Abstract
Primary biliary cirrhosis (PBC) is an autoimmune chronic cholestatic liver disease with a strong genetic susceptibility due to the high concordance in monozygotic (MZ) twins and a striking female predominance. Women with PBC manifest an enhanced X monosomy rate in peripheral lymphocytes and we thus hypothesized an X chromosome epigenetic component to explain PBC female prevalence. While most genes on the female inactive X chromosome are silenced by promoter methylation following X chromosome inactivation (XCI), approximately 10% of X- linked genes exhibit variable escape from XCI in healthy females. This study was designed to test the hypothesis that susceptibility to PBC is modified by one or more X-linked gene with variable XCI status. Peripheral blood mRNA and DNA samples were obtained from a unique cohort of MZ twin sets discordant and concordant for PBC. Transcript levels of the 125 variable XCI status genes was determined by quantitative RT-PCR analysis and two genes (CLIC2 and PIN4) were identified as consistently downregulated in the affected twin of discordant pairs. Both CLIC2 and PIN4 demonstrated partial and variable methylation of CpG sites within 300 bp of the transcription start site that did not predict the XCI status. Promoter methylation of CLIC2 manifested no significant difference between samples and no significant correlation with transcript levels. PIN4 methylation showed a positive trend with transcription in all samples but no differential methylation was observed between discordant twins. A genetic polymorphism affecting the number of CpG sites in the PIN4 promoter did not impact methylation or transcript levels in a heterozygous twin pair and showed a similar frequency in independent series of unrelated PBC cases and controls. Our results suggest that epigenetic factors influencing PBC onset are more complex than methylation differences at X-linked promoters and variably 3 inactivated X-linked genes may be characterized by partial promoter methylation and biallelic transcription.
Introduction
Clinical features of different autoimmune disease share more commonalities than differences, including late onset, heritability and female predominance.Citation1 Primary biliary cirrhosis (PBC) is characterized by progressive destruction of intrahepatic bile ducts, high-titer serum anti-mitochondrial antibodies (AMA) and circulating autoreactive T cells.Citation2 Similar to most autoimmune diseases, PBC manifests a 10:1 female predominanceCitation3 with a possible involvement of sex chromosomes,Citation4 as suggested by the preferential X chromosome loss observed in peripheral lymphocytes from women with PBC despite random X chromosome inactivation.Citation5,Citation6 Despite a 50–100-fold higher risk for PBC among first degree relatives, the incomplete disease concordance among monozygotic (MZ) twins suggests that PBC results from combined effects of genetics and the environment.Citation2,Citation7 Epigenetic modifications, particularly DNA methylation, have been shown to differ between MZ twinsCitation8 and appear as ideal candidates to explain the environmental influence on individual susceptibility to complex diseases such as PBC.
In females, the inactivation of one parental X chromosome is caused by epigenetic mechanisms, primarily promoter DNA methylation and histone modifications, resulting in dosage compensation. Of particular interest are the approximately 125 different X-linked genes that comprise almost 10% of the X chromosome and exhibit widely variable patterns of X chromosome inactivation (XCI) between healthy female individuals.Citation9 We therefore hypothesized that PBC susceptibility could be influenced by epigenetic differences of specific genes on the X chromosome, either by protective genes being silenced or susceptibility X chromosome genes variably escaping XCI.
In this study we investigated a unique cohort of MZ twins discordant or concordant for PBC for transcription differences of 125 X chromosome genes and the promoter methylation patterns of consistently differentially expressed genes. Two genes CLIC2 and PIN4 had consistently decreased transcript levels in the PBC-affected twins compared to healthy twins. Bisulfite sequencing of CLIC2 and PIN4 promoter regions revealed both genes to show partially variable methylation patterns that did not separate into active versus inactive alleles and did not significantly correlate with transcript levels. These results provide two novel genes that may be relevant to understanding PBC pathogenesis and suggest that additional epigenetic studies outside of the promoter regions are needed to understand the differential transcript levels between MZ twin pairs.
