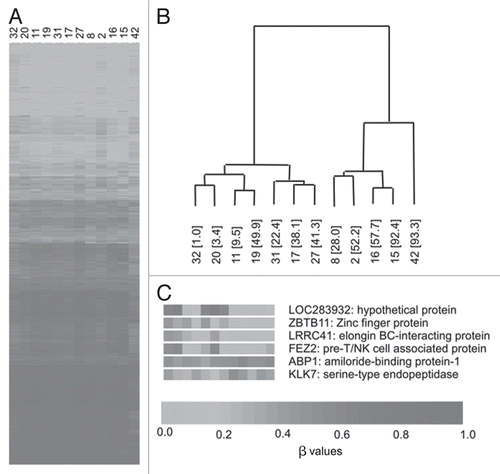
Figure 3 Venn diagram showing number of autosomal CpG sites significantly associated with each of the three parameters across the 12 samples; birth weight centile, plasma homocysteine concentration and mean LINE-1 methylation.
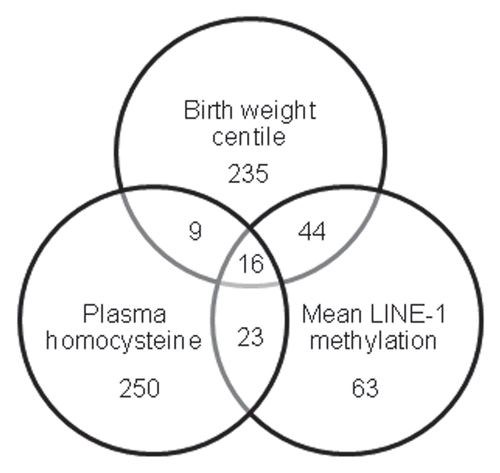
Figure 4 Examples of two genes BMX (left column) and AMN (right column) with significant correlations with each of (A) mean LINE-1 methylation (%), (B) plasma homocysteine (µmol/L) and (C) birth weight centile (%) (see ). BMX was positively correlated with LINE-1 methylation level (r = 0.631, p = 0.028) and birth weight centile (r = 0.760, p = 0.004) but inversely correlated with plasma homocysteine concentration (r = −0.813, p = 0.002) across the 12 samples. In contrast, AMN inversely correlated with LINE-1 methylation level (r = −0.630, p = 0.028) and birth weight centile (r = −0.637, p = 0.026) but directly correlated with plasma homocysteine concentration (r = 0.709, p = 0.015).
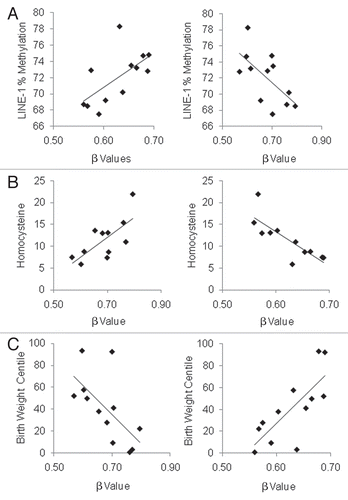
Figure 5 Characteristics of the autosomal CpG sites associated with (A) birth weight centile, (B) plasma homocysteine and (C) mean LINE-1 methylation. Pie charts show proportion of associated CpG sites located within (CGI) and outside CpG islands (non-CGI).
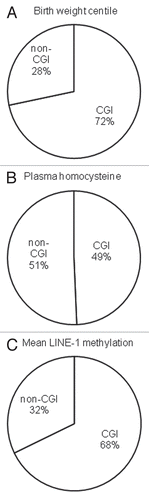
Table 1 Characteristics of the hierarchical clustering-generated groups
Table 2 CpG sites demonstrating significant correlations with plasma homocysteine concentration, birth weight centile and mean LINE-1 methylation level
Table 3 Demographic and biochemical characteristics of the sample cohort
Additional material
Download Zip (362.1 KB)Acknowlegements
We are grateful to Miss Angela Rooney for her invaluable help in recruiting patients for this study and to Dr. Elena Karpova for preparation of the samples for BeadArray analysis. We gratefully acknowledge the financial support of the World Cancer Research Fund (to W.E.F., A.A.F., R.D.E., W.D.C. and K.M.K.I.).
