Abstract
Several studies linking alterations in differential placental methylation with pregnancy disorders have implicated (de)regulation of the placental epigenome with fetal programming and later-in-life disease. We have previously demonstrated that maternal tobacco use is associated with alterations in promoter methylation of placental CYP1A1 and that these changes are correlated with CYP1A1 gene expression and fetal growth restriction. In this study we sought to expand our analysis of promoter methylation by correlating it to gene expression on a genome-wide scale. Employing side-by-side IlluminaHG-12 gene transcription with Infinium27K methylation arrays, we interrogated correlative changes in placental gene expression and DNA methylation associated with maternal tobacco smoke exposure at an epigenome-wide level and in consideration of signature gene pathways. We observed that the expression of 623 genes and the methylation of 1024 CpG dinucleotides are significantly altered among smokers, with only 38 CpGs showing significant differential methylation (differing by a methylation level of ≥ 10%). We identified a significant Pearson correlation (≥ 0.7 or ≤ -0.7) between placental transcriptional regulation and differential CpG methylation in only 25 genes among non-smokers but in 438 genes among smokers (18-fold increase, p < 0.0001), with a dominant effect among oxidative stress pathways. Differential methylation at as few as 6 sites was attributed to maternal smoking-mediated birth weight reduction in linear regression models with Bonferroni correction (p < 1.8x10-6). These studies suggest that a common perinatal exposure (such as maternal smoking) deregulates placental methylation in a CpG site-specific manner that correlates with meaningful alterations in gene expression along signature pathways.
Introduction
In accordance with the developmental origins of disease, an altered in utero environment during critical windows in perinatal and postnatal development influences the risk of later in life health and disease. Which exposures significantly alter the in utero environment and how they manifest long term developmental reprogramming is poorly understood but has recently been attributed in varying measures to altered establishment and modification of the fetal and placental epigenome.Citation1–Citation4 Among the triad of players whose epigenomes are potentially modifiable in the intrauterine environment (e.g., maternal, fetal or placental epigenome), interrogations of placental deregulation are likely to be of high yield since establishment and maintenance of placental integrity and function is critical to fetal growth, development and survival.Citation5 This likely arises from the placenta serving as both a metabolic and endocrine organ, as well as a gateway by which potential toxins and reactive compounds can be converted to less harmful intermediates prior to reaching the fetus.Citation6 In sum, the biochemical and molecular activities of the placenta both respond to and are modified by environmental insults and, thus, the placenta may be considered a “footprint” of the in utero exposures the fetus experiences.Citation6,Citation7 While various compounds have been shown to cross the placenta and effect biochemical activity and gene transcription (i.e., alcohol, nicotine, cocaine, lead and other hazardous air pollutants), tobacco exposure is of singular interest primarily because of its broad-based continued use and effects across populations of diverse racial, ethnic and social demographic levels.Citation1,Citation8–Citation12 In addition, in utero tobacco exposure is an ideal disease model to interrogate the role of environmental exposure and epigenomics.Citation4 First, polycyclic aromatic hydrocarbons (PAH) are a primary component of tobacco smoke but are also common ambient exposures.Citation1,Citation4,Citation6,Citation8–Citation12 They are one of the 33 hazardous air pollutants mandated for evaluation by the Clean Air Act, and are widespread as a result of the petrochemical agency, auto exhaust and burning of fossil fuels. Second, there is now over 40 years of evidence suggesting a causal relationship between tobacco use and delivery of small for gestational age (SGA) infants.Citation1,Citation9–Citation16 While initial clues about the potential genetic, epigenetic and physiologic mechanisms involved in these observations have been reported, these have yet to be fully characterized.Citation13–Citation21
Epigenetic variation of DNA, particularly methyl group modifications to cytosine bases adjacent to guanines (CpG dinucleotides) represent an important form of epigenomic variation during critical windows in development.Citation2,Citation4 Loosely ascribed changes in the levels of DNA methylation are associated with maternal smoking behavior. A report from the National Children's Study showed that DNA methylation of AluYb8 was lower in children (kindergarten and first grade) exposed in utero to maternal smoking.Citation22 Similarly, global DNA methylation of newborn cord blood is reported to have an inverse relationship with cord blood cotinine levels, being lowest in newborns with the highest serum cotinine levels.Citation23 At a single gene level, we have previously reported an increase in expression of the Phase I enzyme CYP1A1 in placental tissue from smoking mothers compared with non-smoking ones.Citation13 Interrogation of the mechanisms underlying this increased transcription with extensive bisulfite sequencing analysis of 59 CpG sites within the CYP1A1 proximal promoter showed that maternal smoking is associated with a decrease in CpG site-specific methylation specifically surrounding the xenobiotic response element (XRE) promoter element(s), which is significantly correlated with the CYP1A1 gene expression regardless of smoking behavior.Citation13
Here we sought to broaden our prior study at a genome-wide scale. Whereas methodological approaches for assessing methylation at candidate loci have proven revealing, epigenome-wide arrays for studying differential methylation with locus-specific resolution have only recently been validated. Using the Illumina genome-wide methylation and gene expression platforms, we report herein our measured alterations in the placental transcriptome and methylome from 18 non-smokers and 18 smokers. Development of an analysis workbench enabled integrated analysis of parallel Illumina-based tiling array-based placental methylome and transcriptome data and querying for significant correlation (r2 > −0.70) between promoter or CGI methylation and gene-specific expression. We report that expression of 623 genes and methylation of 1,024 CpG dinucleotides are significantly altered among smokers, with only 38 CpGs differing by a methylation level of 10% or greater. In correlative analysis with linear regression modeling, altered placental site-specific CpG methylation at as few as six sites along signature pathways is attributed to a significant reduction in infant birth weight among smokers. When considered in the context of the implications for the biology of the development and programming of disease, these studies suggest that a common perinatal exposure (such as maternal smoking) deregulates placental methylation, which correlates with meaningful alterations in gene expression.
