 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
DNA methylation is essential in brain function and behavior; therefore, understanding the role of DNA methylation in brain-based disorders begins with the study of DNA methylation profiles in normal brain. Determining the patterns and scale of methylation conservation and alteration in an evolutionary context enables the design of focused but effective methylation studies of disease states. We applied an enzymatic-based approach, Methylation Mapping Analysis by Paired-end Sequencing (Methyl-MAPS), which utilizes second-generation sequencing technology to provide an unbiased representation of genome-wide DNA methylation profiles of human and mouse brains. In this large-scale study, we assayed CpG methylation in cerebral cortex of neurologically and psychiatrically normal human postmortem specimens, as well as mouse forebrain specimens. Cross-species human-mouse DNA methylation conservation analysis shows that DNA methylation is not correlated with sequence conservation. Instead, greater DNA methylation conservation is correlated with increasing CpG density. In addition to CpG density, these data show that genomic context is a critical factor in DNA methylation conservation and alteration signatures throughout mammalian brain evolution. We identify key genomic features that can be targeted for identification of epigenetic loci that may be developmentally and evolutionarily conserved and wherein aberrations in DNA methylation patterns can confer risk for disease.
Introduction
DNA methylation is an evolutionary ancient epigenetic mark found within most eukaryotic organisms including fungi, plants and animals.Citation1,Citation2 DNA methylation modification involves the addition of a methyl group to cytosine bases in a heritable fashion. Cytosine methylation can be categorized into CG, CHG and CHH methylation (where H refers to either A, C or T nucleotides). In animals, methylation primarily occurs at CG (CpG) dinucleotides, whereas in plants, methylation of cytosines is observed in all three contexts. Cytosine methylation is catalyzed by highly conserved DNA methyltransferases (Dnmts).Citation3,Citation4 As such, DNA methylation is an essential evolutionary process involved in many gene regulatory systems, including genomic imprinting, X-chromosome inactivation, transposon silencing and expression of endogenous genes.
Cross-species comparative epigenomic analyses have revealed intriguing trends in both the conserved and divergent features of DNA methylation in eukaryotic evolution. Cytosine methylation has been detected throughout the majority of repetitive sequences and transposons, but also within the body of protein coding genes. Within gene bodies, CpG methylation appears to be favored in exons over introns. Although the biological function of gene body methylation or mechanisms by which gene bodies are targeted by the methylation machinery are not well-understood, the preferential methylation of exons in plant and animal species appears to be an evolutionary conserved phenomena.Citation5 In mammalian genomes, DNA methylation occurs almost exclusively within CpG dinucleotides, though a quarter of DNA methylation is found in a non-CpG context in embryonic stem cells.Citation6-Citation9 While 70–80% of CpG sites are methylated, the remaining unmethylated CpG sites mostly occur in dense clusters referred to as CpG islands.Citation10,Citation11 CpG islands represent a large fraction of cis-regulatory sequences because they occupy the majority of gene promoters or alternative promoters,Citation12-Citation14 with non-promoter CpG islands functioning as distal regulators (e.g., insulators and enhancers).Citation15 On the other hand, methylated cytosine nucleotides in vertebrates have undergone C-to-T transitions over evolutionary time, which results in a markedly different CpG distribution as compared to genomes of invertebrates that do not have DNA methylation.
The establishment of DNA methylation patterns is correlated with CpG content, and appears to have little to do with sequence conservation from an evolutionary perspective. However, it remains unclear how cross-species DNA methylation conservation and alteration vary in the context of sequence conservation as well as genomic features. In the present study, we show that cross-species genome-wide DNA methylation conservation patterns in human and mouse are only weakly correlated with sequence conservation. Rather, DNA methylation conservation is strongly correlated with CpG content. Genomic regions, such as CpG islands with high CpG density show greater methylation conservation and, by contrast, regions with intermediate-to-low CpG density show substantially less DNA methylation conservation. The data presented here reveal intrinsic patterns of DNA methylation conservation and alteration in different genomic compartments in an evolutionary context. These data also provide insight into how these genome-wide patterns of DNA methylation can guide the design of methylation profiling studies in human disease, especially neuropsychiatric diseases, since they represent the largest and most comprehensive DNA methylation profiles in human and mouse brains.
Results
In this large-scale DNA methylation profiling study, we generated 657,424,756 million sequence reads, mapping DNA methylation for greater than 80% of CpG sites within human and mouse brains. These data are from 10 human cortical specimens (6 ventral prefrontal and 4 auditory), and 5 mouse forebrain specimens (129S6/SvEv strain). Human samples were selected from our postmortem brain collection with extensive neuropathological and psychopathological data, as well as brain toxicology reports, all confirming the absence of neuropsychiatric disorders, pathological lesions and psychoactive substances.Citation16 DNA methylation patterns did not differ significantly by age, sex, pH or postmortem interval within specimens examined. DNA methylation data were generated using the Methylation Mapping Analysis by Paired-end Sequencing (Methyl-MAPS). The Methyl-MAPS method is based on our previously reported enzymatic assay,Citation17 which has been extended to take advantage of vast improvements in sequencing throughput of second-generation sequencing technologyCitation18 (see Supplementary materials on details of experimental and analytical procedures). Methyl-MAPS data were independently validated by Illumina Infinium HumanMethylation27 BeadChip technology,Citation19 showing strong correlation (Pearson Correlation Coefficient r = 0.8) between the two experimental methods across 16,290 overlapping CpG sites, thus demonstrating that the Methyl-MAPS method produces highly robust and reliable DNA methylation data genome-wide. Although our methylation data provide >80% coverage of CpG sites in the genome, examination of methylation levels from biological replicates revealed that genomic coverage of 8x or greater produced robust estimates of methylation states (shown in Supplementary materials). Hence, all analyses were based on these high coverage CpG sites. Using this coverage constraint, we were able to map 10,262,160 (36%) CpG sites in common across all human cortical samples and 19,326,334 (91%) CpG sites within the pooled inbred mouse samples, representing the largest whole-genome methylation profiling effort in primary tissue to date.
