Abstract
Epigenetic mechanisms are frequently deregulated in cancer cells and can lead to the silencing of genes with tumor suppressor activities. The isoform A of the Ras-association domain family member 1 (RASSF1A) gene is one of the most frequently silenced transcripts in human tumors, however, few studies have simultaneously investigated epigenetic abnormalities associated with the 3p21.3 tumor suppressor gene cluster flanking RASSF1 (i.e., SEMA3B, HYAL3, HYAL2, HYAL1, TUSC2, RASSF1, ZMYND10, NPRL2, TMEM115, and CACNA2D2). This study aimed to investigate the role of epigenetic changes to these genes in seventeen breast cancer cell lines and in three non-tumorigenic epithelial breast cell lines (184A1, 184B5, and MCF 10A) and to evaluate the effect on gene expression of treatment with the demethylating agent 5-Aza-2'-deoxycytidine and/or Trichostatin A (TSA), a histone deacetylase inhibitor. We report that, although the RASSF1A isoform was determined to be epigenetically silenced in 15 of the 17 breast cancer cell lines, all the cell lines expressed the RASSF1C isoform. Five breast cancer cell lines overexpressed RASSF1C, when compared to the normal epithelial cell line 184A1. Furthermore, the genes HYAL1 and CACNA2D2 were significantly overexpressed after the treatments. After the combinated treatment, RASSF1A re-expression was accompanied by an increase in expression levels of the flanking genes. The Spearman's correlation coefficient indicated a positive co-regulation of the following gene pairs: RASSF1 and TUSC2 (r=0.64, p=0.002), RASSF1 and ZMYND10 (r=0.58, p=0.07), RASSF1 and NPRL2 (r=0.48, p=0.03), ZMYND10 and NPRL2 (r=0.71; p=0,0004) and NPRL2 and TMEM115 (r=0.66, p=0.001). Interestingly, the genes TUSC2, NPRL2 and TMEM115 were found to be unmethylated in each of the untreated cell lines. Chromatin immunoprecipitation using antibodies against the acetylated and trimethylated lysine 9 of histone H3 demonstrated low levels of histone methylation in these genes, which are located closest to RASSF1. These results provide evidence that epigenetic repression is involved in the down-regulation of multiple genes at 3p21.3 in breast cancer cells.
Introduction
Genetic and epigenetic alterations are well established hallmarks of human cancer that cooperate at tumor development and can result in the silencing of key regulatory genes with demonstrated roles in tumor suppression, DNA mismatch repair and cell cycle regulation.Citation1–Citation3 Specifically, aberrant DNA methylation content (global genome hypomethylation) and patterns of cytosine methylation, especially promoter-specific CpG island hypermethylation, are currently known to be associated with the development of human cancer.Citation4,Citation5
In breast cancer, there is evidence that agglomerative epigenetic aberrations are common events.Citation6,Citation7 This observation has led to the hypothesis that DNA hypermethylation could affect not only discrete genes as a local event but could also span across a large chromosomal region, such as the one observed for the homeobox gene cluster (HOXA and HOXD) Citation6 and the protocadherin family (PHDC genes).Citation7 Long Range Epigenetic Silencing (LRES) is the term proposed to describe the coordinate suppression of neighboring genes based on the interplay between the hypermethylation of clusters of contiguous CpG islands within a genomic region and histone modifications.Citation8 Aberrant DNA methylation associated with LRES has been described for several types of tumors, including colon,Citation9–Citation12 bladder,Citation13 prostateCitation14,Citation15 and WilmsCitation16 and can extend over genomic regions varying from 100–2,400 Kb in size. It has been proposed that epigenetic events associated with LRES can cause the same expected effects of genetic alterations, such as deletions/microdeletions of contiguous genes, due to the functional inactivation of cancer related genes, by epigenetic modifications linked to a repressed chromatin state (such as densely methylated CpG islands and methylation of lysines 9 and 27 of histone H3).
