Abstract
Comprehensive High-throughput Arrays for Relative Methylation (CHARM) was recently developed as an experimental platform and analytic approach to assess DNA methylation (DNAm) at a genome-wide level. Its initial implementation was for human and mouse. We adapted it for rat and sought to examine DNAm differences across tissues and brain regions in this model organism. We extracted DNA from liver, spleen, and three brain regions: cortex, hippocampus, and hypothalamus from adult Sprague Dawley rats. DNA was digested with McrBC, and the resulting methyl-depleted fraction was hybridized to the rat CHARM array along with a mock-treated fraction. Differentially methylated regions (DMRs) between tissue types were detected using normalized methylation log-ratios. In validating 24 of the most significant DMRs by bisulfite pyrosequencing, we detected large mean differences in DNAm, ranging from 33-59%, among the most significant DMRs in the across-tissue comparisons. The comparable figures for the hippocampus vs. hypothalamus DMRs were 14-40%, for the cortex vs. hippocampus DMRs, 12-29%, and for the cortex vs. hypothalamus DMRs, 5-35%, with a correlation of r2 = 0.92 between the methylation differences in 24 DMRs predicted by CHARM and those validated by bisulfite pyrosequencing. Our adaptation of the CHARM array for the rat genome yielded highly robust results that demonstrate the value of this method in detecting substantial DNAm differences between tissues and across different brain regions. This platform should prove valuable in future studies aimed at examining DNAm differences in particular brain regions of rats exposed to environmental stimuli with potential epigenetic consequences.
Introduction
The “methylome,” or genome-wide state of DNA methylation (DNAm), refers to the complete genomic state of DNA, which can vary within an individual based on tissue type and other factors. Despite the availability of an essentially complete genome sequence of numerous organisms for several years, understanding the methylome has progressed more slowly, largely due to limitations in technology affecting sensitivity, specificity, throughput, quantitation and cost among the previously used detection methods. Microarray or sequencing-based methods can interrogate much larger numbers of CpGs than prior approaches that were typically focused on one gene or one CpG island.
Various alternatives for fractionating methylated from unmethylated DNA are available for both microarrays and sequencing. They include methylated DNA immunoprecipitation (MeDIP),Citation1 digestion with Hpa II or similar methylcytosine-sensitive restriction endonucleases followed by ligation-mediated PCR (HELP assay),Citation2 and gel purification of unmethylated DNA after digestion of methylated DNA with the enzyme McrBC.Citation3 We previously compared these methods directly and discovered significant limitations of each: specifically, bias toward CpG islands in MeDIP, relatively incomplete coverage in HELP, and location imprecision using McrBC. However, we found that by using tiling arrays and statistical procedures that average information from neighboring genomic locations, much improved specificity and sensitivity could be achieved, e.g., ∼100% sensitivity at 90% specificity with McrBC. We termed this approach Comprehensive High-throughput Arrays for Relative Methylation (CHARM).Citation4
CHARM combines the following elements: (1) McrBC fractionation—McrBC recognizes methylated DNA of the form mCPuN40-104mCPu, where Pu is any purine; although our array design does not preclude other methods, we use this method of fractionation because it provides the least distortion; (2) array design that includes all clusters of 15 or more CpGs such that no two are >300 bp apart; while this eliminates isolated CpGs it includes almost all low-density CpG methylation as well as CpG islands, and it is agnostic to assumptions about where methylation would be (e.g., promoters or CpG islands); and (3) genome-weighted smoothing of the raw output data, correcting for CpG density-dependent differential hybridization efficiency. Using the NimbleGen 2.1 million feature platform CHARM includes the great majority of potentially methylatable sequences that are missed on promoter or CpG island arrays.Citation4 Thus, the CHARM platform screens the genome in an unbiased way to determine DNA regional methylation with high accuracy.
We previously performed CHARM on three normal tissue types: liver, spleen and brain, obtained from the same five human autopsies, as well as from mice.Citation5 This analysis identified 22,442 tissue specific differentially methylation regions (tDMRs), defined as M values for one tissue consistently different than the others at a false discovery rate (FDR) of 5%. It also showed that the DNAm profile of human brain was more like that of mouse brain than it was like that of other human tissues.
While developing the CHARM assay, we began to investigate the epigenetics of the brain as a necessary step in studies of psychiatric illnesses. For this application, we employed a less comprehensive commercially available strategy. We initially analyzed human brain samples representing cerebral cortex, cerebellum and pons, along with liver samples. There was clustering by percent DNAm of tissue types. By use of comparative marker selection and permutation testing, 156 loci representing 118 genes showed statistically significant differences >17% absolute change in DNAm (p < 0.004) among brain regions. These results were validated for genes tested in a replicate set of samples.Citation6 Our data suggested that DNAm signatures distinguish brain regions and may help account for region-specific functional specialization.