Results
Gene transcription data.
Normalized transcription data were analyzed by and sorted using linearized values from the weakest normalized calibrator to identify consistent genes that were either significantly up or downregulated in the affected twin compared to the healthy twin (Sup. Table 2). While none of the variable XCI genes were consistently dysregulated in all four pairs of discordant PBC twin pairs, two transcripts (CLIC2 and PIN4) were found to be consistently dysregulated in 3 out of 4 discordant sets while with similar transcription profiles in the concordant set of twins ().
CLIC2 promoter methylation.
DNA methylation was examined for the CLIC2 promoter that lacks a CpG island (genome.ucsc.edu, CpG island track). Bisulfite sequencing primers were designed to a 279 bp region 5′ of the transcription start site containing six CpG sites (). CLIC2 bisulfite sequencing data was calculated by five MZ twin sets, one concordant and four discordant for PBC, each with a minimum of 20 clones. Methylation patterns of individually sequenced clones are shown for a representative discordant twin pair in , with circles representing CpG sites that are methylated (filled) or unmethylated (open). Variable methylation patterns were observed between individual clones and individual CpG sites for all samples (Sup. Fig. 1).
Overall promoter methylation was determined as the percentage of methylated CpG sites out of all possible CpG sites while site-specific methylation was calculated for each of the six CpG sites in the CLIC2 promoter region for each sample (). Percent methylation calculations were then grouped according to diagnosis (mean ± SEM) and tested for significance by t-test. CLIC2 showed partial promoter methylation ranging from 44 to 67% of available CpG sites and no significant differences in percent methylation for overall promoter methylation () or site-specific methylation () were observed between healthy and PBC twins. Transcript levels of CLIC2 did not significantly correlate with overall promoter methylation ().
PIN4 promoter genotyping and methylation.
PIN4 lacks a CpG island at its promoter, but bisulfite sequencing primers were designed to span 11–13 CpG sites covering 221 bp including part of exon 1 (). This promoter region contains two linked SNPs at predicted CpG sites 7 and 8. Both SNPs change the predicted CG to AG resulting in 11 CpGs rather than 13 CpGs for the less common allele (polymorphic sites are colored green and red in ). PIN4 alleles are defined as allele 1 (including 11 CpG sites) or allele 2 (13 CpG sites) based on ss10563357, dbSNP (). All twin pair DNA samples were genotyped and were homozygous for allele 1 except for twin set 54, 55 who were heterozygous for both alleles (). The genotype frequencies observed were consistent with the NCBI database with the majority of the population homozygous for allele 1, a small percentage heterozygous and zero of the population homozygous for allele 2 ().
PIN4 promoter methylation was analyzed separately for the two alleles since the polymorphisms affect CpG sites. Only one of the PBC twin pairs was heterozygous for ss10563357, but no apparent or significant difference was observed in methylation patterns between the healthy and PBC twin ( and E). Grouped overall percent methylation of both alleles were determined as the total number methylated sites out of the total possible sites ( for allele 1, for allele 2). Individual CpG site percent methylation of the two alleles in the PIN4 promoter also showed no significant differences between healthy and PBC subjects ( for allele 1, for allele 2). Conversely, transcription analysis of PIN4 demonstrated a trend for increased transcription correlating with increased methylation in allele 1 () that was not statistically significant.
CLIC2 and PIN4 X chromosome inactivation status.