In this context, the short arm of human chromosome 3 has attracted special interest for harboring multiple genes that exhibit varying degrees of tumor suppressor activity. In breast cancer, allelic losses were detected along 3p by loss of heterozygosity (LOH) analysis in 80% of the cases and were associated with early tumor stages and poor prognostic parameters.Citation17 The LOH pattern of these losses both in breast carcinomas and preneoplastic epithelial foci was revealed to be discontinuous and was characterized by at least nine small discrete intervals of common microdeletions. The 3p21.3 region, where the RASSF1 gene is mapped, was the most frequently region involved in LOH.Citation18
The gene RASSF1 is a member of the Ras-Association Domain Family (RASSF) and encodes seven distinct transcripts generated by the differential usage of two promoters and alternative splicing. However, only two of these transcripts (RASSF1A and RASSF1C) have been attributed with a relevant biological role in cancer cells.Citation19 These isoforms are transcribed from the upstream and downstream promoter, respectively, and each one is located within a CpG island. Previous studies have demonstrated that only the upstream promoter is frequently hypermethylated in different tumor types and cancer cell lines.Citation20 Interestingly, the RASSF1 gene is found in the central position of the 3p21.3 tumor suppressor cluster,Citation21,Citation22 identified by the overlapping regions of minimal deletions detected by LOH analysis as well as by the occurrence of homozygous deletions in both primary tumors and cancer cell lines.Citation23,Citation24 These studies have indicated that at least 19 genes are located in the approximately 630 Kb region at 3p21.3. An approximately 120 Kb long subregion within this area contains the following eight genes flanking RASSF1: CACNA2D2 (calcium channel, voltage-dependent, α2/delta subunit 2), TMEM115 (transmembrane protein 115 or placental protein 6, PL6), CYB561D2 (cytochrome b-561 domain containing 2/101F6), NPRL2 (nitrogen permease regulator-like 2, primed named tumor suppressor candidate 4 or TUSC4), ZMYND10 (zinc finger, MYND-type containing 10, BLU or FLU), RASSF1, TUSC2 (tumor suppressor candidate 2, also named FUS1), HYAL2 and HYAL1 (hyaluronidase type 2 and type 1) and the genes HYAL3 and SEMA3B (semaphorin 3B, originally named LUCA1) immediately adjacent to them. Genetic analysis of lung and breast tumors indicated that none of these candidate tumor suppressor genes (TSG) were frequently mutated.
These putative tumor suppressor genes may affect and regulate many important biological processes, such as cell proliferation, cell cycle kinetics, signal transduction, ion exchange and apoptosis and could have a synergistic effect in cancer cells. Therefore, and considering that the genes in 3p21.3 tumor suppressor cluster are rarely inactivated in human cancer by somatic mutations, we have designed this study to investigate whether alternative epigenetic mechanisms may play a role in the downregulation/inactivation of resident candidate TSG flanking RASSF1 in breast cancer cell lines.
Results
Gene transcript expression, HRM and DNA copy number analysis in untreated breast cell lines.
To achieve our goal, we initially evaluated the expression levels of transcripts from RASSF1 and the neighboring genes mapped within a genomic region of approximately 236 Kb, flanked by the SEMA3B (position chr3: 50,305,040–50,314,570, UCSC Genome Browser on Human, GRCh37/hg19 Assembly) and CACNA2D2 (position chr3: 50,400,233–50,540,892, UCSC Genome Browser on Human, GRCh37/hg19 Assembly) genes, which contains nine genes (SEMA3B, HYAL3, HYAL1, HYAL2 and TUSC2, located upstream of RASSF1 and ZMYND10, NPRL2, TMEM115 and CACNA2D2, located downstream) (). These genes were evaluated in three non-malignant cell lines derived from human breast epithelial cells (184A1, 184B5) and fibrocystic disease (MCF 10A) as well as in a series of 17 breast cancer cell lines. The expression levels of the RASSF1 gene were assessed by real-time PCR using a set of primer and probes targeting specifically the exons 5 and 6 (common to all isoforms) and the RASSF1A isoform. Although only two breast cancer cell lines expressed the RASSF1A isoform (MDA-MB-415 and Hs 578T), transcripts of the RASSF1 gene were detected in all of the cell lines analyzed. Interestingly, when the non-tumorigenic and tumorigenic cell lines were compared, a trend of high RASSF1 transcript levels was observed in the breast cancer cell lines compared with the normal epithelial cell line 184A1 (Mann Whitney test, p = 0.0720) (). Expression levels of the RASSF1 gene were found to be elevated in five cell lines (BT-20, BT-549, MDA-MB-231, MDA-MB-468 and T-47D), by at least 2-fold, compared with the normal epithelial cells 184A1. In addition, among the other nine genes evaluated, only the HYAL2, TUSC2 and CACNA2D2 exhibited differential expression levels in the malignant compared with the non-malignant cell lines; these transcripts were detected at higher levels in cancer cell lines (Mann Whitney test, p = 0.0199, p = 0.0443 and p = 0.0262, respectively) ().