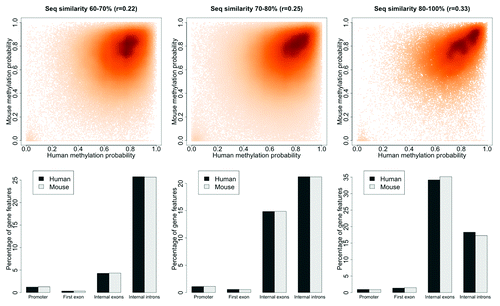
Cross-species DNA methylation conservation. Data from whole genome DNA methylation profiling of human and mouse brains allowed us to determine whether there is a relationship between sequence conservation and DNA methylation. We examined methylation patterns within evolutionary conserved regions (ECRs),Citation20 positing that conservations at these regions likely have a biologically significant function. Human-mouse cross-species methylation comparison of such regions showed that methylation correlates weakly with sequence conservation (r = 0.25; Fig. S1). Detailed examination of ECRs with varying levels of sequence similarity (60–100%) also showed a weak correlation within genic compartments including promoters, exons and introns (). These data were comparable with correlations from random selections of syntenic regions from human-mouse pair-wise alignments (r = 0.15). Additionally, DNA methylation patterns were compared within promoters of human-mouse orthologous genes since promoters are regions where DNA methylation confers a functional effect in the repression of gene expression. Here also, we observed a weak correlation in human and mouse DNA methylation patterns within 8,956 orthologous gene promoters (r = 0.16; Fig. S2).
Figure 1. DNA methylation conservation is weakly correlated with sequence similarity. Human-mouse ECRs were split into three groups based on sequence similarity, with sequence similarity >60 to ≤70%, >70 to ≤80% and >80%. Top part: human-mouse DNA methylation patterns for ranges of sequence similarity, 60–70%, 70–80% and >80% with corresponding human-mouse correlation coefficients (r), respectively. Bottom part: percentage of gene features in both human and mouse within the three compartments.
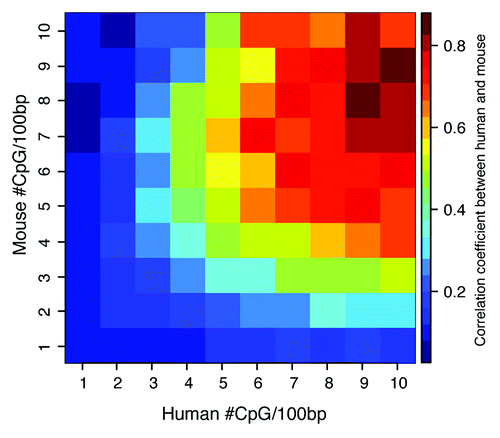
If sequence conservation is not the driving force underlying DNA methylation conservation, then what is? This prompted us to ask whether evolutionary conservation of DNA methylation is driven by CpG density rather than sequence homology. Since CpG dinucleotides are the targets of DNA methyltransferase enzymes with the propensity to methylate CpG clusters,Citation21,Citation22 it is highly plausible that CpG density may be the key factor. To allow for direct comparison of results, we used the sequences from the human and mouse ECRs to examine DNA methylation profiles within distinct ranges of CpG density and found that, indeed, DNA methylation conservation increases with increasing CpG content (). Extending our analyses beyond the ECR sequences to encompass all Human-Mouse syntenic regions, we still detected similar trends (Fig. S3). The patterns of DNA methylation are quite striking, becoming increasingly polarized towards extreme hypo- and hyper-methylated states with increased CpG density ( and S3 top part). The density ranges represented in these figures denoted as poor, intermediate and rich were selected arbitrarily for illustrative purposes. While we find that there is a greater accumulation of internal introns in the CpG-poor compared to CpG-rich compartments, gene promoters and first exons appear to be depleted in CpG-poor compartments and overrepresented in CpG-rich compartments ( and S3 bottom part). Quantitative analysis of DNA methylation profiles of Human-Mouse syntenic regions also showed consistently that DNA methylation conservation is highly correlated with CpG content (r = 0.88; ) for regions with CpG density of 5-CpG dinucleotides or greater, while by comparison, the genome average is approximately 1 CpG per 100 base pairs (bp). To ensure that these comparative analyses are not confounded by possible enrichment of CpG dinucleotides within evolutionary conserved regions, we determined the average CpG density within the ECRs (Fig. S4). The extent of sequence similarity of ECRs ranged from intermediate (60%) to highly conserved (100%), with average CpG density of 1-to-2 CpGs per 100 bp (Fig. S4). This demonstrated that, in human and mouse genomes, CpG density is independent of cross-species sequence conservation and, importantly, established that the correlation between DNA methylation and CpG content is highly robust. These observations prompted us to re-examine the promoters of orthologous genes (Fig. S2) in context of CpG density.