We are further studying DNAm in brain and other tissues with an emphasis on the epigenetic impact of a variety of interventions related to neuropsychiatric illnesses and metabolic disorders. Because these can most efficiently be accomplished in an animal model, we are using rodents for this purpose. In this paper we focus on the rat, a commonly used model for neuroscience and behavioral work. We developed CHARM for the rat, using the same principles as were used for mouse and human CHARM, but with several modifications designed to improve the signal-to-noise ratio, and to increase coverage in genes known to be involved in psychiatric and metabolic processes. Our adaptation of the CHARM array for the rat genome yielded highly robust results that demonstrate the value of this method in detecting substantial DNAm differences among tissues and discrete brain regions, and lay the groundwork for future studies of the epigenetic impact of environmental stimuli in this model organism.
Results
Significant methylation differences among the cerebral cortex, liver and spleen.
We found 8,684 statistically significant (p ≤ 0.05) differentially methylated regions in our across-tissue comparisons (Tables S1–3). As expected, differences were large, ranging from 21.9%–57.6%, among the most robustly differing regions in these comparisons. When the 5,000 most variable probes were analyzed in a clustering analysis, the samples from a given tissue across animals were found to be more similar to each other than they were to samples from other tissues within the same animal ().
When comparing tissue-specific DMRs (tDMRs) between the liver and the spleen, we observed an average methylation difference of 45.7% (min: 32.4%, max: 55.3%). Of the top 20 most significant tDMRs, 18 showed higher DNAm levels in spleen than in liver (). We also analyzed significant DMRs that occur between the liver and the cerebral cortex, and we observed an average methylation difference of 48.4% (min: 40.7%, max: 57.6%), with 10 out of 20 tDMRs showing higher methylation in cortex than in liver (). Last, we compared the DNAm profiles between spleen and cerebral cortex to identify not only the regions that are differentially methylated between these organs, but also DMRs that are preferentially hypomethylated in only one of the three tissue types queried. We observed three tDMRs that show higher DNAm in the cerebral cortex ().
Several genes include tDMRs appearing on more than one of the across-tissue comparisons. For example, tDMRs associated with Pck1, Ahsg and Tymp all appear to be highly methylated in cerebral cortex and spleen as compared to liver. In contrast, tDMRs associated with Nr1d1, Zfhx2 and Amac1 all have lower methylation levels in cortex as compared to liver and spleen. tDMRs associated with Hoxa1 and Hoxa5, members of the Hox gene family that are critical in determining the body plan, show relatively lower DNAm in both cortex and liver, as compared to spleen.
Significant methylation differences among the cerebral cortex, hypothalamus and hippocampus.
In the next set of comparisons, we analyzed three different regions within the forebrain, and asked whether substantial DNAm differences exist within these regions that perform distinct and diverse functions. While we previously found evidence of significant DNAm differences among the cortex, cerebellum and the pons,Citation6 here we chose three regions of particular interest for psychiatric and metabolic research: the cortex, hypothalamus and hippocampus. We found 1,994 FDR-significant (q ≤ 0.05) differentially methylated regions in our brain region-specific comparisons (Tables S4–6).
A clustering analysis of the 5,000 most variable probes showed that DNAm across animals in particular brain regions was more similar than it was between regions for the same animal (). Differences in DNAm for the top 20 most significant hippocampus vs. hypothalamus DMRs were 14.8–40.6% (), for the cortex vs. hippocampus DMRs, 15.1–30.9% (), and for the cortex vs. hypothalamus DMRs, 19.2–33.0% (). Examples of four DMRs (Nr4a2, Ntrk2, Tcf4 and Tcfap2c) from the brain comparisons are shown in .