While the individual clones from bisulfite sequencing of both CLIC2 and PIN4 promoters did not appear to distinguish active and inactive X chromosome alleles, we tested this hypothesis formally using an approach previously described for promoters of XCI subject genes MECP2 and AR.Citation12 Bisulfite sequencing of AR was used as a control for XCI status in twins, as this is a known gene subject to XCI in females and used for clinical XCI assays.Citation13 The XCI status of each allele was illustrated with the comparison of the number of methylated CpG sites per clone compared to the percentage of the total clones. XA (active X) represents the frequency of bisulfite clones with no methylated CpG sites while XI (inactive X) represents the bisulfite clones with greater than 50% methylation of the CpG sites in a given clone designated as the inactivated alleles. X? represents bisulfite clones with greater than 0% and less than 50% methylated CpGs that cannot be classified as activated or repressed (). In addition to the MZ twin cohort, two healthy female and two health male controls with no familial history of PBC or other autoimmune disease history were included. Both males showed no XCI for AR as expected, with 90% of the clones having zero methylation and only 10% with one methylated site. Female controls showed the typical bi-modal distribution of XA and XI alleles of AR seen in , with very few clones in the X? category. The healthy and PBC female twins show a similar pattern of XCI status as the female controls and do not present aberrant XCI at the AR promoter. and C illustrates the XCI status of the CLIC2 and PIN4 promoter regions in which a bimodal distribution into XA and XI alleles is not observed. Unlike AR that contains a CpG island and is always subject to XCI, CLIC2 and PIN4 both show approximately one third of bisulfite sequenced clones as of X? alleles that cannot be designated as active or inactive.
Discussion
PBC is an autoimmune disease commonly referred to as a model for understanding the pathogenesis of autoimmunity based on its uniform features, including female prevalence and clinical presentation. While genetic factors are of seminal importance in determining disease susceptibility, these can be confirmed only in subgroups of patientsCitation14 and could not explain the imbalanced sex ratio. We previously observed that women with PBC manifest an enhanced rate of preferential X monosomy but random XCI in their peripheral lymphocytes compared to age-matched women.Citation5,Citation6 In this study we thus tested the hypothesis that X-linked promoters, particularly those of variable XCI genes may be dysregulated through aberrations in promoter methylation and utilized the unique model of clinically discordant MZ twin pairs. From this analysis, we identified two candidate genes CLIC2 and PIN4 that exhibited decreased transcription in 3 out of 4 affected compared to healthy twins in discordant twin pairs. However, for both CLIC2 and PIN4 promoters, methylation was partial, variable and did not predict either XCI status or transcript levels. These results are informative both for future studies into understanding genetics and epigenetics of PBC as well as the epigenetic characteristics of genes that escape XCI in human females.
Although DNA methylation has been associated with a variety of diseases and disorders, specific patterns of DNA methylation in gene promoters and nonrepetitive genomic regions are remarkably similar between unrelated individuals.Citation15–Citation18 In addition, methylation profiles between male and female autosomes are also highly similar.Citation17 A recent large scale sequencing of the genome and epigenome of twin pairs with mutiple sclerosis has also demonstrated that methylated CpG sites within CpG islands (MspI fragments) are nearly identical in the same tissue (CD4+ T cells) compared between twins, only slightly more variable between unrelated individuals and most variable between different tissues or tumor versus normal tissue.Citation19 Moreover, Javierre and colleagues recently studied the methylation status of a cohort of MZ twins discordant for three autoimmune diseases: systemic lupus erythematosus (SLE), rheumatoid arthritis and dermatomyositis; only MZ twins discordant for SLE featured widespread changes in the DNA methylation status of a significant number of genes.Citation20 However, while most DNA methylation studies focus on CpG island and promoters, genome-wide epigenomic studies have reinstated that gene promoters are vastly de-enriched for DNA methylation, while methylation of intergenic and repetitive regions is extensive.Citation21–Citation24
Several reports have linked epigenetics to disease phenotypes in humans. One example includes the incidence of discordant MZ twins for the X-linked dominant disorder Rett syndrome where discordance was due to differences in skewed XCI.Citation25,Citation26 Methylation at one particular CpG site upstream of the MECP2 promoter has been shown to be significantly increased in male autism subjects compared to controls.Citation27 In schizophrenia samples, hypermethylation of the reelin gene (RLN) promoter CpG island has been observed.Citation28 It is also noteworthy that discordance in MZ twin pairs for Beckwith-Wiedemann syndrome is due to differences in imprinting of the KCNQ1OT1 gene.Citation29 In addition, differences to methylation patterns to the imprinted gene H19 correlates to discordance of MZ twins for Silver-Russell syndrome.Citation30 While there is also evidence of epigenetic mechanisms in animal models for the autoimmune disease systemic lupus erythematosusCitation31,Citation32 we note that cells to be investigated for epigenetic changes may include target or effecter cell populations thus introducing an additional confounding factor.