Figure 2. DNA methylation conservation is correlated with CpG dinucleotide density. Human-mouse ECRs were split into three groups based on CpG density of each region. The three parts correspond to poor, intermediate and rich CpG densities, defined as, Poor: ≤3, Intermediate: >3 to ≤5 and Rich: >5 CpG sites. Top part: human-mouse DNA methylation patterns for poor, intermediate, and rich CpG densities with corresponding correlation coefficients (r), respectively. Bottom part: percentage of gene features in both human and mouse within the three compartments.
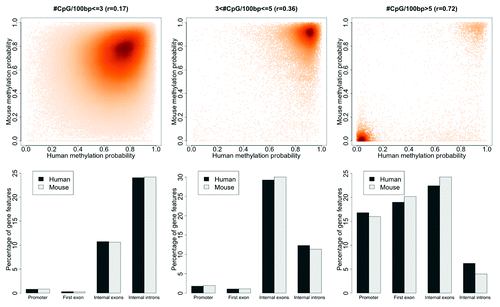
Figure 3. Heatmap of human-mouse DNA methylation conservation with respect to CpG density. Human-mouse pairwise alignments were obtained from UCSC Genome Bioinformatics Browser, with DNA methylation correlation computed for ranges of CpG density depicted as number of CpG sites within 100 bp windows of aligned sequences. The observed correlation coefficients varied in range from 0.02 to 0.88, demarcating regions of ≥5 CpG sites with high methylation correlation.
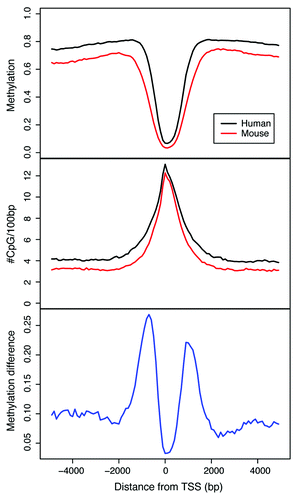
Within CpG-rich gene promoters, DNA methylation patterns are highly conserved, proximal to transcription start sites (TSS) (). The extent of DNA methylation conservation appears to be confined to [-200 bp to +500 bp] around the TSS. This pattern tracks with CpG density revealing that, as expected, these CpG-rich promoters are hypo-methylated and are ultra-conserved across species (). Remarkably, the shores of these CpG-rich regions show elevated DNA methylation variability that extends up to 2 KB upstream and downstream of the TSS ( bottom part). These shores demarcate transitions in CpG density. A salient characteristic of these shores is that they retain an intermediate CpG density cross-species ( and middle part), which might be important for the establishment of tissue-specificCitation23 and perhaps species-specific methylation profiles. Beyond these shores, DNA methylation patterns resume basal levels ( and top part) and do not appear to vary substantially cross-species ( and bottom part). Furthermore, gene ontology analysis identified seven significant functional groups with genes that appear to have species-specific DNA methylation profiles (Supplementary materials). Taken together, these results underscore the significance of CpG density in sustained DNA methylation conservation and alteration throughout evolution as a fundamental component of mammalian epigenetic programming.
Figure 4. Methylation patterns at promoter regions for human/mouse orthologous genes. A total of 9,926 orthologous genes with CpG-rich promoters were examined in the analysis. Regions of 5 kb-upstream and 5 kb-downstream from TSS were split into 100 bp non-overlapping windows, where each window is represented by average methylation across all genes. (Top part) CpG methylation level, (Middle part) CpG density and (Bottom part) CpG methylation difference in human and mouse.
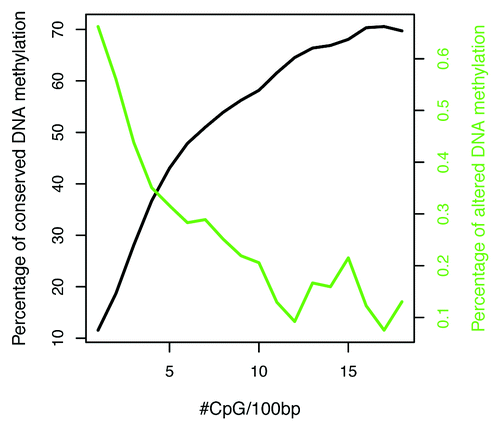
Intra-species DNA methylation conservation and alteration in cerebral cortex. In human cortex, patterns of DNA methylation conservation and alteration are also highly dependent on CpG density. In comparing DNA methylation signatures within prefrontal cortex (PFC) and auditory cortex (AC) across individuals, we found that DNA methylation patterns are highly conserved among 26% of CpG sites and only significantly altered in 1% of CpG sites, with the remaining having variable CpG methylation (See definition of conserved, altered and variable methylation in Materials and Methods). These conserved, altered and variable methylated domains were represented within all genomic features (Fig. S5). Further examination of the conserved and altered methylation distributions relative to CpG density revealed that, in human cortex, DNA methylation is conserved in CpG-rich regions and altered in CpG-depleted regions of the genome (). The CpG density transition associated with conserved methylation is similar to what we observed cross-species (approximately 5-CpG sites within 100 bp region; ). This might demarcate a potential genomic signature for evolutionary conservation of DNA methylation and maintenance.