Among these significant brain region-specific DMRs or brDMRs, were a number located within or near genes of interest for brain development and/or for the etiopathogenesis of psychiatric and behavioral disorders. One brDMR is located near Tcfap2c, which encodes the transcription factor AP-2 gamma ( and ). It plays a role in the region- and layer-specific regulation of basal progenitor neurons in the developing cortex.Citation7 A second brDMR was located upstream of Neurog2 which encodes neurogenin 2 (), a transcription factor with a major role in cortical neuron migration,Citation8 and a particular role in the development of midbrain dopaminergic neurons.Citation9 A third brDMR of interest was near Emx2 or empty spiracles homeobox/homolog 2 (), the protein product of which is a transcription factor expressed in the dorsal encephalon during development that is critical to patterning the neocortex into defined functional areas.Citation10 Fourth, a brDMR was observed in Ptch1 or patched 1 (), which encodes the receptor for sonic hedgehog that regulates neural progenitor development in the cortex.Citation11 Increased DNAm at the promoter of this gene has been shown in medulloblastomas.Citation12 Finally, a brDMR was located in Dlgap2 (Disks Large-Associated Protein 2), a membrane-associated kinase localized at the postsynaptic density in neurons (). Copy number variation in this gene is suggested to play a role in autism,Citation13 and DNAm in the gene has been implicated in a rat model of post-traumatic stress disorder.Citation14
Validation of DMRs by pyrosequencing.
Using bisulfite pyrosequencing, we attempted to validate a few select regions from the top 20 DMRs in each comparison, using DNA from a new cohort of rats. We validated three candidate tDMRs from each of the comparisons involving the liver, spleen, and the cerebral cortex (9 tDMRs, ), and at least five DMRs from those involving the three brain regions (15 brDMRs, ). Due to the amplicon size restriction imposed on primer design by the pyrosequencing protocol, we assayed only a fraction (∼250 bp) of each of the approximately 1.5 kb regions annotated by CHARM. Validation across all 24 of the most significant DMRs showed significant differences that were in the predicted direction, with an overall correlation of 0.92 (p = 2.6 × 10−13) when compared to the corresponding differences found by CHARM. For example, the DMR associated with Nr4a2, encoding a nuclear receptor implicated in dopamine neuron developmentCitation15 and possibly schizophrenia,Citation16 is predicted by CHARM to have 71.6% DNAm in the hippocampus vs. 39.9% DNAm in the hypothalamus, or an average difference of 31.6% across a 2,069 bp region that encompasses two small CpG islands ( and ). Pyrosequencing of 11 of the 68 CpGs in the region revealed a mean DNAm of 77.1% for the hippocampus vs. 57.7% for the hypothalamus, or a difference of 19.4%.
Other DMRs we validated included a couple in or near genes of interest for psychiatric disorders. One such gene was Ntrk2, or neurotrophic tyrosine kinase receptor type 2 (TrkB receptor), a membrane-bound receptor that binds to brain-derived neurotrophic factor or Bdnf. Dysregulation of Ntrk2 function has been implicated in depression and antidepressant response as well as in energy balance regulation.Citation17,Citation18 We saw that the cortex was 20.0% less methylated than the hypothalamus by CHARM and 35.0% less methylated by bisulfite pyrosequencing ( and data not shown). Another gene of interest is Tcf4 or transcription factor 4, found to include a common variant conferring risk of schizophrenia.Citation19 We saw that the hippocampus was 29.9% less methylated than the hypothalamus by CHARM and 38.4% less methylated by bisulfite pyrosequencing ( and data not shown). We also examined the methylation status of a brDMR associated with the circadian clock gene Per3, and found a modest DNAm difference of 11.7% by pyrosequencing between the cortex and the hippocampus, which is approximately half of the 21.0% difference predicted by CHARM (). VNTRs (variable-number tandem-repeat) in the coding region and SNPs in the gene have been implicated in bipolar disorder by modest association findings.Citation20–Citation22 Last, we proceeded to validate a DMR at the 3′ end of the Tcfap2c gene, based on our prior interest in this gene in the context of mood disorders. CHARM predicted a 25.3% higher DNAm in the cortex than in the hippocampus (Table S5). Upon validation, we found a 25.1% mean difference between the two corresponding tissues (data not shown).
Locations of DMRs to genes and CpG islands.
Given the discovery of thousands of potentially interesting p-value and FDR-significant DMRs in the across-tissue and brain-region comparisons, respectively, we investigated their genomic locations in relation to their associated genes and nearby CpG islands. In the across-tissue comparisons looking at differences among liver, spleen and cortex, we found that a plurality of DMRs (25.0%) fell within or overlapped a CpG island, with the majority falling on the CpG island “shores” (60.8%), within 3,000 bp proximity of a CpG island (). In the brain-region comparisons, a more substantial percentage of DMRs (38.8%) were associated with CpG islands with a lower percentage of DMRs (46.3%) falling into the “shore” category ().