While CLIC2 and PIN4 are dysregulated in most discordant PBC twin pairs, the mechanism behind the variant transcript levels does not appear to be promoter methylation. Genomic evidence has suggested that X-linked genes lacking a CpG island may not be ‘classically’ regulated by promoter methylation.Citation33 In addition, PCR-based methylation analyses of the known XCI escape gene MAOA, encoding monoamine oxidase A, has shown a similar pattern of variable promoter methylation not associated with transcription or XCI status.Citation34 The bisulfite sequencing based XCI status categorization approach performed here and described for XCI subject genes previouslyCitation12 may be useful for future studies investigating promoter methylation patterns for genes that escape XCI. While genetic polymorphisms within or close to promoters have been hypothesized to be responsible for the variability in XCI status of specific genes, our preliminary analysis of a known polymorphism creating two additional CpG sites in the PIN4 promoter did not apparently affect overall promoter methylation or transcription. Perhaps polymorphic repetitive elements may be an unexplored genetic component affecting transcription and XCI status of X-linked genes in human.
CLIC2, chloride intracellular channel gene 2, was first suggested as a possible candidate for one of the many diseases linked to chromosomal region XqCitation28 because previous findings showed other human chloride channel genes had a direct association to a wide range of hereditary diseases.Citation35 More recent work demonstrated that CLIC2 modulated intracellular Ca2+ homeostasis through ryanodine receptors (RyR), suggesting that CLIC2 may alter the calcium homeostasis of any tissue through RyR.Citation36CLIC2 dysregulation could possibly be causing aberrations in calcium levels in the liver resulting in the degradation of the tissue, ultimately leading to disease.
PIN4 is part of the parvulin family of peptdylprolyl cis/trans isomerases and has been shown to be involved in mitotic regulatory mechanisms and cell proliferation.Citation37,Citation38 More interesting, however, is a previous study has showing PIN4 is involved in chromatin remodeling, an epigenetic mechanism involving ATP dependent changes to histone modifications.Citation39PIN4 dysregulation could therefore affect chromatin remodeling, altering transcription and regulation of multiple susceptibility or protective genes for PBC.
The major challenges for the future of epigenetic studies in human disease outcome is in identifying regions of the genome to investigate for DNA methylation differences relevant to gene transcription and to develop novel methods for identifying DNA methylation patterns that correlated with disease outcome. MZ twin studies of discordant disease are expected to continue to be of use in separating genetic from epigenetic variables in such studies.
Methods
Biological samples.
Peripheral blood samples were collected and processed from 5 MZ twin sets (one concordant and 4 discordant for PBC) identified in our previous study.Citation7 The age and pairing of the utilized subjects are illustrated in . Genomic DNA from whole blood was isolated using Qiagen Blood Mini kits (Qiagen, Valencia, CA) while Tempus tubes (Applied Biosystems, Foster City, CA) were utilized and mRNA isolated using an automated ABI 6100 Nucleic Acid PrepStation. Obtained mRNA was then translated into cDNA using previously described methods.Citation10 We used internal quality control thresholds obtained with species specific TaqMan assays to assess the quality of bulk samples. Stored DNA samples from 114 female patients with PBC and 118 age-matched controls and 20 discordant pairs of sisters of similar age (within 5 years) were utilized for single nucleotide polymorphism (SNP) analysis. The protocol was approved by the IRB of the University of California at Davis and all subjects provided written informed consent.
Transcription analysis.