Figure 5. Distribution of conserved and altered DNA methylation patterns in prefrontal and auditory cortices. DNA methylation conservation increases with increasing CpG density, where as DNA methylation alteration increases with decreasing CpG density.
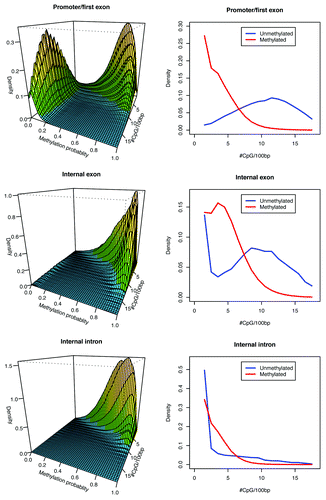
Investigation of conserved and altered CpG sites within different genomic compartments showed that, in addition to CpG density, genomic context determines DNA methylation profiles. Adopting a gene-centric view, we found that across different gene features, DNA methylation patterns vary markedly relative to CpG density (). Promoters and first exons demonstrate the expected pattern of methylation, showing less methylation in CpG-dense regions associated with CpG islands and more methylation in CpG-poor regions. The internal exons and introns tend to be more methylated with greater CpG depletion as compared to promoters and first exons. Thus, the difference in methylation patterns between promoters/first exons and internal exons/introns correlates with different patterns of CpG-density distributions. The transition between unmethylated and methylated domains appears to have similar CpG transition density as observed previously ( and ), which is consistent with our genomic signature hypothesis. These patterns are also evident in the mouse genome (Fig. S6).
Figure 6. Human cortex patterns of DNA methylation and CpG density for CpG sites within genic compartments. Left part shows that the extent of CpG methylation depends on CpG density and genomic context. Right part shows the shift in transition from unmethylated to methylated states relative to CpG density. CpG sites with ≤0.2 and ≥0.8 methylation were assigned to the unmethylated and methylated groups, respectively.
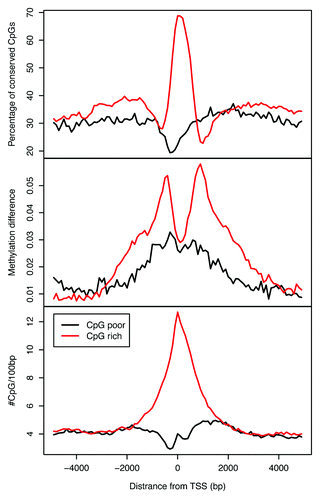
We next focused our attention on the promoter and TSS regions, considering whether differences in DNA methylation can be detected when comparing two human cortical regions for two distinct classes of CpG-rich and -poor promoters. We addressed this question in the context of DNA methylation conservation. We found that for CpG-rich promoters, DNA methylation patterns are highly conserved within approximately -200 bp and +500 bp of the TSS (). Beyond this range, we detected elevated methylation differences for up to ±2 kb, akin to our previous observations with the “shores” (). Although the magnitude of DNA methylation differences is relatively small, the patterns are highly consistent.
Figure 7. Comparative analysis of patterns of DNA methylation conservation at the TSS of refSeq genes within human prefrontal and auditory cortices. The top part shows the percentage of conserved CpGs in CpG-poor (black) and CpG-rich (red) promoters; the middle part shows methylation difference between prefrontal and auditory cortices for CpG-poor and -rich promoters; the bottom part illustrates CpG density in the two types of promoters of refSeq genes.
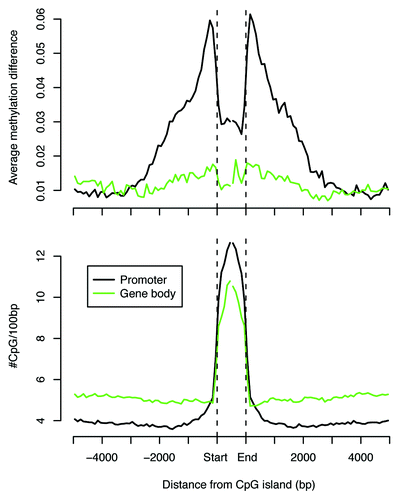
We also examined the DNA methylation patterns at shores of CpG islands. In the two cortical regions, we examined gene promoter-associated CpG island shores, as well as CpG island shores contained within gene bodies. We found that promoter-associated islands show differential methylation patterns at the shores (), consistent with our observations at the promoter TSS region (). We also examined the patterns of CpG island shore methylation within gene bodies and found that the two cortical regions did not differ in their methylation patterns at these shores (). This is perhaps not unexpected, since gene body CpG islands tend to be highly methylated as part of the evolutionary conserved feature of gene body methylation.Citation5 Indeed, the average length of gene body islands is half the length of those in gene promoters.Citation18 This is attributed to cytosine methylation resulting in increased C-to-T transitions after deamination of methylated cytosines and, hence, depletion of CpG sites from gene-body CpG islands. However, a small fraction of gene-body CpG islands remain unmethylated within each cortical region (Fig. S7). We also found that a small proportion of promoter-associated CpG islands are methylated. Examination of these CpG island shores revealed that unmethylated CpG islands within promoters and gene bodies show striking similarity in their shores, in that they exhibit increased methylation differences between PFC and AC in the immediate shores of the CpG islands (Fig. S8 top part). In contrast, the methylated CpG islands show no such differences at the shores (Fig. S8 middle part). These findings indicate that although unmethylated CpG islands within gene bodies may be few in number, they still may harbor regulatory regions with the potential for tissue-specific gene expression regulation.Citation24 Taken together, these results underscore the importance of CpG content and context in patterns of DNA methylation conservation and alteration in mammalian brain evolution.