We then analyzed the location of the DMRs with respect to the associated gene. A surprisingly significant portion of tDMRs was located upstream (22.1%), in introns (30.3%) or downstream (20.3%) for across-tissue comparisons (), with only 10.5% of tDMRs occupying the promoter areas (<2,500 bp upstream from transcription start site). A similar distribution of genomic locations was observed for brain-region comparisons, where we found most of the brDMRs were located upstream (25.0%), in introns (25.2%) or downstream (29.6%) of genes. Only 5.9% of the brDMRs were associated with the promoter ().
Discussion
In this genome-wide assessment of DNA methylation in the rat, we explored the rat methylome by adapting the CHARM technology. We first examined tissue-specific DMRs within three tissues of distinct lineages, the liver, spleen and the cerebral cortex, where we expected to find large differences in DNAm. CHARM did indeed predict large differences among the three tissues nearby or within genes involved in development.
Given this result we then attempted to assess the sensitivity of CHARM by exploring distinct sub-regions within an organ. We chose three unique regions in the forebrain, namely the cortex, hippocampus and the hypothalamus. Once again, we detected highly significant DNAm differences among the three regions. Many of the genes associated with these DMRs are involved in brain development. However, regions of DNAm variability that we have seen in this study are potentially interesting not only because of their relationship to differentiation and development, but perhaps also because they may be related to pathological processes. Such was the case for DMRs identified in a study using the CHARM platform in human samples. Some of the same regions that distinguished tissue types from each other also distinguished colon cancer from normal colon tissue.Citation5 Therefore, it may be of interest to search for DNAm variability in the orthologous regions of the brDMRs associated with Ntrk2, Tcf4, Nr4a2 and Dlgap2, in human studies of psychiatric disorders.
We also analyzed the genomic locations of these DMRs in relation to CpG islands and nearby genes. As observed previously with the human and mouse CHARM, we found a majority of the DMRs in the “shores,” although DMRs that are incident upon CpG islands constituted a significant portion of brDMRs in our brain-region comparisons. These findings further strengthen the notion of much epigenetic variability occurring outside of CpG islands, which in turn validates our rat CHARM method in the context of the other two. We also observed most of the DMRs occurring upstream, in introns and downstream of genes rather than in the promoter region. This finding is consistent with the existence of tissue-specific enhancers, repressors and insulators in non-promoter regionsCitation23,Citation24 and supports non-promoter regions mediating and conferring tissue-specific expression of genes. For instance, our group has found that white blood cells and neurons exposed to glucocorticoids undergo loss of DNA methylation at different intronic enhancers, and not in promoter regions, to increase expression levels of the stress-response gene Fkbp5.Citation25 Nonetheless, it remains to be determined whether the DMRs identified in this study influence expression of the associated genes, and whether these expression differences might be tissue-specific.
Tissue-specific differences in DNAm were first identified in the late 70s. For example, a CpG in the rabbit β-globin gene was found to be more methylated in sperm and brain than in spleen, bone marrow or blood.Citation26 Differences between brain regions in DNAm were first identified more recently in a paper comparing cerebral cortex, pons and cerebellum across ∼1,500 CpGs.Citation6 Other studies have also shown differences between brain regions, with two focusing on the DNAm distinctions between cerebellum and the rest of the brain.Citation27,Citation28 Ours is the first experiment to show broad differences in methylation among hippocampus, hypothalamus and cerebral cortex. These results are not unexpected given prior demonstration of gene expression differences among these regions.Citation29,Citation30
This study has several limitations. First, while our validation assays showed good consistency with CHARM results, they were not in perfect agreement, with the magnitude of difference in DNAm varying between the methods. This likely reflects the fact that CHARM averages across a broad region of ∼1,500 bp, typically including more than 30 CpGs, whereas the bisulfite pyrosequencing assays focus on a narrow subset of ∼10 of these. Thus, what is being measured varies slightly between the methods. Second, while CHARM covers a significant portion (∼70%) of the CpG-dense regions of the genome, there remain many more that are not covered, where relevant variation between tissues might exist. These will likely only be covered when whole genome sequencing for methylation status becomes feasible and affordable.Citation31 Third, there are likely many more brain region-specific DNAm variations that will become evident when additional regions are assayed.
This platform should prove valuable in future studies aimed at examining DNAm differences in particular brain regions of rats exposed to environmental stimuli with potential epigenetic consequences. These stimuli include stress, high fat diet and psychiatric medications. This platform also provides a means for identifying novel DMRs that will expand our understanding of modulation of gene expression by DNA methylation.
Materials and Methods
Animals.