Results
Characteristics of the study population.
All placental specimens were obtained from term singleton gestations. Consistent with a nested cohort design, matching was performed by virtue of maternal characteristics and without a priori knowledge of differential gene expression, methylation or consideration of fetal factors (beyond gestational age) including fetal weight, length or neonatal outcome. Thus, 36 matched subjects were analyzed with minimized potential for selection bias. As anticipated,Citation8,Citation12,Citation20–Citation28 we did observe a significant decrease in infant birth weight among smokers (3,059 g ± 107 versus 3,460 g ± 93, p = 0.008; ). The remainder of the clinical outcome variables was not significantly different among cases and controls (), and subjects were excluded by virtue of significant maternal or fetal comorbidities (see Methods).
Differential placental gene expression occurs in association with maternal smoking.
Placental high-purity mRNA from 36 samples was hybridized to the Illumina Human HG-12 Expression array. Hierarchical analysis revealed that the groups did not distinctly cluster solely by virtue of maternal tobacco smoke exposure, as 6 distinct clusters emerged (). This was not unexpected, as Bruchova et al. similarly demonstrated a failure of only two distinct hierarchical clusters to emerge, likely as a reflection of multiple interactions among smokers.Citation14 However, in R package comparisons of smokers with non-smokers, employing a cut-off p value < 0.05 revealed 622 genes that were differentially expressed (Table S1 and ). To confirm these findings, RNA was extracted from a validation cohort of smokers (n = 9) and non-smokers (n = 9) and subjected to RT-qPCR, calculating fold change by the ΔΔCt method.Citation15 We were able to validate our array findings among a number of genes of interest, demonstrating significant fold change in placental gene expression ( and ).
Site-specific placental differential methylation occurs in association with maternal smoking.
The Illumina Infinium Array (HumanMethylation27 BeadChip) enables site-specific distinction of methylation status of more than 27,000 CpG sites across the human genome, which are known to lie in gene promoter CpG “islands” and “shores”. Because of its relative ease of use, high quality and data reproducibility, the Infinium platform is emerging as a primary genome-wide technology for the discrimination of methylation states at a given CpG locus. While the technique is relatively straightforward, it deserves a few comments important for the interpretation of the data we report herein. Briefly, extracted genomic DNA is bisulfite converted so that unmethylated CpG dinucleotides are converted while methylated ones are not. Bisulfite treated DNA is thereafter hybridized to BeadChips so that annealing occurs at locus-specific oligomers linked to a methylated or an unmethylated bead. Quantification of signal intensity after allele-specific priming and single base extension of fluorescently labeled ddNTPs allows for intensity measures; the average methylation per sample is reported as a number between −1 and 1 (termed the “beta value”). A beta value of 0 represents a site that is unmethylated, while a beta value of 1 would indicate the CpG is completely methylated. Median, interquartile ranges and linear regression can therefore be used to assess the likelihood of a significant distribution.Citation23,Citation29
As shown in , median beta values at a given placental CpG site were derived and a “Delta Beta” score is calculated as the attributed difference between smokers and nonsmokers to reveal that 1,024 placental CpGs are significantly differentially methylated. Where a positive delta beta score indicates the smokers had a higher beta score than the non-smokers, a negative delta beta indicates the non-smokers had the higher beta score. In contrast to our transcriptome analysis of these same samples (), hierarchical clustering revealed the samples clustered into two groups, with majority smokers in one group, and majority non-smokers in the other group (). When we further filtered CpGs with a delta beta score corresponding to a 10% or greater change in methylation and a Diff score >20 (equivalent to a p value < 0.05), only 38 CpG sites passed these criteria (Table S2). In validation studies, bisulfite converted placental DNA was amplified, cloned and sequenced with primers designed to flank the specific CpG sites detected by the oligo on the Illumina array—confirmation is as presented in and .
Maternal smoking is associated with functional alterations in differential site-specific CpG methylation in placenta.