RNA was extracted using a 6100 Nucleic Acid PrepStation (Applied Biosystems) according to the manufacturer's instructions. The cDNA was synthesized as described in Clay et al.Citation10 Real time (RT)-PCR was performed at the Lucy Whittier Molecular & Diagnostic Core Facility at UC Davis using Applied Biosystem TaqMan 384 low density arrays custom-designed to include primers for the 125 differentially inactivated genes, five housekeeping genes to maximize reliability (18S, B2M, GAPDH, HPRT1, RPLPO) and three copies of each of the internal controls androgen receptor (AR-fully inactivated) and XIST (fully escaping XCI). The assays were designed using AB Primer Express Software 3.0 with the sequences from the listed genebank accession numbers (Sup. Table 1). Each PCR reaction contained 20X primers and probe for the respective TaqMan® system with a final concentration of 400 nM for each primer and 80 nM for the TaqMan® probe and commercially available PCR mastermix (Applied Biosystems) containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 5 mM MgCl2, 2.5 mM deoxynucleotide triphosphates, 0.625 U AmpliTaq Gold DNA polymerase per reaction, 0.25 U AmpErase UNG per reaction and 5 µl of the diluted cDNA sample in a final volume of 12 µl. The samples were placed in a 384-well plate and amplified in an automated fluorometer (ABI PRISM 7900 HTA FAST, ABI). ABI's standard amplification conditions were used: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C and 60 s at 60°C. Control wells without reverse transcriptase were included. Fluorescent signals were collected during the annealing temperature and CT values were exported with a threshold of 0.06 and a baseline of 3–15 for the genes of interest. The intra-assay coefficient of variation of the threshold cycle (CT) ranged from 0.1–3%. Final quantitation was done using the comparative Ct method, and is reported as relative transcription or the n-fold difference relative to a calibrator cDNA. In brief, the weakest-expressing housekeeping gene, beta2-microglobin (B2M), was used to normalize the Ct values of the target genes (ΔCT). The ΔCT was then calibrated against the average of the healthy control for each target gene. The relative linear amount of target molecules relative to the calibrator, was calculated by 2 elevated to the absolute value of ΔΔCt. Therefore, all gene transcription is expressed as an n-fold difference relative to the calibrator. The linearized value of the calibrator is 1. Eleven/125 analyzed genes did not provide sufficient amplification to obtain transcription data; only genes for which at least three twin pairs provided transcripts were included for consistency comparisons (genes not fulfilling this criterion are highlighted in Sup. Table 2).
Bisulfite sequencing of PIN4, CLIC2 and AR.
Bisulfite treatment of 500 ng of gDNA from each sample was performed using the Zymo Methylation Direct Kit (Zymo Research) following manufacturer's instructions. Converted DNA was stored at −20°C until used. Converted bisulfite DNA was PCR-amplified using primers for the promoter regions of CLIC2, PIN4 and AR designed by online MethPrimer software (www.urogene.org/methprimer/index.html).Citation11 Primers for were as follows: CLIC2 () F: CTC AAT CTC AGG GTC CAC TTG AGT GC, R: CCA AAG GGG TGT GTA TAA ACT GCC TGG; PIN4 () F: GTC TGC CCC AAG CTG TGC CTG CTT C, R: GAA GGG AGG GAA CAG ACA GTC CAT CT; AR F: GAG CTT TCC AGA ATC TGT TCC AGA G, R: TAG AGG CCC CAC AGG CTA CCT GGT C. These primers were designed to recognize regions without CpG sites to avoid amplification bias of methylated versus unmethylated sequences. PCR products were purified using a gel extraction kit (Qiagen, Valencia, CA) and then cloned into a pGEM-T easy plasmid and JM109 competent cells (Promega). Using blue/white color screening on ampicillin plates, 20 white transformed colonies from each sample and each PCR amplicon were sequenced and analyzed for the percentage of methylated cytosine at each sample.
PIN4 genotyping.