Nine-week old (N = 8) male Sprague Dawley rats (Charles River Laboratories) were housed in conventional polycarbonate rat cages in a temperature and humidity-controlled room on a 12-h light, 12-h dark cycle with light onset at 0600 h. All animals were received ad libitum access to water and standard rodent chow (Harlan Teklad, #2018) for 1 wk after arrival in the laboratory. Animals were subsequently euthanized by decapitation and relevant tissues and brain regions were collected, frozen on dry ice and stored at −80°C until processing for CHARM analysis and subsequent validation. A second cohort of nine-week old rats (N = 12) was similarly euthanized and tissues were reserved for validation of CHARM results by bisulfite pyrosequencing. All procedures were approved by the Institutional Animal Care and Use Committee at the Johns Hopkins University School of Medicine and were performed in accordance with guidelines established in the National Research Council's Guide for the Care and Use of Laboratory Animals.
Samples.
All frozen tissue was stored at −80°C. DNA was extracted using the MasterPure DNA Purification kit (Epicentre Biotechnologies, #MCD85201).
Rat CHARM array design.
The array was designed as described previously in reference Citation4, with several modifications aimed at increasing coverage and accuracy. These include giving priority to regions and genes implicated in neuropsychiatric illnesses and metabolic disorders, eliminating duplicate probes and adding additional control probes from regions unaffected by McrBC. Further, CpG content of control probes was increased to reflect that of the genome, where first versions of CHARM arrays had none.
CHARM assay.
The CHARM assay was performed as described previously in reference Citation4. Briefly, DNA was extracted from tissues using the MasterPure DNA Purification kit (Epicentre Biotechnologies) and 10 µg of DNA was sheared in 100 µl using a Hydroshear device (Digilab) into 1.6 kb–3 kb fragments. Sheared DNA was then divided into two fractions. One fraction was digested overnight at 37°C with the methyl-sensitive enzyme McrBC (New England Biolabs, #M0272L). Following digestion, cut and uncut fractions from the same sample were electrophoresed in adjacent wells of a 1% agarose gel. Areas corresponding to the 1.6 kb–3 kb regions were excised and purified using QIAquick Gel Extraction columns (Qiagen, #28704). The gel-purified DNA was quantified on a NanoDrop 1000 Spectrophotometer (Thermo Scientific) and 30 ng of DNA from each fraction was amplified using a GenomePlex Whole Genome Amplification Kit (Sigma-Aldrich, #WGA2). The amplified DNA was then isolated with a Qiagen PCR Purification column, then quantified on NanoDrop. The untreated total DNA fraction was labeled with Cy3 and the methyl-depleted DNA fraction was labeled with Cy5 and hybridized onto the custom NimbleGen 2.1 M feature CHARM microarray (Roche-NimbleGen).
Pyrosequencing.
500 ng of genomic DNA was bisulfite-treated using the EpiTect Bisulfite kit (Qiagen, #59104). CpG unbiased primers were designed to PCR amplify 162 CpG sites in 24 genes by PCR. Nested PCR was performed using 2 µL of the outside reaction. Amplicons were analyzed on a PSQ HS 96 pyrosequencer (Qiagen) and CpG sites were quantified, from 0% to 100% methylation, using Pyro Q-CpG software.Citation32
Microarray data preprocessing.
Hybridization quality was assessed by comparing the untreated fraction signal intensity for each genomic probe to that of background (anti-genomic) probes, with the expectation that the genomic probes should register significantly higher signals. Poor hybridization was indicated by genomic probe signal levels not being significantly higher than background probe levels.
Detection of differentially methylated regions (DMRs).
Normalized methylation log-ratios were smoothed using a weighted sliding window as previously described in reference Citation4. For each probe, the average log-ratio and standard deviation were computed for various tissue comparisons allowing a Z-score to be calculated for each probe. Under the assumption that most regions are not differentially methylated, the median absolute deviation of t-scores across all probes was used to determine the standard deviation of the null distribution. Contiguous regions of ≥6 smoothed Z-scores with p < 0.005 were identified as candidate DMRs. For these regions, a novel Bayesian model was used to convert log ratios of intensities to estimated percent methylation.Citation33
Clustering analysis.
Preprocessed methylation estimates were smoothed using a 10-probe moving average sliding window. Average linkage hierarchical clustering was performed on the 5,000 probes with highest variability across all samples.
Analysis of pyrosequencing data.
For each of the 24 most differentially methylated regions, we assessed pyrosequencing data based on primers designed across the most CpG dense part of the region identified by CHARM. Statistical analysis was performed in Microsoft Excel (Microsoft, Office 2007).