The integrated analysis of parallel Illumina platforms enabled the correlation of differential methylation at a given CpG site in a described promoter or enhancer and the associated level of expression in the corresponding gene. Specifically, we used Pearson coefficients of −0.7 ≥ r ≥ 0.7. In the absence of stratification by virtue of maternal smoking, in toto analysis reveals that only 13 genes show a significant correlation (either inverse or directional) between expression and methylation (Tables S1 and S2). However, when stratified by maternal smoking, among non-smokers, 25 CpGs significantly correlated with gene expression (Tables S1 and S3). However, among smokers, an 18-fold increase in correlative placental gene expression occurs (25 versus 438 genes, p < 0.0001; Table S1 and Table S4). Three of these 438 genes were chosen at random for validation and plotted ().
We thereafter employed two independent pathway analysis tools to determine which signature placental transcriptome pathways were epigenetically modified in association with maternal smoking. Ingenuity Pathway Analysis (IPA) of the 438 differentially methylated genes among smokers revealed that the top canonical pathways include oxidative phosphorylation, mitochondrial dysfunction and HIF1α signaling with molecular enrichment along cell death, morphology and cell signaling signatures (). Wholly independent analysis employing DAVID yielded oxidative phosphorylation signature pathways (p = 0.015; data not shown).
Linear regression analysis controlling for potential interactions.
As shown in and as previously reported, maternal smoking is associated with a significant decrease in fetal birth weight and renders risk of SGA birth across maternal strata.Citation8,Citation12,Citation20 However, large population-derived coefficients have previously demonstrated that birth weight is similarly influenced by maternal ethnicity, age, parity, BMI, gestational age and fetal gender, with females weighing, on average, 120–200 grams less than male infants.Citation30 For the study reported herein, subjects were matched in a nested cohort design by virtue of maternal age (±1 year), race/ethnicity, BMI and gestational age (±1 week), but without regard to infant outcome, including birth weight and gender. Thus, it remained a formal possibility that unmatched potential confounders of birth weight (i.e., fetal gender) could similarly be in association with an altered placental methylome and be misattributed to an effect of smoking. In order to interrogate for potential interactions, a linear regression analysis was performed.Citation16,Citation29
As a first pass analysis, the beta value for each individual CpG site in the methylation array was modeled as the dependent variable in a multiple linear regression model and maternal smoking status, infant gender and infant birth weight served as independent variables. shows the distribution of linear regression p value for each locus for infant gender variable. After conservative adjustment for multiple testing using the Bonferroni method, variation of methylation level at 162 CpG sites could be explained by infant gender (cut-off for genome-wide significance of p = 1.81 × 10−6) (Table S5). Subjects were thereafter stratified by the infant gender with smoking status, infant weight and smoking status by infant weight interaction as independent variable fitting to multiple linear regression model. After Bonferroni correction, the variation of methylation at 6 CpG sites in female newborn samples could be explained by smoking status by infant weight interaction, revealing that the interaction effect between smoking status and infant birth weight could potentially significantly modify the methylation of six essential sites in the placental methylome (Table S6).
Discussion
Up to 20% of women smoke during pregnancy and, although many fetuses are exposed to tobacco smoke in utero, not all experience similar adverse outcomes.Citation1,Citation8–Citation12 This discrepancy cannot be accounted for by dose effect alone and, despite decades of research, the mechanisms leading to attenuation of birth weight and related adverse outcomes are still largely unknown—likely because they are complex, involving interaction between epidemiologic, genetic, epigenetic and socio-demographic factors.
Of note and with respect to both our current and prior work, evidence to date suggests that these factors converge on a limited number of metabolic pathways that convert the vast majority of over 4,000 compounds found in tobacco smoke to reactive, potentially harmful and, in some instances, excretable intermediates.Citation1–Citation4,Citation8–Citation14 Potentially harmful DNA adducts (metabolic products of polycyclic aromatic hydrocarbons; PAH) are known to cross or collect in the placenta of smokers.Citation1 PAH compounds together with nitrosamines comprise likely carcinogenic species in tobacco smoke, and are metabolized in a sequential series of two-phase enzymatic metabolic reactions.Citation1–Citation4 Phase I enzymes (such as CYP1A1) metabolically activate PAH compounds into oxidized derivatives, resulting in reactive oxygen intermediates capable of covalently binding DNA to form adducts. In turn, these reactive electrophilic intermediates can be detoxified by phase II enzymes, such as the glutathione S-transferase (GSTT1), via conjugation with endogenous species to form hydrophilic glutathione conjugates, which are then readily excretable. Thus, the coordinated expression of these enzymes and their relative balance may determine the extent of cellular DNA damage and related development of adverse outcomes.