The DNA methylation status of the RASSF1A promoter region was measured by High Resolution Melting (HRM) analysis. As expected, the silencing of this isoform was correlated with DNA hypermethylation of CpG island 1 (CpG island 84, nucleotide position chr3: 50377804-50378540, UCSC Genome Browser on Human, assembly GRCh37/hg19): concordant results were observed in 13/17 (76%) breast cancer cell lines, which showed 100% methylation and an absence of RASSF1A gene expression. On the other hand, approximately 50% of methylation was detected in the MDA-MB-453 and 25% in the BT-20 cell lines. Furthermore, the two cell lines expressing the A isoform of RASSF1 showed 0% and 25% of methylation (MDA-MB-415 and Hs 578T, respectively). Interestingly, despite the lack of methylation observed in 184A1 and 184B5 cells and the 25% of methylation in MCF 10A cells, these cells did not express RASSF1A. When the copy number of the RASSF1 locus was evaluated, 11/20 cell lines showed allelic losses. The comparison between the expression level of the RASSF1 transcript and gene copy number revealed a significant correlation (r2 = 0.44, p = 0.0015). In contrast, the two breast cell lines that expressed RASSF1A, MDA-MB-415 and Hs 578T, presented no alterations in the copy number at this locus.
Gene transcript expression analysis in 5-Aza-dC and TSA treated breast cell lines.
In a second step, the panel of 20 epithelial breast cell lines was treated with the demethylating nucleoside analog 5-Aza-2′-deoxycytidine (5-Aza-dC) and the histone deacetylase inhibitor Trichostatin A (TSA), either alone or in combination. These treatments were used to evaluate whether these drugs were able to induce the reactivation of the RASSF1A isoform and to evaluate the drugs' effect on the expression levels of the neighboring genes. The treatment with TSA alone was not able to significantly modulate the expression levels (). The treatment with 5-Aza-dC alone or in combination with TSA led to the re-expression of RASSF1A in 18 breast epithelial cell lines (). However, the two cell lines expressing RASSF1A showed differential responses to the treatments, as RASSF1A was upregulated in Hs 578T cells and remained unchanged in MDA-MB-415 () compared with the untreated controls (). The non-tumorigenic 184A1 cell line showed the lowest variation in the expression levels of all transcripts evaluated after treatments with 5-Aza-dC and 5-Aza-dC/TSA.
To evaluate the effects of the demethylating agent and the histone deacetylase inhibitor on the transcription of the genes flanking RASSF1, the expression levels were determined as fold-change relative to the respective untreated controls. In addition, an unsupervised hierarchical clustering analysis was performed based on the Euclidean distance to calculate the average linkage. A single cluster was obtained with the untreated controls and the cell lines treated only with TSA and another cluster obtained separated those with the cell lines treated with 5-Aza-dC or 5-Aza-dC/TSA (). The gene expression profiles in Hs 578T, MDA-MB-415 (25% methylated and unmethylated at the RASSF1A locus, respectively) and BT-20 cells (25% methylated) did not significantly change upon the treatments and were distinct, compared with the other cell lines. A group of five genes were found to be more similar to the RASSF1A/RASSF1 expression levels before and after the treatments, including SEMA3B, TMEM115, HYAL2, TUSC2 and NPRL2 (). Although all genes were positively regulated by the epigenetic drugs, their effect was highest for the HYAL1 and CACNA2D2 genes ( and Table S1).
A pairwise Spearman correlation coefficient was then calculated to create a correlation coefficient matrix for each pair of genes (). After 5-Aza-dC treatment, a positive correlation was observed between RASSF1, NPRL2 and TMEM115 and between ZMYND10 and NPRL2. Moreover, after the combined treatment with 5-Aza-dC and TSA, RASSF1 expression levels were positively correlated with TUSC2 and ZMYND10. Because TUSC2 expression was positively correlated with ZMYND10, which in turn correlated with NPRL2, and this gene also positively correlated with TMEM115, the contiguous genes mapped in the central portion of the 3p21.3 cluster flanking RASSF1 (i.e., from telomere to centromere, TUSC2, RASSF1, ZMYND10, NPRL2 and TMEM115) were selected for DNA methylation analysis and chromatin immunoprecipitation (ChIP).
In contrast to the high hypermethylation frequencies observed in RASSF1A after the HRM approach, the CpG islands in the promoter regions of the CACNA2D2, TUSC2, NPRL2 and TMEM115 genes were unmethylated in all 20 cell lines analyzed. For ZMYND10, two cell lines were 100% methylated (MDA-MB-436 and ZR-75-30) and one was 25% methylated (MDA-MB-453). In contrast, the non-CpG promoter of HYAL1 gene was 25% methylated in all cell lines with exception of three (BT-20, MCF7 and MDA-MB-415) that were unmethylated. shows the enrichment status over the input fraction for the five genes evaluated after ChIP using antibodies to detect two modifications of histone H3 (H3K9ac and H3K9me3). Marked differences in the levels of histone H3 acetylation and methylation at the promoters of the TUSC2, RASSF1, ZMYND10, NPRL2 and TMEM115 genes were detected, with a clear predominance of acetylation. H3K9 acetylation and the absence of trimethylation at the same site were observed at five loci in the two cell lines with unmethylated RASSF1A (). In contrast, the non-tumorigenic cell lines (184A1, 184B5 and MCF 10A) manifested enrichment of H3K9me3, which could explain the inactivation of the RASSF1A gene despite the absence or low levels of DNA methylation.