Abbreviations
| CHARM | = | comprehensive high-throughput arrays for relative methylation |
| DNAm | = | DNA methylation |
| DMRs | = | differentially methylated regions |
| tDMRs | = | tissue specific DMRs |
| brDMRs | = | brain region-specific DMRs |
Figures and Tables
Figure 1 Clustering analysis based on 5,000 most variably methylated probes. The brain, liver and spleen are clearly distinguished from each, falling into separate branches. Within the branch for brain, the cortex, hippocampus and hypothalamus all cluster separately.
Figure 2 Methylation plots generated by CHARM for 4 brDMRs: (A) Nr4a2, (B) Ntrk2, (C) Tcf4 and (D) Tcfap2c. Chromosomal locations are shown at the top of each part. The upper portion for each panel displays the approximate methylation percentage for all five tissues, represented by the five plot curves with different colors. Although CHARM generates and displays plot curves for all of the five tissues queried, the DMR is chosen and ranked based on the significant differences in DNAm between two selected tissues, represented by black arrows in each part. Displayed below the plot curves are the CpG density, and the genomic organization of the genes associated with the brDMRs. Black boxes represent exons, whereas white boxes represent intronic regions. For instance, brDMRs for Ntrk2 and Tcf4 fall within an intronic region of both genes.
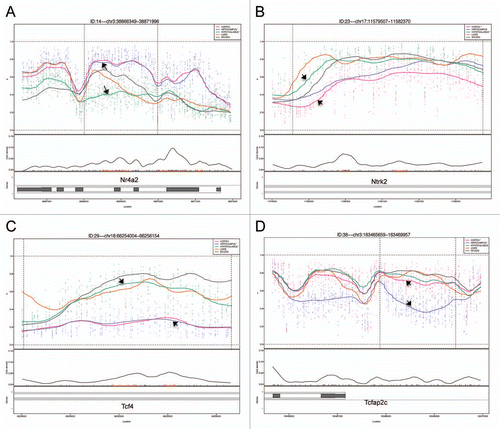
Figure 3 Boxplot of pyrosequencing data obtained from liver (LIV), spleen (SPL) and cerebral cortex (CTX) tDMRs. We assessed and validated percent DNA methylation of three unique tDMRs from each pair of tissues compared by CHARM. For each tDMR, the number of CpGs assayed varied from 4 to 8 CpGs, thus a small subset of the region assayed by the CHARM array. Average percent methylation was determined from all of the assayed CpGs for each tDMR in each tissue type and displayed as a boxplot. Stippled horizontal lines represent the percent DNAm predicted by CHARM for the particular tissue for the entire tDMR region. Types of tissue being compared and the nearest genes associated with the tDMRs are indicated below the X-axis.
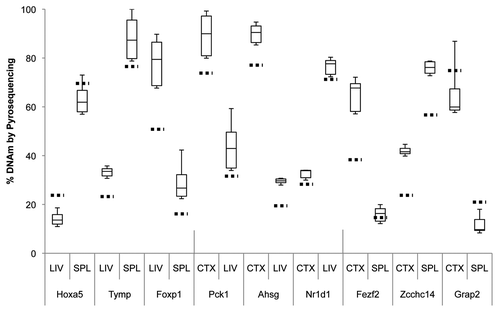
Figure 4 Boxplot of pyrosequencing data obtained from the cerebral cortex (CTX), hippocampus (HIP) and hypothalamus (HYP) brDMRs. We assessed and validated percent DNA methylation of five unique DMRs from each pair of brain tissues compared by CHARM. For each brDMR, the number of CpGs assayed varied from 4 to 16 CpGs, thus a small subset of the region assayed by the CHARM array. Average percent methylation was determined from all of the assayed CpGs for each brDMR in each brain tissue type and displayed as a boxplot. Stippled horizontal lines represent the percent DNAm predicted by CHARM for the particular brain tissue for the entire brDMR region. Types of brain tissues being compared and the nearest genes associated with the brDMRs are indicated below the X-axis.