In an effort to understand the relationship between epigenetic regulation and genetic susceptibility to in utero tobacco exposure from a systems biology approach, we previously characterized known metabolic functional candidate polymorphisms along well-described metabolic pathways using a targeted-genomic approach. We demonstrated that while deletion of fetal GSTT1 significantly modified birth weight in smokers, it did not fully account for growth restriction per se.Citation8 However, further interrogations demonstrated that tobacco exposure significantly increases placental expression of a phase I metabolite gene (CYP1A1) in association with differential promoter methylation at a critical XRE binding element.Citation13 Importantly, the methylation status of this region was correlated with the expression level of CYP1A1, irrespective of maternal smoke exposure. In this study, we sought to extend our previous analysis and set out to determine if site-specific CpG methylation changes are associated with maternal smoking on a genome-wide level and whether these changes correlated with gene transcription.
Our work presented herein is the first to undertake a rigorous genome-wide approach to relate site-specific alterations in the methylome with meaningful changes in gene expression revealing signature pathways that are associated with smoking-mediated fetal growth attenuation. Using the Illumina genome-wide methylation and gene expression platforms in a well-matched nested cohort, we have described our measured alterations in the placental transcriptome and methylome. Development of an analysis workbench enabled interrogation of significant correlation (r > 0.70) between a given gene's promoter or CGI methylation and its expression. We demonstrate that expression of 623 genes and methylation of 1,024 CpG dinucleotides are significantly altered among smokers, with only 38 CpGs differing by a methylation level of 10% or greater. When we further apply linear regression models, altered placental site-specific CpG methylation at as few as 6 sites is attributed to a significant reduction in infant birth weight among smokers.
Our findings are consistent with those of other investigators who similarly reported that maternal smoking is associated with modified placental gene expression.Citation14,Citation21 A study of five control placentas and five placentas from smoking mothers reported differential expression of 174 genes, including changes in the level of Phase I enzymes that metabolize polycyclic aromatic hydrocarbons from tobacco smoke.Citation21 A similar study including 12 smokers and 64 non-smokers characterized differential expression of 241 genes including genes involved in xenobiotic processing and coagulation.Citation14 At a functional level, we and other investigators have similarly demonstrated an association between maternal active and passive smoking and evidence of oxidative stressCitation16,Citation20,Citation24,Citation31 and hypoxia inducible factors (such as HIF1α).Citation25 In this study, we expand upon these findings to correlate site-specific CpG methylation with meaningful alterations in oxidative stress and hypoxia pathways.
There are several methodological strengths in our study. We utilized the Illumina platform to run concomitant genome-wide gene expression and DNA methylation analysis, allowing us to determine differentially expressed genes and differentially methylated CpG sites, and correlating the two to discover meaningful alterations in placental signature pathways that occur in association with maternal smoking. Specifically, we calculated Pearson coefficients (significance at −0.7 < r < 0.7) to determine whether a significant correlation of gene expression and site-specific CpG methylation within the gene's promoter or enhancer exists. In the absence of stratification by virtue of maternal smoking, in toto analysis (n = 36) reveals that only 13 genes demonstrate a significant correlation (either inverse or directional) between expression and methylation (Table S1). However, when stratified by maternal smoking distinct and impressively significant variance in correlative methylation and gene expression occurs with an observed 18-fold increase in correlative placental gene expression (25 versus 438 genes, p < 0.0001; and Table S1). We conclude that exposure to maternal smoking is associated with neither a global nor indiscriminate change in placental DNA methylation but rather occurs at specific CpG dinucleotides, which deregulate a significant number of genes in the transcriptome.
An additional strength to our study arises from the employment of linear regression analysis to control for potential interactions. This was needed because, although maternal smoking is associated with a significant decrease in fetal birth weight and renders risk of SGA birth across maternal strata,Citation8,Citation12,Citation20,Citation28 large population-derived studies have also shown that female infants weigh on-average 120 to 200 grams less than males.Citation30 After Bonferroni correction, the variation of methylation level at 6 CpG sites in female newborn samples could be explained by smoking status to infant weight interactions, revealing that as few as 6 essential sites in the placental methylome are modified in association with maternal smoking to significantly influence birth weight. OthersCitation29 have similarly employed integrated computational and multivariate analysis approaches.