The PIN4 coupled polymorphism that removes two adjacent CpG sites is designated as SNP: ss10563357 (Rs7058353) according to NCBI dbSNP. Primers for PIN4 genotyping were designed using Primer3 online software F: CAG ACA TCT TCA GCC CCA TT, R: CTC TAC CTT TCC CCG CTT TT. The polymorphism was genotyped in all twins' DNA as well as in 114 unrelated PBC cases and 118 controls for further confirmation.
Statistical analysis.
Consistency for transcription data among MZ twin sets was defined as a consistent up or downregulation of specific genes observed in 3 out of 4 affected twins compared to their healthy counterpart among discordant sets. We also analyzed the transcription profiles between concordant twins and 20 pairs of PBC discordant sisters as a stringent criterion for consistency (see example in Sup. Fig. 1). SNP frequencies were compared between patients with PBC and controls using the chi-square test. All statistical analyses were performed using Intercooled Stata 8.0 (Stata Corporation, College Station, TX) and SAS (SAS Institute Inc., Cary, NC).
Figures and Tables
Figure 1 CLIC2 bisulfite sequencing for promoter methylation analysis. (A) Chromosomal sequence location of the region of the CLIC2 promoter that was analyzed by bisulfite sequencing. This region contains 6 CpG sites (circles) within a 279 bp region overlapping the transcription start site as shown. (B) Representative bisulfite sequencing data from a PBC discordant twin pair. Each circle represents a CpG site and each line represents an individually sequenced clone. Filled circles represent methylated sites (protected from bisulfite conversion) while unfilled circles represent unmethylated CpG sites (converted). (C) Overall percent methylation was determined by the total number methylated sites out of the total CpG sites in all clones and graphed as mean ± SEM for PBC (n = 6) versus healthy (n = 4) samples. (D) Percent methylation was calculated for each individual site within the CLIC2 promoter region for all clones and represented as mean ± SEM for PBC (n = 6) versus healthy (n = 4) samples. (E) Analysis of CLIC2 promoter methylation versus transcription for each individual sample. Each twin pair is represented by an individual symbol, as shown, with filled symbols representing PBC and open symbols representing healthy individuals of each twin pair. No significant correlation was observed between CLIC2 promoter methylation and transcription.
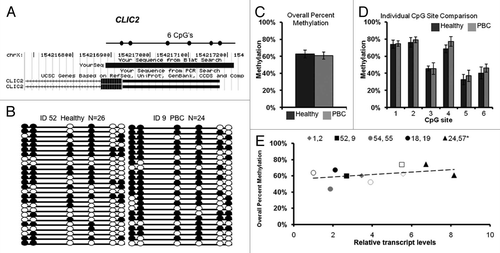
Figure 2 Bisulfite sequencing analysis of PIN4 promoter methylation and genotype. (A) Chromosomal sequence location of the region of the PIN4 promoter that was analyzed by bisulfite sequencing. This region contains 11 or 13 CpG sites dependent on SNP: ss10563357. All samples were homozygous for allele 1 (11 CpG sites, AG at sites 7 and 8) except for one discordant twin set heterozygous for alleles 1 and 2 (allele 2 has 13 CpG sites). (B and E) Bisulfite sequencing results for the heterozygous twin pair 54 and 55. Results are exhibited as in (1B) except red and green filled circles represent CG sites absent in allele 1 and the two alleles are grouped separately as allele 1 (B) and allele 2 (E). DNA methylation patterns were variable between clones, alleles and twins, but no apparent association was observed between PBC status or allele status and DNA methylation. (C and F) Overall percent methylation of allele 1 (C) or allele 2 (F) was determined and graphed as mean ± SEM of all available samples. (D and G) Site specific analysis of individual CpG sites in the PIN4 promoter of allele 1 (D) or allele 2 (G) was determined and graphed as mean ± SEM of all available samples. (H) Analysis of PIN4 promoter methylation versus transcription for each individual sample. Each twin pair is represented by an individual symbol, as shown, with filled symbols representing PBC and open symbols representing healthy individuals of each twin pair; black symbols representing allele 1 and red symbols representing allele 2. Allele 1 of PIN4 showed a trend for increased transcription with increased methylation that was not siginificant, while allele 2 did not show an apparent correlation between transcription and methylation.