Discussion
Recently, “omics”-based integrative analysis has predicted regions of the human genome that are probably subjected to mechanisms of coordinated epigenetic suppression in different tumor types.Citation13,Citation15,Citation25 In fact, some of these predictions have been experimentally validated on the chromosome regions 2p14.2,Citation9,Citation11,Citation12 3p22.3,Citation13,Citation26 5q31,Citation7,Citation16 7p15.2,Citation6 7q31.1-q31.3 Citation15 and 16p11.2.Citation25 These studies have provided new approaches for the characterization of discrete regions of LRES, supporting the hypothesis that this alternative mechanism may be a common phenomenon in cancerCitation8 and could lead to the identification of new genes or genomic regions that are involved in tumor progression and can be potentially targeted for epigenetic therapy.
Genetic and epigenetic abnormalities at 3p21.3 are frequently observed in several tumor types, and a particular focus has been given to RASSF1A promoter hypermethylation, which is one of the most frequent epigenetic alterations in human cancers. However, few studies have been dedicated to the investigation of the DNA methylation patterns of the other genes mapped to the 3p21.3 tumor suppressor cluster and only few reports have simultaneously analyzed the epigenetic status of more than two genes at this locus.Citation26–Citation30
In the present study, the expression levels of ten contiguous genes mapped to the 3p21.3 tumor suppressor cluster were compared among non-tumorigenic and tumorigenic epithelial breast cell lines, before and after treatment with the epigenetics drugs 5-Aza-dC and TSA (alone or in combination). Initially, the expression levels of each gene were compared among the different breast cell lines studied. The RASSF1 gene, and specifically the RASSF1A isoform, received a special focus because epigenetic modification at this isoform, i.e., DNA methylation of the promoter-associated CpG island 1, has frequently been reported in breast cancer.Citation31 Our data showed that, although epigenetic silencing of RASSF1A was detected in 18/20 epithelial breast cell lines, all of them expressed the RASSF1 gene. This gene encodes seven isoforms (RASSF1A-G), of which RASSF1A and RASSF1C are ubiquitously expressed in normal tissues and are controlled by two distinct promoter regions associated with CpG islands.Citation19 RASSF1B isoform is expressed in hematopoietic cells and has a different 5′ exon (exon 1β); RASSF1D-G isoforms, which are splice variants of RASSF1A, are transcribed from the promoter associated with the first CpG island. The biological function of these additional transcripts is not clear; however, all the RASSF1 isoforms that are transcribed from the first CpG island are frequently missing in a variety of tumors as a result of epigenetic inactivation of the RASSF1A promoter. Additionally, because the second CpG island (CpG island 139, nucleotide position chr3: 50,374,265–50,375,629, UCSC Genome Browser on Human, assembly GRCh37/hg19) associated with the RASSF1C promoter did not present abnormal DNA methylation in cancer cells,Citation19 the RASSF1 transcripts detected in the panel of 20 epithelial breast cells are probably from the RASSF1C isoform. Thus, this present study has demonstrated that, in breast cell lines, although RASSF1A is commonly epigenetically silenced, RASSF1C remains expressed. Interestingly, although all cell lines expressed RASSF1C, this transcript was differentially expressed in the non-tumorigenic and tumorigenic cell lines, with 5/17 breast cancer cell lines (BT-20, BT-549, MDA-MB-231, MDA-MB-468 and T-47D) overexpressing this isoform compared with the 184A1 expression levels. Our findings are in accordance with those recently reported for the MDA-MB-231 and T-47D cell lines, in which the downregulation of caspase 3 via overexpression of RASSF1C was demonstrated to reduce the sensitivity of breast cancer cells to apoptosis. RASSF1C overexpression also enhanced T-47D cell invasion/migration in vitro.Citation32 A study from Estrabaud et al. demonstrated that, when the balance of RASSF1C and RASSF1A is disrupted by the absence of RASSF1A or by RASSF1C overexpression, the function of SCFβTrCP is inhibited, leading to the accumulation of the β-catenin protein. In this sense, it is possible that RASSF1C could exert an opposite effect than RASSF1A in the process of carcinogenesis. Although previous reports have attributed RASSF1C with tumor suppressor activity, other studies have suggested that RASSF1C could activate osteoblast cell proliferation.Citation19 Together, these data suggest that RASSF1C, unlike RASSF1A, is not a tumor suppressor but instead may play a role in stimulating proliferation and invasion in breast cancer cells.