As further evidence of our attempt to be rigorous in our analyses, we employed two independent pathway tools (i.e., IPA and DAVID). Ingenuity Pathway Analysis (IPA) of the 438 differentially methylated genes among smokers reveals that the top canonical pathways include oxidative phosphorylation, mitochondrial dysfunction and HIF1α signaling with molecular enrichment along cell death, morphology and cell signaling signatures; we have independently confirmed that these pathways are functionally disrupted at the level of cellular physiology employing immunohistochemistry and in situ analysis (In press).Citation28
Our observed gene signature pathways are of likely biological and clinical significance. First, our data is consistent with our functional cellular analysis of placentas, demonstrating the significant increased presence of markers of oxidative damage among smokers, namely 8-OHdG and 4-HNE.Citation16,Citation24,Citation28 Second, HIF1α is a transcription factor that senses hypoxia to ultimately regulate transcription of these same pathways.Citation25 Given that chronic fetal hypoxia due to utero-placental insufficiency in tobacco-exposed fetuses has long been hypothesized as a potential underlying physiologic mechanism that plays a role in growth attenuation, our findings allow for the convergence of multiple lines of data.Citation8–Citation12,Citation24–Citation28
Employing these methodologies in a nested cohort design, we have completed a robust analysis and demonstrated that biologically relevant and statistically significant deregulation of placental methylation correlates with gene expression. Development of a comprehensive analysis workbench enabled integrated analysis of parallel Illumina-based tiling array-based placental methylome and transcriptome data to reveal that altered placental site-specific CpG methylation at as few as 6 sites along signature pathways may contribute to a significant reduction in infant birth weight among smokers. When considered in the context of the implications for the biology of the development and programming of disease, these studies suggest that a common perinatal exposure (such as maternal smoking) deregulates placental methylation, which correlates with meaningful alterations in gene expression. We speculate that our methodologies and observations will lay the groundwork for further interrogations into the role of epigenomic deregulation of common perinatal exposures, which in turn have the potential to profoundly impact the health across that same individual's lifespan.
Materials and Methods
Study population and sample collection.
Placental samples (n = 36) for this study were obtained from subjects selected from a well-described total cohort of 28 self-reported smokers alongside 53 non-smoking controls; this has been previously validated as an accurate measure of maternal tobacco exposure.Citation8 Of this cohort, 18 smokers and 18 non-smokers were employed in the discovery arrays and 9 smokers and 9 non-smokers comprised the validation cohort. The Institutional Review Board of Baylor College of Medicine and its affiliated institutions approved this study, and written informed consent was obtained from each participant at the time of enrollment. Data collected from each patient included age, ethnicity, height and weight, past obstetrical history, gestational age at delivery and potential maternal comorbidities. Data collected from the newborns included gender, Apgar scores, weight and length, and level of resuscitation interventions if any. Exclusion criteria included multiple gestation, known fetal anomalies and maternal hepatic, hypertensive or endocrine disorders. For the analysis reported herein, subjects were matched in a nested cohort design by virtue of maternal age (±1 year), race/ethnicity, BMI and gestational age (±1 week). Consistent with a nested cohort design, matching was performed prior to knowledge of the primary outcomes and without consideration of fetal factors (beyond gestational age) including fetal weight, length or neonatal outcome. In such a manner, an initial 36 matched subjects were analyzed with minimized potential for selection bias. This is as noted in .
In consideration of the potential for heterogeneity within any given individuals placenta, great care was taken to systematically and uniformly sample each subjects placenta. Briefly, for each subject we sampled a total of six placental sites in a uniform manner. This is accomplished by sampling at a designated 4 cm from the cord insertion in a graduated circumferential manner. For each subject, segments of each of these six segments comprise the source for the genomic DNA for that subject. Using this approach, we have not previously noted significant clonal distinctions with respect to site-specific DNA methylationCitation13 or clear distinctions of markers of oxidative stress by immunohistochemistry.Citation28 Placental specimens were collected immediately after delivery, flash frozen on dry ice and held at −80°C until use.
DNA and RNA extraction.
Genomic DNA was extracted from each subject's placental specimen(s) using the Puregene Kit (Qiagen, cat # 158667). Approximately 500 ng DNA per sample was bisulfite treated using the EZ DNA methylation kit (Zymo Research, cat # D5001), eluted in 10 µL sample buffer and immediately delivered to the core facility after elution. RNA was extracted from each sample using the Nucleospin II columns (Macherey Nagel, cat # 740609.250) eluted in 50 µL of RNase free buffer and snap frozen until submitted to the core.
Illumina bead array.
Bisulfite treated DNA was processed according to the manufacturers conditions and hybridized to the Infinium Human Methylation 27 bead chip (Illumina, cat # WG-311-2201), which is designed to cover over 27,000 CpG sites located within the proximal promoter region of over 14,000 consensus coding sequences (CCDS) genes throughout the genome. RNA was processed according to the manufacturer conditions and hybridized to the Human HG-12 Expression (Illumina, cat # BD-103-0204) array. The methylation status at each interrogated CpG site is estimated by measuring the intensity of a pair of probes (methylated and unmethylated). Illumina's Genome Studio program was used to analyze Bead Array data to assign site-specific DNA methylation beta value to each CpG site. The beta value for each CpG site is calculated by first subtracting the background signal intensity of negative controls from both the methylated and unmethylated signals, then the ratio of the methylated signal intensity to the sum of both methylated and unmethylated signals was taken as the beta value. Thus, the beta value is a continuous variable ranging between 0 and 1, with zero indicating no methylation detected and one indicating every copy of the site was methylated.
Gene expression analysis.