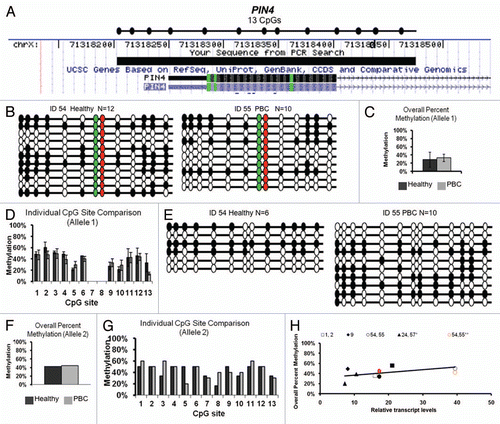
Figure 3 Determination of XCI allele status from bisulfite sequencing analyses. (A–C) Each clone from bisulfite sequencing was analyzed for the number of CpG sites methylated in each clone. Histograms represent the distribution of percentage of clones based on degree of methylation. The bimodal peaks observed at all three loci are consistent with X chromosome inactivation. Clones with no sites methylated are designated as clear active alleles (XA), clones with >50% of maximum number of methylated sites are designated as inactive (XI) and the intervening clones as potentially undefined XCI status (X?). (A) The control X inactivated gene AR has a maximum of 9 possible methylation sites. Both male controls had no XI clones, as expected, with 90% of the clones showing no methylated sites and only one clone showing one methylated CpG site. Female controls showed the typical bimodal distribution into active and inactive alleles. Individuals within the PBC twin pairs showed a similar bimodal distribution with no apparent differences between XA and XI alleles at the AR promoter. (B) Unlike AR, the XCI allele distribution of CLIC2 methylation patterns did not follow a bimodal distribution typical of promoters subject to XCI. Approximately 40% of clones at CLIC2 fell into the X? status category that could not be designated as active or inactive. No apparent differences were observed between healthy and PBC individuals in the XCI status distribution. (C) The PIN4 methylation pattern also did not fit the typical XCI distribution pattern, as 20–30% of the clones fell within the X? category. Both healthy and PBC subjects showed similar frequencies of X? clones.
Table 1 Transcription and promoter methylation analyses of CLIC2 and PIN4 in PBC twin pairs
Table 2 PIN4 genotyping of SNP Rs7058353 in unrelated patients with PBC and controls and in 20 sets of sisters of similar age discordant for PBC
Additional material
Download Zip (234.7 KB)Acknowledgements
This work was supported by the American Liver Foundation (C.S.), NIH R21DK075400 (C.S.) and R01ES015171 (J.M.L.). The authors thank the PBCers Organization for their continuous support.
References
- Shoenfeld Y, Selmi C, Zimlichman E, Gershwin ME. The autoimmunologist: geoepidemiology, a new center of gravity and prime time for autoimmunity. J Autoimmun 2008; 31:15 - 18
- Selmi C, Zuin M, Gershwin ME. The unfinished business of primary biliary cirrhosis. J Hepatol 2008; 49:451 - 460
- Lleo A, Battezzati PM, Selmi C, Gershwin ME, Podda M. Is autoimmunity a matter of sex?. Autoimmun Rev 2008; 7:626 - 630
- Invernizzi P, Pasini S, Selmi C, Gershwin ME, Podda M. Female predominance and X chromosome defects in autoimmune diseases. J Autoimmun 2009; 33:12 - 16
- Invernizzi P, Miozzo M, Battezzati PM, Bianchi I, Grati FR, Simoni G, et al. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet 2004; 363:533 - 535
- Miozzo M, Selmi C, Gentilin B, Grati FR, Sirchia S, Oertelt S, et al. Preferential X chromosome loss but random inactivation characterize primary biliary cirrhosis. Hepatology 2007; 46:456 - 462
- Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics and environment. Gastroenterology 2004; 127:485 - 492
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 2005; 102:10604 - 10609
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005; 434:400 - 404
- Clay CC, Rodrigues DS, Brignolo LL, Spinner A, Tarara RP, Plopper CG, et al. Chemokine networks and in vivo T-lymphocyte trafficking in nonhuman primates. J Immunol Methods 2004; 293:23 - 42
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002; 18:1427 - 1431