HYAL1 and CACNA2D2 were the most positively modulated genes after treatments with 5-Aza-dC alone or with TSA. Non-tumorigenic cell lines were found to have even higher expression levels of these genes after the treatments. HYAL1 belongs to the hyaluronidase (HAases) family of enzymes that degrade hyaluronic acid (HA). There are six hyaluronidase genes in the human genome: three are arranged at the 3p21.3 suppressor tumor cluster (HYAL1, HYAL2 and HYAL3). HAase levels were demonstrated to be elevated in breast tumors, and RT-PCR analysis has detected the expression of HYAL2 and HYAL3 in breast cancer tissues,Citation34 suggesting an association between HAase and the tumor invasive/metastatic phenotype.Citation35,Citation36 In accordance with the data from the present study demonstrating higher levels of HYAL2 in breast cancer cell lines compared with non-tumorigenic cell line 184A1, previous reports have shown that, whereas less invasive breast cancer cells expressed HYAL3, highly invasive cells expressed HYAL2.Citation34 Although the promoter region of these genes is characterized by a CpG island, their expression levels were not significantly increased after the treatments with the demethylating agents in our study. However, it is of note that the promoter region of the HYAL1 gene has a CpG-rich promoter that does not meet the established criteria for a CpG island, similar to that observed in the MASPIN gene (which encodes a serine protease inhibitor with tumor-suppressor activity) and the MAGE family (for which promoter hypermethylation is responsible for the restricted expression of the tumor-associated MAGE antigens).Citation3 Like MASPIN and MAGE, the present study suggests that the HYAL1 gene is clearly upregulated by epigenetic therapy. Recently, it was described that HYAL1 expression is regulated by methylation of cytosine at positions -71 and -59, which constitute portions of the binding sites for the transcription factors SP1/Egr-1.Citation37 Thus, in the present study we have included the methylation analysis by melting curve of the non-CpG promoter region of HYAL1 gene. Low levels of DNA methylation (approximately 25%) were detected in 17/20 cell lines.
The HYAL1 gene encodes the major tumor-derived HAase. In bladder, prostate and head and neck carcinomas, higher levels of HA and HAases were found in tumor cells, tissues and related blood fluids and serve as biomarkers to detect high-grade bladder tumors.Citation38 In breast cancer cells, HAase levels were associated with the ability to invade through matrigel, and silencing of HYAL1 gene expression by iRNA induced cell cycle arrest and inhibition of proliferation in vitro. Tan et al. showed that HYAL1 was overexpressed in the breast cancer cell lines MDA-MB-231 and MCF7, in invasive ductal cancer tissues and in metastatic lymph nodes, compared with the nonmalignant breast cell line HBL-100 and normal breast tissues. In our study, although slightly higher levels of HYAL1 transcript were detected in the MDA-MB-231 and MCF7 cell lines, the majority of breast cancer-derived cells showed lower expression levels of this gene. Furthermore, after treatments with 5-Aza-dC and TSA, the non-tumorigenic cell lines showed a 50–150 fold increase in the expression of HYAL1 mRNA, whereas the breast cancer cell lines manifested a change of up to 50-fold relative to untreated controls. These data are relevant and indicate that epigenetic modifications of HYAL1, like the methylation levels found in our study, could affect HYAL1 expression in epithelial breast cells. At present, it is unknown whether HYAL1 functions as a tumor-promoter or tumor-suppressor gene. These controversial cellular roles are highlighted by previous studies demonstrating that silencing of HYAL1 reduced tumor growth and invasion, suggesting a tumor promoting function. In contrast, the overexpression of HYAL1 in tumor cells induced apoptosis and inhibited tumor formation.Citation38 More studies are clearly necessary to establish a correlation between the regulation of tissue-specific expression and methylation at non-CpG islands and to explain how this correlation plays a role in breast cancer cells.
The CACNA2D2 gene was also downregulated in breast cancer cells, and expression levels were reversed by treatment with 5-Aza-dC both in the presence or absence of TSA. Nevertheless, all cell lines were unmethylated at the CpG island promoter of CACNA2D2. This gene encodes a member of the α-2/delta subunit family, a protein of the voltage-dependent calcium channel complex. Initial studies demonstrated frequent loss of CACNA2D2 expression in lung cancer cell lines.Citation20,Citation21 In nasopharyngeal and glioma cell lines, loss of CACNA2D2 expression occurred concomitantly with DNA methylation. However, DNA methylation was less frequently observed in primary tumors.Citation39,Citation40 Overexpression of CACNA2D2 induced apoptosis in lung cell lines, in which the levels of intracellular free Ca2+ were elevated in AdCACNA2D2-transduced cells compared with the controls, followed by depolarization of the mitochondrial membrane.Citation41 These results led to the suggestion that re-expression of this gene could induce apoptosis. The findings of the present study provide additional evidence that CACNA2D2 may be a putative tumor suppressor gene that is epigenetically regulated in breast cancer cells.