Figure 8. Methylation differences between human prefrontal and auditory cortices for CpG islands in promoters and gene bodies. The top part shows methylation differences in promoter (black) and gene-body (green) CpG islands; the bottom part shows CpG density for all annotated islands in promoters and gene bodies.
Discussion
The data represent the first comprehensive map of the DNA methylation landscape of the human cortex to our knowledge, substantially expanding on previous work on a part of 1,500 CpG sites.Citation25 In this study, we have demonstrated that CpG methylation patterns have a distinct evolutionary signature that is both content- and context-dependent. We find that in the brain, CpG methylation is conserved in CpG dense regions of the genome, and such methylation conservation is independent of sequence conservation. The conservation of DNA methylation extends to both unmethylated and methylated compartments of the genome. On the other hand, regions with intermediate-to-low CpG density show substantially less DNA methylation conservation. These findings are supported by previous observations, in which genomic regions with relatively low CpG density, as CpG island shores, show greater DNA methylation variability across multiple tissue types.Citation23 Furthermore, our study indicates that CpG island shores may harbor regulatory elements that, in addition to determining tissue- and species-specific methylation profiles, may reveal DNA methylation difference specific to human cortical specialization and function. It is possible that the extent of DNA methylation differences at the shores might be a reflection of functional and evolutionary divergence. Also, the genomic environment plays a critical role in defining DNA methylation patterning throughout the mammalian genome. Within promoter CpG islands, the CpG depleted shores may contain cis-acting regulatory elements with important roles in transcriptional regulation. Additionally, gene promoters with low CpG density also show high methylation variability and, correspondingly, tissue-specific methylation and gene expression. These methylation signatures provide important insight into the role of DNA methylation in brain development and disease.
Genomic regions, like CpG islands with conserved DNA methylation signatures cross-species, may be important in basic cellular function and development of brain neural circuitry. Although conserved in the adult brain, DNA methylation patterns at these regions may be highly dynamic during neurodevelopment. Differential DNA methylation patterns have been well established in imprinted loci during early development,Citation26,Citation27 and many loss of imprinting disorders, including Prader-Wili and Angelman Syndromes, are associated with significant neurological abnormalities, indicating the importance of proper DNA methylation patterning in neural development and function.Citation28 It is likely that methylation changes at non-imprinted loci can also play an important role in neuronal development and differentiation. One can speculate that aberrant changes in DNA methylation in epigenetically conserved regions would result in drastic phenotypic abnormalities during early stages of development in a manner similar to genetic mutations in some cases. Hence, it is plausible to posit that DNA methylation changes during key neurodevelopmental trajectories may lead to such severe early-onset neurodevelopmental disorders as autism and childhood schizophrenia. Autism is an early-onset disorder typically diagnosed before the age of 3 and is accompanied by significantly altered neurodevelopment. Individuals with the disorder exhibit global cerebral gray matter hyperplasia in the first two years of lifeCitation29 and larger frontal and temporal gray matter volumes by four years of age, followed by a slower rate of growth in these regions by seven years.Citation30,Citation31 Childhood-onset schizophrenia, with a mean age of onset around ten years, is associated with striking parietal gray matter loss, which progresses anteriorly during adolescence.Citation32 Alterations in DNA methylation patterns that can influence the degree or timing of basic brain maturational patterns may at least partially underlie these neurodevelopmental disorders and are active areas of current research. Based on our study of brain methylation profiles, we hypothesize that variation in methylation patterns at evolutionary conserved CpG-rich genomic regions will involve loci where alterations in DNA methylation may lead to neurodevelopmental disorders, and such regions should be the focus of investigations in elucidating the developmental origins of such disorders.
While CpG-rich genomic regions with evolutionary conserved DNA methylation patterns may be essential in normal brain development and function, they might not be the likely loci for DNA methylation abnormalities associated with adult onset neuropsychiatric disorders, which may result from accumulating environmental insults during the lifespan. Unlike dramatic methylation changes that are typically observed in most cancers, DNA methylation changes for such disorders are likely to be subtle. Thus, we speculate that changes in genomic regions with intermediate or poor CpG density, as in CpG island shores, are likely the targets. In contrast to CpG islands that are generally protected,Citation33,Citation34 CpG dinucleotides at these regions are most likely targets of epigenetic modifications that are induced by accumulating exposures to environmental stressors. DNA methylation modifications at these regions may be the consequence of the neuropathology associated with the disease, or the consequence of environmental stressors that increase disease risk. For instance, adult-onset schizophrenia (the more typical form) is more strongly associated with deficits in later-maturing temporal and frontal regions,Citation35-Citation37 and is associated with selective abnormalities of the heteromodal regions.Citation38 In major depression, decreased volumes of cortical and subcortical regions have been reported. Patients with major depressive disorder show reduced gray matter concentration in the left inferior temporal cortex, the right orbitofrontal and the dorsolateral prefrontal cortex.Citation39 Such brain abnormalities in schizophrenia and depression may, in addition to genetic factors, be due to epigenetic factors that undergo alterations in response to the changes in the environment. In disease studies of schizophrenia and depression in postmortem samples, it is not possible to separate the influence of genetics and environment on disease neuropathology and associated epigenetic alterations. However, epigenetic studies provide crucially important insight on regulatory signatures that may be aberrant in these disorders.