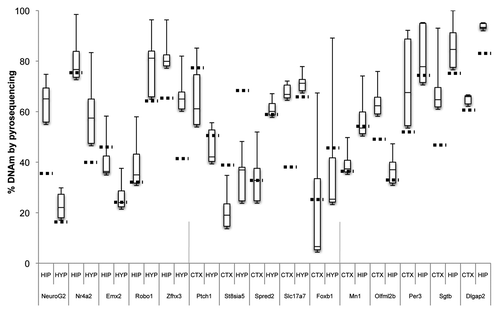
Figure 5 Histogram of the distance of tDMRs and brDMRs to the nearest CpG island. (A) tDMRs annotated by CHARM in liver, spleen and cortex comparisons were tallied and their distance to the nearest CpG island calculated. Distances of DMRs to CpG islands were separated by different distance categories or bins and graphed as a percentage of the total DMRs analyzed. Red bar represents the percentage of DMRs that fall on a CpG island. Distances greater than 50 bp and less than or equal to 3,000 bp from a CpG island are collectively considered as “CpG shores.” (B) Similar analysis was performed for brDMRs annotated by CHARM in cortex, hippocampus and hypothalamus.
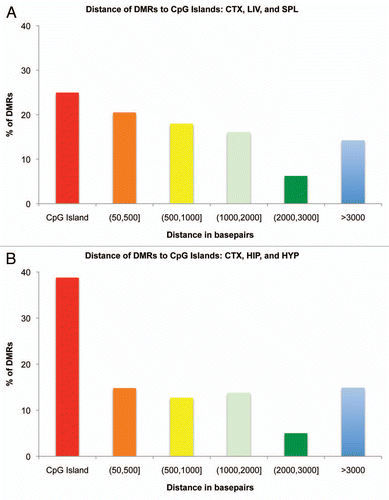
Figure 6 Histogram of genomic locations of tDMRs and brDMRs in relation to associated genes. (A) tDMRs annotated by CHARM in the liver, spleen and cortex comparisons were categorized based on their locations to genes, tallied and graphed as a percentage of the total DMRs analyzed. Promoter and “Close to 3′” categories represent DMRs that fall within 2,500 bp of the first and last exons, respectively. (B) Similar analysis was performed for brDMRs annotated by CHARM in the cortex, hippocampus and the hypothalamus.
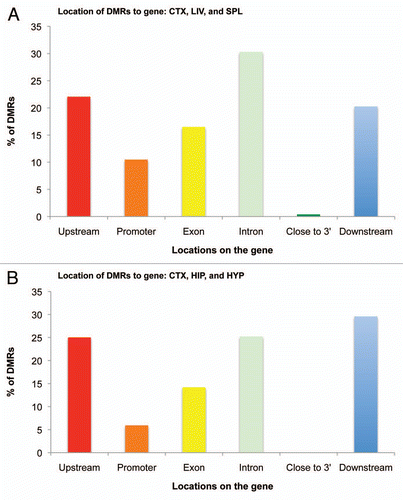
Table 1 Differentially methylated regions in the liver vs. spleen comparison
Table 2 Differentially methylated regions in the liver vs. cerebral cortex comparison
Table 3 Differentially methylated regions in the spleen vs. cerebral cortex comparison
Table 4 Differentially methylated regions in the hippocampus vs. hypothalamus comparison
Table 5 Differentially methylated regions in the hippocampus vs. cerebral cortex comparison
Table 6 Differentially methylated regions in the cerebral cortex vs. hypothalamus comparison
Additional material
Download Zip (23.3 MB)Acknowledgements
The investigators report no competing interests. This work was supported by grants from the NIDDK to Dr. Moran (R01DK077623), and the NICHD to Dr. Tamashiro (R00HD055030), by T32MH015330 supporting Dr. Lee and by The Margaret Ann Price Investigatorship for Bipolar Research supporting Dr. Potash.