Among the genes located between HYAL1 and CACNA2D2, expression of three genes (TUSC2, ZMYND10 and NPRL2) positively correlated with RASSF1 or with the closest gene (i.e., ZMYND10 positively correlated with NPRL2 and NPRL2 with TMEM115) after the combined treatment with the DNA demethylating and the histone deacetylase inhibitor drugs. These findings indicate that the genes flanking RASSF1 were simultaneously upregulated by the epigenetic treatment. As previously suggested, epigenetic changes in cancer cells could occur at the neighborhood level, in which the expression of genes residing in the same vicinity could be affected by hypermethylation of a flanking gene, leading to coordinate downregulation or inactivation mediated by chromatin modifications. However, in our study, it was observed that the promoter-associated CpG islands of the TUSC2, NPRL2 and TMEM115 genes were unmethylated and showed varying degrees of enrichment of the H3K9me3 fraction after ChIP in the untreated cells. Similar findings were first reported by Frigola et al. in colorectal cancer in which genes located at an approximately 4 Mb cluster on chromosome 2q14.2 were epigenetically suppressed even though the involved CpG islands remained unmethylated. Despite the unmethylated CpG islands, the repression of these genes on 2q14.2 was relieved by demethylating drug treatment. The effect of the 5-Aza-dC on global gene expression showed that this drug can result in variable changes in gene expression depending on the functional categories of the genes and that most genes that demonstrated altered expression levels were not regulated by promoters displaying DNA methylation prior to the treatment.Citation42
Furthermore, the fact that the expression levels of the unmethylated genes located close to RASSF1 were upregulated in tumor cell lines treated with 5-Aza-dC suggests an effect of this nucleoside analog on histone modification.Citation43 In light of the results obtained from the ChIP assays in our study, which showed less enrichment of the H3K9me3 in the breast cancer cells when compared with non-malignant cell lines, it is possible that other histone repressive marks could modulate the expression levels that were detected before the set of breast cancer cell lines studied were treated. Distinct repressive histone modifications have been frequently investigated in human cancer, H3K9me2, H3K9me3 and H3K27me. Besides the interactions between DNA methylation and H3K9me, which may establish and reinforce a silencing loop, polycomb group proteins can modify the methylation status of histone H3 on lysine 27 and recruit other members of repressor complex, including PRC1, which is able to prevent transcriptional activation.Citation44 In a recent report by Mayo et al.Citation45 the epigenetic profile of two chromosomal regions undergoing long range epigenetic silencing was investigated in colorectal cancer (2q14.2 and 5q35.2). After the treatment with 5-Aza-dC or TSA, alone or in combination, the authors classified the genes into three groups according the expression levels and the epigenetic states of their promoters: (1) silenced genes exhibiting methylated CpG island promoters and retention of bivalent chromatin epigenetic marks (i.e., H3K4me3 and H3K27), termed MBV genes (methylated and bivalent); (2) downregulated unmethylated CpG island-promoter genes and (3) low expressed and non-CpG island promoter. Interestingly, six MBV genes were reactivated after treatment with 5-Aza-dC or TSA, and a combination of both. However, the expression of these genes was restored at low levels, similar to findings observed in the present study. Alternatively, it is possible that changes in gene expression observed in the treated cells could be the result of a secondary effect, instead of direct promoter demethylation. According to this idea, genes encoding transcription factors could became demethylated and as result upregulated. In turn, these transcription factors could initiate a cascade of transcriptional changes.Citation42
Previous studies have identified genomic regions showing coordinated epigenetic deregulation of neighboring genes in cancer cells as an event independent from copy number alterations. However, allelic losses and deletions at the 3p21.3 region are frequently observed in the most common human tumors, including breast cancer. Therefore the parallel analysis of epigenetic and copy number alterations, as it was performed in this study, is imperative and have demonstrated for our cell lines that DNA copy number changes are implicated in the impairment of the corresponding expression levels of the genes studied. However, although not completely characterized, epigenetic changes at 3p21.3 can also contribute to the repression of several TSG. Altogether, these data suggest that genetic and epigenetic changes in this tumor suppressor cluster can affect a wide spectrum of relevant biological pathways. Further studies could lead to the characterization of the new deregulated networks in human cancer.
Materials and Methods
Cell lines and cell culture.