Discovery of DNA methylation alterations associated with neurodevelopmental and neuropsychiatric disorders will depend on CpG content and context in the genome. It is critical to recognize the potential role that these factors play in brain DNA methylation signatures, and to profile these methylation patterns within relevant brain regions. The comparison between human cerebral cortex and mouse forebrain in the present study shows that cross-species methylation conservation is correlated with CpG density. However, DNA methylation alterations observed at CpG island shores between human and mouse may be attributed to DNA methylation differences in different brain regions since anatomically the brain regions used in the present study may not be precisely homologous across the human and mouse species.
This was largely due to practical limitations associated with the small size of mouse cortex and consequently the availability of sufficient quantities of brain tissue to perform the epigenetic experiments. Furthermore, in the brain, DNA methylation patterns are region specificCitation25,Citation40 and cell-specific. Though this study examines DNA methylation patterns within specific cortical regions, it does not capture the DNA methylation complexity within specific cell populations. This is partly due to limitations in current approaches, which requires relatively large amounts of starting DNA to perform such whole-genome DNA methylation profiling. However, with improvements in methylation sequencing technology, interrogation of cell-specific epigenetic profiles across multiple samples will be feasible in the near future. Hence, depending on the nature of the brain-based disorder, specific brain regions, cell populations and genomic regions (i.e., conserved or altered regions) may be targeted for epigenetic studies. This work highlights the evolutionary signatures of DNA methylation patterning in the brain, which will both inform our understanding of DNA methylation programming in mammalian genomes and offer insight into study-design considerations for future epigenetic studies of neurodevelopmental and neuropsychiatric disorders.
Materials and Methods
Ethics statement.
All human and rodent procedures were approved by the Institutional Review Boards of the New York State Psychiatric Institute/Columbia University Department of Psychiatry and the School of Medicine, University “Ss. Cyril & Methodius,” or by the Institutional Animal Care and Use Committee, respectively.
Samples and subjects.
Human brain specimens were obtained from the New York State Psychiatric Institute (NYSPI) and the Macedonia brain collection. Normal human tissue specimens were obtained with institutional review board approval and anonymous individual identifiers. A total of 10 cortical specimens were dissected frozen, including six specimens from the rostral portion of the right orbital gyrus, and four specimens from the right primary auditory cortex. For three subjects, tissue specimens from both prefrontal and auditory cortices were collected. Samples consisted of 4 males, average age 51 ± 5 years, and 3 females, average age 45 ± 5. Brain pH ranged from 6.3 to 6.7 with average postmortem interval (PMI) of 9.4 ± 5.1 hours. The subjects had died suddenly and were autopsied in the Institute for Forensic Medicine at the School of Medicine, University “Ss. Cyril & Methodius,” Skopje, Macedonia. Cases were chosen that were without psychopathology or history of psychoactive drugs (as determined by psychological autopsy interviews with their survivors), without significant abnormalities on neuropathological examination, and with negative screening of brain and body fluids for psychoactive drugs, including therapeutic levels in brain. The right cerebral hemisphere was sliced coronally at intervals of 2–4 cm. The slices were rapidly frozen in Freon 134a (1,1,1,2-tetrafluoroethane) and stored at -80°C until used. For the current study, slices were warmed to -20°C, and cortical samples of ~200–500 mg were cut, with a scalpel chilled in dry ice, from the rostral portion of the orbital gyrus (BA47), taking care to avoid visible white matter. Primary auditory cortex (BA41) was similarly dissected from the caudal portion of Heschl’s gyrus.Citation41 Five mouse brains (129S6/SvEv inbred strain from Taconic) were collected from 6-month-old male mice and samples were stored at -80°C until further processing. Genomic DNA was isolated from the entire left cerebral hemisphere with the cerebellum removed.
Methyl-MAPS procedure.
Brain DNA from human and mouse was fractionated into methylated and unmethylated compartments. Paired-end libraries were constructed, subsequently sequenced, and then mapped onto the human genome. DNA fractionation and library preparation methods have been previously described in reference Citation18 (and detailed in Supplementary materials). Briefly the Methyl-MAPS procedure is described as follows. Seven micrograms of DNA is digested with McrBC and RE in parallel. McrBC endonuclease generates the unmethylated compartment and is able to interrogate the methylation state of more than 74% of the CpG sites in the genome. The methylated compartment is generated by digestion with a part of all known methylation-sensitive tetranucleotide restriction enzymes termed RE (HpaII, HhaI, AciI, BstUI and HpyCH4IV), each of which cuts at a specific 4-bp sequence only if the CpG in the recognition site is unmethylated. By using such a cocktail, sequence-specific biases of the enzyme recognition site are minimized, and we are able to interrogate the methylation state of 38% of CpG sites genome-wide. The strength of this approach is that each strategy augments the other and combined, they provide greater overall coverage, permitting the assessment of >80% of all CpG sites genome-wide. Sequencing libraries of fragments greater than 700 bp in size were constructed utilizing the unique digestion properties of EcoP15I. Deep sequencing of digested sequence fragments was performed on the SOLiD sequencing platform from Applied Biosystems.Citation42 Mapping of paired-end sequenced fragments were performed with the SOLiD software analysis package, and all data have been deposited in Gene Expression Omnibus (GEO Accession GSE32647).