Human HG-12 Bead Chip data files were analyzed with Genome Studio gene expression module and R-based Bioconductor package to find out gene expression values. The raw files of Illumina HG-12 expression array were extracted and corrected by background subtraction in Genome Studio Module. The lumi module in Bioconductor package was used to normalize the expression value followed by quality control. Then the Limma package in Bioconductor was used to find out the differential expression genes. Limma adopts an empirical Bayes approach to estimate a standard error and has improved performance when an experiment has a limited number of samples. Since one of our goals was to investigate if differential expression of genes arises from methylation, a generous nominal p value cut-off of 0.05 was used to correlate gene expression with methylation intensity.
Expression and methylation analysis.
Probes from the Infinium 27K methylation array and the HG-12 expression array that interrogated the same genomic locus were determined based on the symbol annotation included in the array descriptions. The beta values or average signal values were averaged across multiple probes interrogating the same locus based on the symbol annotation. Pearson's correlation was calculated between the average beta and signal values using R and Excel.
Validation of differential expression and methylation.
In order to validate the expression array data, RNA was extracted from non-smoker (n = 9) and smoker (n = 9) placentas. RNA was used as a template to make cDNA (Invitrogen, 11752), and cDNA was used as a template for quantitative qPCR using commercially available TaqMan primer and probe sets from Applied Biosystems (Table S3). Fold change comparing smokers to non-smokers was calculated using the ΔΔCt method.Citation15 For validation of CpG methylation, DNA was extracted from non-smokers and smokers (n = 9/group), bisulfite treated, PCR amplified, cloned, purified and sequenced, as previously described in reference Citation13. The PCR primers used for validation flanked the specific CpG identified by the Illumina array (Table S4). Sequences were analyzed using BiQ Analyzer.
Canonical pathway analysis.
A dataset containing genes that showed a significant correlation between expression and DNA methylation was uploaded for Ingenuity Pathway Analysis (IPA). IPA determines the canonical pathways that were most significant to the data set. A ratio of number of molecules from the data set that mapped to the pathway compared with the total number of molecules in the canonical pathway is reported. Furthermore, Fisher's exact test was used to calculate a p value. For the functional analysis, IPA determines the biological functions most significant to the uploaded data set. Right-tailed Fisher's exact test was used to calculate a p value, determining the probability that each biological function and/or disease assigned to that data set is due to chance alone.
Linear regression analysis.
The beta value for each individual CpG site in the methylation array was modeled as a dependent variable in a multiple linear regression model, while smoking status, infant gender and infant weight were used as independent variables. Weight was categorized as either greater or less than 3,200 grams. The linear regression was performed for each CpG site using R. p values of infant gender association with beta value for each locus are shown in : the x axis indicates −log10 (rank of p value) and the y axis indicates −log10 (p value); a straight line would indicate the lack of association between methylation level and infant gender.Citation29 The Bonferroni correction for multiple comparisons on type I error rate of 0.05 gives a cut-off rate of p = 1.81 × 10−6,Citation29 which is used as genome-wide significance cut-off to discover significant locus. Similar analyses were done for samples stratified by infant gender, with beta value as dependent variable and smoking status, infant birth weight and smoking status by infant birth weight interaction as independent variables.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 Analysis of the placental transcriptome in association with in utero tobacco smoke exposure. (A) A heat map of the differentially expressed genes between smokers and non-smokers demonstrating multiple clustered associations. Human HG-12 Bead Chip data files were analyzed with Genome Studio gene expression module and R-based Bioconductor was employed in determining gene expression values. The samples are listed at the bottom of the heat map. “S” signifies smoker while “C” is non-smoker. (B) Array data was validated using qPCR. Fold change of smokers compared with non-smokers for the array was reported using the analysis package R. Fold change was calculated using the ΔΔCt method of the qPCR data comparing smokers to non-smokers.
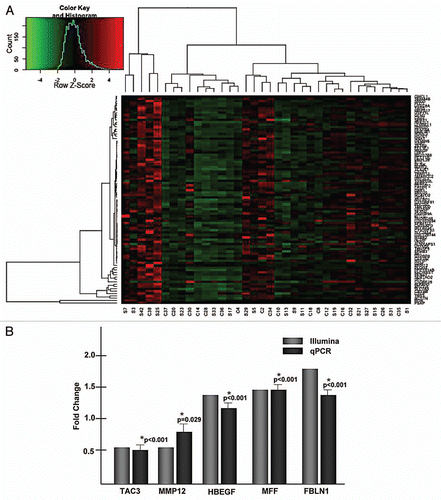
Figure 2 Genome wide DNA methylation is modestly altered in placenta from mothers who smoke. (A) A heat map generated from the differentially methylated CpG sites between smokers and non-smokers. Bisulfite treated DNA was processed according to the manufacturers conditions and hybridized to the Infinium Human Methylation27, which is designed to cover over 27,000 CpG sites located within the proximal promoter region of over 14,000 consensus coding sequences (CCDS) genes throughout the genome. The methylation status at each interrogated CpG site is estimated by measuring the intensity of a pair of probes (methylated and unmethylated) with beta values for each CpG represented as a continuous variable ranging between 0 and 1, with zero indicating no methylation detected and one indicating every copy of the site was methylated. The sample designations are listed at the bottom of the heat map. “S” signifies smoker while “C” is non-smoker. (B) Validation of the array employing bisulfite conversion, cloning and sequencing, as previously described in reference Citation3 and Citation13. A positive delta beta score indicates the smokers had a higher beta score than the non-smokers. A negative delta beta indicates the non-smokers had the higher beta score.