Seventeen human breast cancer cell lines (BT-20, BT-474, BT-483, BT-549, Hs 578T, MCF7, MDA-MB-134-IV, MDA-MB-231, MDA-MB-361, MDA-MB-415, MDA-MB-436, MDA-MB-453, MDA-MB-468, SK-BR-3, T-47D, ZR-75-1 and ZR-75-30) and three non-tumorigenic epithelial breast cell (184A1, 184B5 and MCF 10A) were obtained from the Tissue Culture Shared Resource (TCSR) at the Lombardi Comprehensive Cancer Center, Georgetown University, Washington DC. Cell lines were cultured at 37°C in a humidified environment containing 5% CO2. DMEM medium containing 10% fetal bovine serum (FBS) was used for the MDA-MB-134-IV, SK-BR-3, MDA-MB-453, MDA-MB-231, MDA-MB-361, MDA-MB-415, MDA-MB-436 and MDA-MB-468 cell lines. This medium was supplemented with 1% Non-Essential Amino Acids (NEAA) for the BT-20 and MCF7 cells and with 10 µg/ml insulin for the Hs 578T cells. RPMI medium containing 10% FBS was used in cultures of T-47D cells and was supplemented with 1% pyruvate for the ZR-75-1 and ZR-75-30 cells. The BT-549, BT-483 and BT-474 cells were maintained in RPMI medium containing 10% FBS supplemented with 10 µg/ml insulin and 1% pyruvate. The 184A, 184B5 and MCF 10A cells were cultured in serum-free Mammary Epithelial Basal Medium (MEBM) supplemented with Epidermal Growth Factor (EGF), hydrocortisone, L-glutamine and insulin using Singlequot reagent packs from Lonza, along with 0.005 mg/mL transferrin for 184A1 and 5% horse serum for MCF 10A.
5-Aza-2′-deoxycytidine and trichostatin A treatment of cells.
The breast cell lines were seeded at a density of 1 × 106 cells in 25 cm3 culture flask. After 24 h of growth, 1 µM 5-Aza-2′-deoxycytidine (5-Aza-dC; Sigma) was added to the medium, and the cells were incubated for 96 h. Due to its chemical instability, 5-Aza-dC was added to the fresh medium every 24 h. Alternatively, the cells were treated with 150 ng/mL trichostatin A (TSA; Sigma) for 24 h. To assess the combined effect of both drugs, we performed the co-treatment of cells with 5-Aza-dC and TSA as follows: 5-Aza-dC was added initially for 96 h, after which it was removed, and TSA was added for an additional period of 24 h. All experiments were performed in duplicate. A mock treatment was done with DMSO. The cells were then allowed to recover for 24 h prior to harvesting. The genomic DNA and total RNA were extracted from the cell lines after the drug treatments and were used for HRM analysis and reverse transcription-PCR (RT-PCR).
Total RNA extraction and quantitative real-time RT-PCR.
The expression of the SEMA3B, HYAL3, HYAL1, HYAL2, TUSC2, RASSF1, ZMYND10, NPRL2, TMEM115 and CACNA2D2 genes in the breast cell lines were analyzed by quantitative real-time RT-PCR. Total RNA was extracted from the untreated and 5-Aza-dC and TSA treated cultured breast cell lines using the RNeasy Mini Kit (Qiagen) and subsequently treated with DNase I to degrade any contaminating genomic DNA. mRNA was reverse transcribed from 1 µg of total RNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The reaction was primed with random primers in a 20 µL volume, according to the manufacturer's instructions. The reverse transcription reaction was diluted 1:20 with sterile H2O before addition to the RT-PCR reaction. The diluted reverse transcription reaction (1 µL) was used in a 10 µL reaction with 2x TaqMan Universal PCR Master Mix and 0.5 µL of the 20X TaqMan Gene Expression Assay (primer/probe sets) (Applied Biosystems) for SEMA3B (Hs01090156_m1), HYAL3 (Hs00185910_m1), HYAL1 (Hs00201046_m1), HYAL2 (Hs00186841_m1), TUSC2 (Hs00200725_m1), RASSF1 (Hs00200394_m1 all transcripts and Hs00945257_m1 for isoform A), ZMYND10 (Hs00210720_m1), NPRL2 (Hs00198012_m1), TMEM115 (Hs00183656_m1), and CACNA2D2 (Hs01021049_m1). The reactions were performed in duplicate in the StepOne Real-time PCR System (Applied Biosystems), and the results were calculated using the ΔΔCt-method normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression levels. This endogenous control was experimentally chosen based on amplification of cDNA obtained from treated and untreated MCF7 and MDA-MD-231 cells in the TaqMan® Human Endogenous Control Plate (Applied Biosystems). The data were evaluated using GeNorm software to detect the most stable genes under these experimental conditions.
Chromatin immunoprecipitation assays.