References
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853 - 862
- Khulan B, Thompson RF, Ye K, Fazzari MJ, Suzuki M, Stasiek E, et al. Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res 2006; 16:1046 - 1055
- Ordway JM, Bedell JA, Citek RW, Nunberg A, Garrido A, Kendall R, et al. Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis 2006; 27:2409 - 2423
- Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA, Jeddeloh JA, et al. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res 2008; 18:780 - 790
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 186
- Ladd-Acosta C, Pevsner J, Sabunciyan S, Yolken RH, Webster MJ, Dinkins T, et al. DNA methylation signatures within the human brain. Am J Hum Genet 2007; 81:1304 - 1315
- Pinto L, Drechsel D, Schmid MT, Ninkovic J, Irmler M, Brill MS, et al. AP2gamma regulates basal progenitor fate in a region- and layer-specific manner in the developing cortex. Nat Neurosci 2009; 12:1229 - 1237
- Heng JI, Nguyen L, Castro DS, Zimmer C, Wildner H, Armant O, et al. Neurogenin 2 controls cortical neuron migration through regulation of Rnd2. Nature 2008; 455:114 - 118
- Kele J, Simplicio N, Ferri AL, Mira H, Guillemot F, Arenas E, et al. Neurogenin 2 is required for the development of ventral midbrain dopaminergic neurons. Development 2006; 133:495 - 505
- Bishop KM, Goudreau G, O'Leary DD. Regulation of area identity in the mammalian neocortex by Emx2 and Pax6. Science 2000; 288:344 - 349
- Shikata Y, Okada T, Hashimoto M, Ellis T, Matsumaru D, Shiroishi T, et al. Ptch1-mediated dosage-dependent action of Shh signaling regulates neural progenitor development at late gestational stages. Dev Biol 2011; 349:147 - 159
- Diede SJ, Guenthoer J, Geng LN, Mahoney SE, Marotta M, Olson JM, et al. DNA methylation of developmental genes in pediatric medulloblastomas identified by denaturation analysis of methylation differences. Proc Natl Acad Sci USA 2010; 107:234 - 239
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010; 466:368 - 372
- Chertkow-Deutsher Y, Cohen H, Klein E, Ben-Shachar D. DNA methylation in vulnerability to post-traumatic stress in rats: evidence for the role of the post-synaptic density protein Dlgap2. Int J Neuropsychopharmacol 2010; 13:347 - 359
- Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science 1997; 276:248 - 250
- Rojas P, Joodmardi E, Hong Y, Perlmann T, Ogren SO. Adult mice with reduced Nurr1 expression: an animal model for schizophrenia. Mol Psychiatry 2007; 12:756 - 766
- Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci 2003; 23:349 - 357
- Noble EE, Billington CJ, Kotz CM, Wang C. The lighter side of BDNF. Am J Physiol Regul Integr Comp Physiol 2011; 300:1053 - 1069
- Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, et al. Common variants conferring risk of schizophrenia. Nature 2009; 460:744 - 747
- Dallaspezia S, Lorenzi C, Pirovano A, Colombo C, Smeraldi E, Benedetti F. Circadian clock gene Per3 variants influence the postpartum onset of bipolar disorder. Eur Psychiatry 2011; 26:138 - 140
- Nievergelt CM, Kripke DF, Barrett TB, Burg E, Remick RA, Sadovnick AD, et al. Suggestive evidence for association of the circadian genes PERIOD3 and ARNTL with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet 2006; 141:234 - 241
- Benedetti F, Dallaspezia S, Colombo C, Pirovano A, Marino E, Smeraldi E. A length polymorphism in the circadian clock gene Per3 influences age at onset of bipolar disorder. Neurosci Lett 2008; 445:184 - 187
- Martin D, Pantoja C, Fernandez Minan A, Valdes-Quezada C, Molto E, Matesanz F, et al. Genome-wide CTCF distribution in vertebrates defines equivalent sites that aid the identification of disease-associated genes. Nat Struct Mol Biol 2011; 18:708 - 714
- Nuber UA, Kriaucionis S, Roloff TC, Guy J, Selfridge J, Steinhoff C, et al. Upregulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum Mol Genet 2005; 14:2247 - 2256
- Lee RS, Tamashiro KL, Yang X, Purcell RH, Harvey A, Willour VL, et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology 2010; 151:4332 - 4343
- Waalwijk C, Flavell RA. DNA methylation at a CCGG sequence in the large intron of the rabbit beta-globin gene: tissue-specific variations. Nucleic Acids Res 1978; 5:4631 - 4634
- Xin Y, Chanrion B, Liu MM, Galfalvy H, Costa R, Ilievski B, et al. Genome-wide divergence of DNA methylation marks in cerebral and cerebellar cortices. PLoS One 2010; 5:11357
- Ghosh S, Yates AJ, Fruhwald MC, Miecznikowski JC, Plass C, Smiraglia D. Tissue specific DNA methylation of CpG islands in normal human adult somatic tissues distinguishes neural from non-neural tissues. Epigenetics 2010; 5:527 - 538
- Stansberg C, Vik-Mo AO, Holdhus R, Breilid H, Srebro B, Petersen K, et al. Gene expression profiles in rat brain disclose CNS signature genes and regional patterns of functional specialisation. BMC Genomics 2007; 8:94
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007; 445:168 - 176
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 2011; 43:768 - 775
- Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques 2003; 35:146 - 150
- Aryee MJ, Wu Z, Ladd-Acosta C, Herb B, Feinberg AP, Yegnasubramanian S, et al. Accurate genome-scale percentage DNA methylation estimates from microarray data. Biostatistics 2011; 12:197 - 210