Methyl-MAPS estimation of methylation state.
DNA methylation state estimation was carried out by the Methyl-Analyzer software package.Citation43 In brief, the methylation probability of a CpG dinucleotide was estimated by the genomic coverage of the methylated and unmethylated sequence fragments from the RE and McrBC library, respectively. The RE fragments contribute to the methylation coverage and the McrBC fragments contribute to the unmethylated coverage. For each CpG, we defined two variables to represent coverage corresponding to methylated coverage (n1 for RE fragments) and unmethylated coverage (n2 for McrBC fragments). To correct intra-individual sampling bias between the methylated and unmethylated compartments, we estimated the ratio (⌊) of sampling probabilities for the McrBC and RE library with
where is the global methylation level estimated experimentally using a highly reliable method, known as the LUminometric Methylation Assay (LUMA).Citation44 Therefore, the methylation probability
of each CpG was computed using the following equation
Methyl-MAPS data analysis. Only CpGs with genomic coverage of 8x or greater were used in the analysis for both the human and mouse data. The average methylation probability for each CpG in the human genome was used to form the representative methylation profile of human. The representative methylation profile of mouse was generated based on pooled reads from the five genetically identical mice. Similarly, representative methylation profiles of human prefrontal cortex and auditory cortex were generated using average methylation of samples from the two regions, respectively. To characterize methylation patterns between the two human brain regions, we defined (1) DNA methylation conservation, with CpG i methylation being where the sample j and k correspond to the maximum and minimum observed methylation levels respectively at CpG i; (2) DNA methylation alteration, with CpG i methylation being
, IQRi,a ≤ 0.2 and IQRi,p ≤ 0.2 where
and
correspond to average methylation in auditory cortex (a) and prefrontal cortex (p), respectively, and IQRi,a and IQRi,p correspond to the interquartile range of auditory cortex and prefrontal cortex, respectively; (3) variable methylation refer to CpG sites with either
, or
, or IQRi,f > 0.2, or IQRi,o > 0.2.
Genomic feature annotations were downloaded from the UCSC Bioinformatics website (genome.ucsc.edu), including CpG islands and RefSeq gene annotations. All annotations and methylation data were indexed by CpG site and were stored in a MySQL database for use in subsequent analyses. CpG dinucleotides were overlapped with the following mutually exclusive genomic features, i.e., promoters, first exons, first introns, internal exons, internal introns and last exons. RefSeq gene annotationsCitation45 were based on the Human Genome NCBI Build 36 and the Mouse Genome NCBI Build 37. Only genes with complete start and end coding sequences were used in our analysis. Orthologous gene annotation for human and mouse was obtained from the HomoloGene database (www.ncbi.nlm.nih.gov/homologene) and formed a subset of the RefSeq genes. For promoter analyses, promoter regions referred to 1 KB upstream of the TSS. Classification of CpG-poor and CpG-rich promoters was determined by observed CpG density within ±500 bp centered at TSS of RefSeq genes. Both human and mouse exhibited expected bimodal distributions for CpG density. These data were used to determine cutoff values for CpG-poor and CpG-rich promoters with <0.07 and >0.07 CpG densities, respectively. Here CpG density is defined as (2x #CpG sites)/fragment length. For ease of representation, we also report the number of CpG sites within a 100 bp sequence fragment to describe CpG density. Finally, evolutionary conserved regions, ECRsCitation20 were used to estimate correlations between DNA methylation conservation and sequence similarity. ECRs are sequence fragments that are greater than 70 bp in length with greater than 60% sequence identity between human and mouse. For genome-wide sequence alignments, we use pair-wise human-mouse alignments.Citation46
| Abbreviations: | ||
| AC | = | auditory cortex |
| ECR | = | evolutionary conserved region |
| Methyl-Maps | = | methylation mapping analysis by paired-end sequencing |
| PFC | = | prefrontal cortex |
| TSS | = | transcription start site |
Additional material
Download Zip (2.1 MB)Acknowledgments
We like to thank Dr. J. John Mann for his effort in conducting psychological autopsies of the human samples and his valuable comments in preparation of the manuscript. Also, Dr. John Smiley for his efforts in dissection of auditory cortex specimens.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Financial Support
This work was supported by grants from the National Institute of Mental Health (MH048514) and the National Human Genome Research institute (HG002915). A.H.O. was supported by an NRSA F30 fellowship from the NIMH (MH085471). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Note
Supplemental material can be found at: www.landesbioscience.com/journals/epigenetics/article/17876
References
- Chan SWL, Henderson IR, Jacobsen SE. Gardening the genome: DNA methylation in Arabidopsis thaliana.. Nat Rev Genet 2005; 6:351 - 60
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 2010; 11:204 - 20