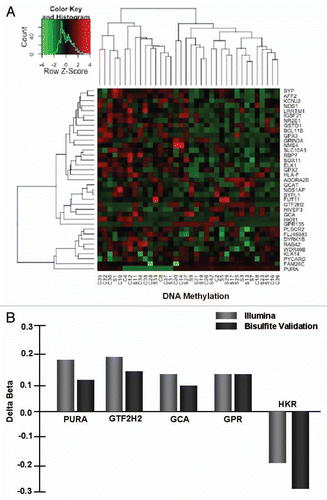
Figure 3 Deregulation of the placental methylome and transcriptome occurs in association with maternal smoking. The values for methylation (Average Beta) and gene expression (Average Signal) are plotted for each individual. Probes from the Infinium 27K methylation array and the HG-12 expression array that interrogated the same genomic locus were determined based on the symbol annotation included in the array descriptions. The beta values or average signal values were averaged across multiple probes interrogating the same locus based on the symbol annotation. Pearson's correlation was calculated between the average beta and signal values using R and Excel. As three examples of the 438 deregulated genes, significant correlation was observed for (A) ADA; (B) PTPN22 and; (C) LRFN3 methylation and gene expression by virtue of maternal smoking.
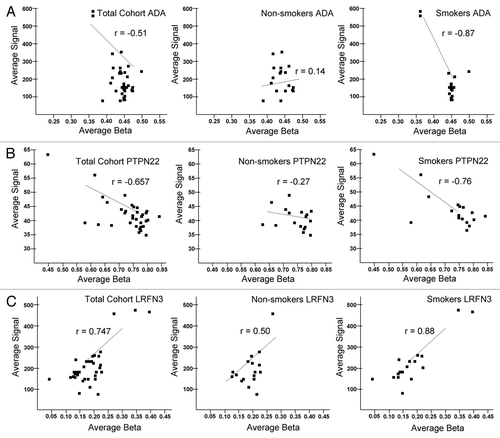
Figure 4 p values for the infant gender-methylation association for all CpG sites. Linear regression analysis was performed for each CpG site using R in an effort to control potential interactions among variables. The p values for infant gender association with methylation level are as depicted with the x axis indicating log10 (rank of p value) and the y axis indicating −log10 (p value).
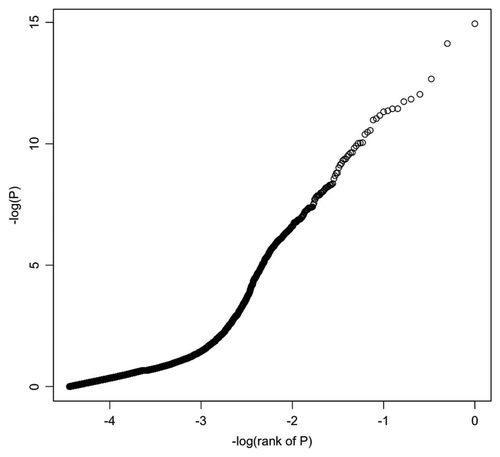
Table 1 Characteristics of the study population
Table 2 Validation of differentially expressed placental genes by virtue of maternal tobacco smoke exposure
Table 3 Validation of site-specific CpG differential placental methylation by virtue of maternal smoking
Table 4 Maternal smoking deregulates placental methylation that correlates with significant alterations in gene expression along signature pathways
Additional material
Download Zip (230.5 KB)Acknowledgments
Support for this work came from the NIH Director New Innovator Pioneer Award DP21DP2OD001500-01 (KAT) and REACH IRACDA K12 GM084897 (M.S.). We are grateful to members of the Aagaard and Hawkins laboratories for helpful discussion and Dr. John Belmont for assistance with the Genome Studio Software.