- Nagarajan RP, Patzel KA, Martin M, Yasui DH, Swanberg SE, Hertz-Picciotto I, et al. MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res 2008; 1:169 - 178
- Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992; 51:1229 - 1239
- Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. Primary biliary cirrhosis associated with HLA, IL12A and IL12RB2 variants. N Engl J Med 2009; 360:2544 - 2555
- Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, et al. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet 2009; 18:4808 - 4817
- Grunau C, Hindermann W, Rosenthal A. Large-scale methylation analysis of human genomic DNA reveals tissue-specific differences between the methylation profiles of genes and pseudogenes. Hum Mol Genet 2000; 9:2651 - 2663
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science 2007; 315:1141 - 1143
- Ladd-Acosta C, Pevsner J, Sabunciyan S, Yolken RH, Webster MJ, Dinkins T, et al. DNA methylation signatures within the human brain. Am J Hum Genet 2007; 81:1304 - 1315
- Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 2010; 464:1351 - 1356
- Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 2010; 20:170 - 179
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 186
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315 - 322
- Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer GP. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci USA 2009; 106:671 - 678
- Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, et al. Large-scale structure of genomic methylation patterns. Genome Res 2006; 16:157 - 163
- Bruck I, Philippart M, Giraldi D, Antoniuk S. Difference in early development of presumed monozygotic twins with Rett syndrome. Am J Med Genet 1991; 39:415 - 417
- Migeon BR, Dunn MA, Thomas G, Schmeckpeper BJ, Naidu S. Studies of X inactivation and isodisomy in twins provide further evidence that the X chromosome is not involved in Rett syndrome. Am J Hum Genet 1995; 56:647 - 653
- Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 2006; 1:1 - 11
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA 2005; 102:9341 - 9346
- Weksberg R, Shuman C, Caluseriu O, Smith AC, Fei YL, Nishikawa J, et al. Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum Mol Genet 2002; 11:1317 - 1325
- Yamazawa K, Kagami M, Fukami M, Matsubara K, Ogata T. Monozygotic female twins discordant for Silver-Russell syndrome and hypomethylation of the H19-DMR. J Hum Genet 2008; 53:950 - 955
- Richardson B. Primer: epigenetics of autoimmunity. Nat Clin Pract Rheumatol 2007; 3:521 - 527
- Sekigawa I, Kawasaki M, Ogasawara H, Kaneda K, Kaneko H, Takasaki Y, et al. DNA methylation: its contribution to systemic lupus erythematosus. Clin Exp Med 2006; 6:99 - 106
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853 - 862
- Pinsonneault JK, Papp AC, Sadee W. Allelic mRNA expression of X-linked monoamine oxidase a (MAOA) in human brain: dissection of epigenetic and genetic factors. Hum Mol Genet 2006; 15:2636 - 2649
- Heiss NS, Poustka A. Genomic structure of a novel chloride channel gene, CLIC2, in Xq28. Genomics 1997; 45:224 - 228
- Board PG, Coggan M, Watson S, Gage PW, Dulhunty AF. CLIC-2 modulates cardiac ryanodine receptor Ca2+ release channels. Int J Biochem Cell Biol 2004; 36:1599 - 1612
- Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 1996; 380:544 - 547
- Uchida T, Takamiya M, Takahashi M, Miyashita H, Ikeda H, Terada T, et al. Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cell proliferation. Chem Biol 2003; 10:15 - 24
- Surmacz TA, Bayer E, Rahfeld JU, Fischer G, Bayer P. The N-terminal basic domain of human parvulin hPar14 is responsible for the entry to the nucleus and high-affinity DNA-binding. J Mol Biol 2002; 321:235 - 247