- Cheng XD, Blumenthal RM. Mammalian DNA methyltransferases: A structural perspective. Structure 2008; 16:341 - 50
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem 2005; 74:481 - 514
- Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA 2010; 107:8689 - 94
- Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 1982; 10:2709 - 21
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315 - 22
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA 2000; 97:5237 - 42
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10:295 - 304
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008; 9:465 - 76
- Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci USA 1993; 90:11995 - 9
- Bajic VB, Tan SL, Christoffels A, Schonbach C, Lipovich L, Yang L, et al. Mice and men: Their promoter properties. PLoS Genet 2006; 2:614 - 26
- Ioshikhes IP, Zhang MQ. Large-scale human promoter mapping using CpG islands. Nat Genet 2000; 26:61 - 3
- Tanay A, O’Donnell AH, Damelin M, Bestor TH. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc Natl Acad Sci USA 2007; 104:5521 - 6
- Kelly MP, Johnson CT, Govern JM. Recognition memory test: validity in diffuse traumatic brain injury. Appl Neuropsychol 1996; 3:147 - 54
- Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, et al. Large-scale structure of genomic methylation patterns. Genome Res 2006; 16:157 - 63
- Edwards JR, O’Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res 2010; 20:972 - 80
- Bibikova M, Zhou L. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics 2009; 1:177 - 200
- Loots G, Ovcharenko I. ECRbase: database of evolutionary conserved regions, promoters and transcription factor binding sites in vertebrate genomes. Bioinformatics 2007; 23:122 - 4
- Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng XD. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007; 449:248 - 13
- Glass JL, Fazzari MJ, Ferguson-Smith AC, Greally JM. CG dinucleotide periodicities recognized by the Dnmt3a-Dnmt3L complex are distinctive at retroelements and imprinted domains. Mamm Genome 2009; 20:633 - 43
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 86
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010; 466:253 - 7
- Ladd-Acosta C, Pevsner J, Sabunciyan S, Yolken RH, Webster MJ, Dinkins T, et al. DNA methylation signatures within the human brain. Am J Hum Genet 2007; 81:1304 - 15
- Dulac C. Brain function and chromatin plasticity. Nature 2010; 465:728 - 35
- Wilkinson LS, Davies W, Isles AR. Genomic imprinting effects on brain development and function. Nat Rev Neurosci 2007; 8:832 - 43
- Robertson KD. DNA methylation and human disease. Nat Rev Genet 2005; 6:597 - 610
- Courchesne E, Carper R, Akshoomoff N. Evidence of brain overgrowth in the first year of life in autism. JAMA 2003; 290:337 - 44
- Carper RA, Moses P, Tigue ZD, Courchesne E. Cerebral lobes in autism: early hyperplasia and abnormal age effects. Neuroimage 2002; 16:1038 - 51
- Saitoh O, Courchesne E. Magnetic resonance imaging study of the brain in autism. Psychiatry Clin Neurosci 1998; 52:219 - 22
- Thompson PM, Vidal C, Giedd JN, Gochman P, Blumenthal J, Nicolson R, et al. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc Natl Acad Sci USA 2001; 98:11650 - 5
- Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 2010; 464:1082 - 6
- Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res 2010; 38:4246 - 53
- DeLisi LE, Stritzke P, Riordan H, Holan V, Boccio A, Kushner M, et al. The timing of brain morphological changes in schizophrenia and their relationship to clinical outcome. Biol Psychiatry 1992; 31:241 - 54
- Gur RE, Cowell P, Turetsky BI, Gallacher F, Cannon T, Bilker W, et al. A follow-up magnetic resonance imaging study of schizophrenia. Relationship of neuroanatomical changes to clinical and neurobehavioral measures. Arch Gen Psychiatry 1998; 55:145 - 52
- Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res 2001; 49:1 - 52
- Buchanan RW, Francis A, Arango C, Miller K, Lefkowitz DM, McMahon RP, et al. Morphometric assessment of the heteromodal association cortex in schizophrenia. Am J Psychiatry 2004; 161:322 - 31
- Vasic N, Walter H, Hose A, Wolf RC. Gray matter reduction associated with psychopathology and cognitive dysfunction in unipolar depression: a voxel-based morphometry study. J Affect Disord 2008; 109:107 - 16
- Xin Y, Chanrion B, Liu MM, Galfalvy H, Costa R, Ilievski B, et al. Genome-wide divergence of DNA methylation marks in cerebral and cerebellar cortices. PLoS ONE 2010; 5:11357
- Dwork A, Smiley J, Colizbazzi T, Hoptman M. Postmortem and in vivo structural pathology in schizhoprenia. In: Nestler E, Charney D, Eds. Neurobiology of Mental Illness: Oxford University Press 2008; 201-320.
- Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005; 309:1728 - 32
- Xin Y, Ge Y, Haghighi F. Methyl-analyzer—whole genome DNA methylation profiling. Bioinformatics 2011; 27:2296 - 7
- Karimi M, Johansson S, Stach D, Corcoran M, Grander D, Schalling M, et al. LUMA (LUminometric Methylation Assay)—a high throughput method to the analysis of genomic DNA methylation. Exp Cell Res 2006; 312:1989 - 95
- Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 2005; 33:501 - 4
- Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, et al. Human-mouse alignments with BLASTZ. Genome Res 2003; 13:103 - 7