References
- Suter M, Abramovici A, Aagaard-Tillery K. Genetic and epigenetic influences associated with intrauterine growth restriction due to in utero tobacco exposure. Pediatr Endocrinol Rev 2010; 8:94 - 102
- Aagaard-Tillery K, Suter M, Harris A, Abramovici AJC. Epigenetics and reproduction and the developmental origins of health and disease. Animal Reproduction 2010; 7:103 - 116
- Suter M, Bocock P, Showalter L, Hu M, Shope C, McKnight R, et al. Epigenomics: maternal high-fat diet exposure in utero disrupts peripheral circadian gene expression in nonhuman primates. FASEB J 2011; 25:714 - 726
- Suter MA, Aagaard-Tillery KM. Environmental influences on epigenetic profiles. Semin Reprod Med 2009; 27:380 - 390
- Sood R, Zehnder JL, Druzin ML, Brown PO. Gene expression patterns in human placenta. Proc Natl Acad Sci USA 2006; 103:5478 - 5483
- Reamon-Buettner SM, Mutschler V, Borlak J. The next innovation cycle in toxicogenomics: environmental epigenetics. Mutat Res 2008; 659:158 - 165
- Maccani MA, Marsit CJ. Epigenetics in the placenta. Am J Reprod Immunol 2009; 62:78 - 89
- Aagaard-Tillery K, Spong CY, Thom E, Sibai B, Wendel G Jr, Wenstrom K, et al. Pharmacogenomics of maternal tobacco use: metabolic gene polymorphisms and risk of adverse pregnancy outcomes. Obstet Gynecol 2010; 115:568 - 577
- Simpson WJ. A preliminary report on cigarette smoking and the incidence of prematurity. Am J Obstet Gynecol 1957; 73:807 - 815
- Kyrklund-Blomberg NB, Cnattingius S. Preterm birth and maternal smoking: risks related to gestational age and onset of delivery. Am J Obstet Gynecol 1998; 179:1051 - 1055
- Hammoud AO, Bujold E, Sorokin Y, Schild C, Krapp M, Baumann P. Smoking in pregnancy revisited: findings from a large population-based study. Am J Obstet Gynecol 2005; 192:1856 - 1862
- Aagaard-Tillery KM, Porter TF, Lane RH, Varner MW, Lacoursiere DY. In utero tobacco exposure is associated with modified effects of maternal factors on fetal growth. Am J Obstet Gynecol 2008; 198:66
- Suter M, Abramovici A, Showalter L, Hu M, Shope CD, Varner M, et al. In utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism 2010; 59:1481 - 1490
- Bruchova H, Vasikova A, Merkerova M, Milcova A, Topinka J, Balascak I, et al. Effect of maternal tobacco smoke exposure on the placental transcriptome. Placenta 2010; 31:186 - 191
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402 - 408
- Zdravkoci T, Genbacev O, McMaster MT, Fisher SJ. The adverse effects of maternal smoking on the human placenta. Placenta 2005; 26:81 - 86
- Wang X, Zuckerman B, Pearson C, Kaufman G, Chen C, Wang G, et al. Maternal cigarette smoking, metabolic gene polymorphism and infant birth weight. JAMA 2002; 287:195 - 202
- Nukui T, Day RD, Sims CS, Ness RB, Romkes M. Maternal/newborn GSTT1 null genotype contributes to risk of preterm, low birthweight infants. Pharmacogenetics 2004; 14:569 - 576
- Tsai HJ, Liu X, Mestan K, Yu Y, Zhang S, Fang Y, et al. Maternal cigarette smoking, metabolic gene polymorphisms and preterm delivery: new insights on GxE interactions and pathogenic pathways. Hum Genet 2008; 123:359 - 369
- Brunnemann KD, Hoffmann D. Analytical studies on tobacco-specific N-nitrosamines in tobacco and tobacco smoke. Crit Rev Toxicol 1991; 21:235 - 240
- Huuskonen P, Storvik M, Reinisalo M, Honkakoski P, Rysa J, Hakkola J, et al. Microarray analysis of the global alterations in the gene expression in the placentas from cigarette-smoking mothers. Clin Pharmacol Ther 2008; 83:542 - 550
- Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med 2009; 180:462 - 467
- Guerrero-Preston R, Goldman LR, Brebi-Mieville P, Ili-Gangas C, Lebron C, Hernandez-Arroyo M, et al. Global DNA hypomethylation is associated with in utero exposure to cotinine and perfluorinated alkyl compounds. Epigenetics 5
- Aycicek A, Varma M, Ahmet K, Abdurrahim K, Erel O. Maternal active or passive smoking causes oxidative stress in placental tissue. Eur J Pediatr 2011; 170:645 - 651
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 40:294 - 309
- Haustein KO. Cigarette smoking, nicotine and pregnancy. Int J Clin Pharmacol Ther 1999; 37:417 - 427
- Halliday HL. Neonatal management and long-term sequelae. Best Pract Res Clin Obstet Gynaecol 2009; 23:871 - 880
- Sbrana E, Suter MA, Abramovici A, Wahkins HK, Moss JE, Patterson L, et al. Maternal tobacco use is associated with increased markers of oxidative stress in the placenta. Am J Obstet Gynecol In press
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 2011; 88:450 - 457
- Sahota DS, Kagan KO, Lau TK, Leung TY, Nicolaides KH. Customized birth weight: coefficients and validation of models in a UK population. Ultrasound Obstet Gynecol 2008; 32:884 - 889
- Aliyu MH, Lynch O, Wilson RE, Alio AP, Kristensen S, Marty PJ, Whiteman VE, Salihu HM. Association between tobacco use in pregnancy and placenta-associated syndromes: a population-based study. Arch Gynecol Obstet 2011; 283:729 - 734