References
- Suzuki M, Bird A. DNA methylation landscapes: provocative insights from the epigenome. Nature Rev 2008; 9:465 - 475
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415 - 428
- Issa JP. CpG-island methylation in ageing and cancer. Curr Top Microbiol Immunol 2000; 249:101 - 118
- Richardson B. Impact of ageing on DNA methylation. Ageing Res Rev 2003; 2:245 - 261
- Nafee TM, Farrell WE, Carroll WD, Fryer AA, Ismail KM. Epigenetic control of fetal gene expression. Br J Obs Gynaecol 2008; 115:158 - 168
- Waterland RA, Dolinoy DC, Lin JR, Smith CA, Shi X, Tahiliani KG. Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis 2006; 44:401 - 406
- Waterland RA, Jirtle RI. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 2003; 23:5293 - 5300
- Adcock IM, Tsaprouni L, Bhavsar P, Ito K. Epigenetic regulation of airways inflammation. Curr Opin Immunol 2007; 19:694 - 700
- Hollingsworth JW, Maruoka S, Boon K, Garantziotis S, Li Z, Tomfohr J, et al. In utero supplementation with methyl donors enhances allergic airways disease in mice. J Clin Invest 2008; 118:3462 - 3469
- Sinclair KD, Allegrucci C, Singh R, Gardner DS, Sebastian S, Bispham J, et al. DNA methylation, insulin resistance and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc Natl Acad Sci USA 2007; 104:19351 - 19356
- Achon M, Alonso-Aperts E, Varela-Moreinas G. High-dose folic acid supplementation: effects on gestation and the methionine cycle. Br J Nutr 2002; 83:177 - 183
- Niculescu MD, Zeisel SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J Nutr 2002; 132:2333 - 2365
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA 2008; 105:17046 - 17049
- Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA methylation differences after exposure to prenatal famine are common and timing and sex-specific. Hum Mol Genet 2009; 18:4046 - 4053
- Fryer AA, Nafee TM, Ismail KM, Carroll WD, Emes RD, Farrell WE. LINE-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: a preliminary study. Epigenetics 2009; 4:394 - 398
- Kim YI. Will mandatory folic acid fortification prevent or promote cancer. Am J Clin Nutr 2004; 50:1123 - 1128
- Mason JB. Biomarkers of nutrient exposure and status in one-carbon (methyl) metabolism. J Nutr 2003; 133:941 - 947
- Molloy AM, Mills JL, Cox C, Daly SF, Conley M, Brody LC, et al. Choline and homocysteine interrelations in umbilical cord and maternal plasma at delivery. Am J Clin Nutr 2005; 82:836 - 842
- Obeid R, Herrmann W. Homocysteine, folic acid and vitamin B12 in relation to pre- and postnatal health aspects. Clin Chem Lab Med 2005; 42:1052 - 1057
- Yang AS, Estécio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulphite PCR of repetitive DNA elements. Nuc Acids Res 2004; 32:38
- Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, et al. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet 2009; 18:4808 - 4817
- Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium assay. Epigenomics 2009; 1:177 - 200
- Milani L, Lundmark A, Kiialainen A, Nordlund J, Flaegstad T, Forestier E, et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood 2010; 115:1214 - 1225
- Choi SH, Worswick S, Byun HM, Shear T, Soussa JC, Wolff EM, et al. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Intl J Cancer 2009; 125:723 - 729
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 2006; 38:1378 - 1385
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 186
- Kim KC, Friso S, Choi SW. DNA methylation, an epigenetic mechanism connecting folate to healthy embryonic development and aging. J Nutr Biochem 2009; 20:917 - 926
- Wang L, Wang F, Guan J, Le J, Wu L, Zou J, et al. Relation between hypomethylation of long interspersed nucleotide elements and risk of neural tube defects. Am J Clin Nutr 2010; 91:1359 - 1367
- Achon M, Alonso-Aperts E, Varela-Moreiras G. High dietary folate supplementation: effects on diet utilization and methionine metabolism in aged rats. J Nutr Health Aging 2002; 6:51 - 54
- Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect 2006; 114:567 - 572
- Son GH, Kwon JY, Kim YH, Park YW. Maternal serum triglycerides as predictive factors for large-forgestational age newborns in women with gestational diabetes mellitus. Acta Obstet Gynecol Scand 2010; 89:700 - 704
- Martin-Gronert MS, Ozanne SE. Mechanisms linking suboptimal early nutrition and increased risk of type 2 diabetes and obesity. J Nutr 2010; 140:662 - 666
- Stover PJ. One-carbon metabolism-genome interactions in folate-associated pathologies. J Nutr 2009; 139:2402 - 2405
- Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol 2007; 23:297 - 307
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004; 7:847 - 854
- McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 2009; 12:342 - 348
- Kishi T, Fujita N, Eguchi T, Ueda K. Mechanism for reduction of serum folate by antiepileptic drugs during prolonged therapy. J Neurol Sci 1997; 145:109 - 112
- Martin SC, Tsakas-Ampatzis I, Bartlett WA, Jones AF. Measurement of plasma total homocysteine by HPLC with coulometric detection. Clin Chem 1999; 45:150 - 152
- World Health Organisation. WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr Suppl 2006; 450:76 - 85
- Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for annotation, visualization and integrated discovery. Genome Biol 2003; 4:3