Chromatin immunoprecipitation (ChIP) assays were performed on breast cell lines using the MAGnify Chromatin Immunoprecipitation System (Invitrogen), according to the manufacturer's instructions. The fixed chromatin complexes were immunoprecipitated using 1 µg of either an anti-trimethyl-histone H3K9 antibody or an anti-acetyl-histone H3K9 antibody (Invitrogen). Controls were also performed using rabbit and mouse antibodies and showed negligible levels of background precipitation. Immunoprecipitated DNA was quantified by quantitative real-time PCR. Oligonucleotide primer sequences for the TMEM115, TUSC2, NPRL2 and ZMYND10 genes were designed using the Primer Express Software (Applied Biosystems), as indicated in Table S2. The amount of immunoprecipitated target was measured using the StepOne Real-time PCR System (Applied Biosystems). Reactions containing 10 µL volume were set up using 2x SYBR GREEN PCR Master Mix (Applied Biosystems), 200 nM of each primer and 2 µL of immunoprecipitated DNA, no-antibody control, or input chromatin for each PCR. Each experiment was set up in duplicate. The cycling conditions were as follows: 95°C for 10 min, 95°C for 15 sec and 60°C for 1 min, repeated for 40 cycles, followed by a melt curve stage. The data were normalized using the input control and the chromatin enrichment was calculated based on the formula Input Enrichment = 2(Average CtInput - Average CtIP) × dilution factor.
DNA isolation and sodium bisulfite treatment.
Genomic DNA was prepared from untreated breast cell lines by proteinase K digestion followed by conventional extraction using a phenol/chloroform protocol. One microgram of genomic DNA was subjected to sodium bisulfite treatment using the EpiTect® Bisulphite Kit (Qiagen) according to the manufacturer's instructions. Ten nanograms of eluted DNA were used directly for the HRM assays.
High resolution melting (HRM) analysis.
After DNA sodium bisulfate treatment, PCR amplification and HRM were performed on the StepOne Real-time PCR System (Applied Biosystems), according to the manufacturer's instructions. The primer sequences for CACNA2D2, HYAL1, RASSF1, TMEM115, TUSC2, NPRL2 and ZMYND10 are listed in Table S2. PCR was performed in a 20 µL volume containing 1x MeltDoctor HRM Master Mix (Applied Biosystems), 200 nM of each primer and 10 ng bisulfite-treated DNA template. A standard curve with known methylation ratios (100, 75, 50, 25 and 0%) was included in each assay to estimate the methylation ratio of each sample using CpG Methylated HeLa Genomic DNA (New England BioLabs) and Epitect Unmethylated Human Control DNA as references. Methylation standards were performed in duplicate, and cell line samples were performed in triplicate. The cycling conditions were as follows: 1 cycle of 95°C for 10 min, 40 cycles of 95°C for 15 sec, 60°C for 1 min; followed by an HRM step of 95°C for 15 sec, 60°C for 1 min, 95°C for 15 sec, and continuous acquisition from 60 to 95°C at 0.3%. HRM data were analyzed using the High Resolution Melting Software (Applied Biosystems). The data were visualized as normalized melting curves and difference plots.
Analysis of DNA copy number by qPCR.
The primer sequences for the RASSF1, TMEM115, TUSC2, NPRL2 and ZMYND10 genes are the same as used in the ChIP analysis. Reactions of 10 µL were set up using 2X SYBR GREEN PCR Master Mix (Applied Biosystems), 200 nM of each primer, and 10 ng of DNA in each PCR reaction, set up in duplicate. The cycling conditions were as follows: 10 min at 95°C, then 40 cycles at 95°C for 15 sec and 60°C for 1 min, followed by a melt curve stage, in a total volume 10 µl, in duplicate. The qPCR data were analyzed using the 2-ΔΔct method based on the DNA copy number ratio of the target gene compared with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a reference gene in a given cell line sample relative to two standard deviations from the normal pool DNA sample.
Statistical analysis.
All data are presented as the mean ± SD of independent measurements. For statistical comparisons of gene expression levels between two groups (non-tumorigenic vs. breast cancer cells), the non-parametric Mann-Whitney test was used. The Spearman correlation coefficient r was determined for every paired data set after each treatment. All data were analyzed using GraphPad Prism for Windows, version 5.0. p values of < 0.05 were regarded as statistically significant. Data Assist™ software (Applied Biosystems) was used to display graphically the results of the hierarchical clustering analysis of gene expression levels before and after each treatment, based on Euclidean distance and average linkage.
Conclusion
In summary, we have described that, among the candidate TSGs mapped to 3p21.3, the RASSF1A isoform was epigenetically silenced in most of the epithelial breast cell lines analyzed, whereas the RASSF1C isoform was overexpressed only in a subset of breast cancer cell lines. The treatment of the cell lines with the demethylating agent 5-Aza-dC, alone or in combination with TSA, led to a significant increase in the expression levels of the HYAL1 and CACNA2D2 genes. Genes that mapped closest to the RASSF1 showed a more modest increase in expression levels, but pair-wise analysis showed that the RASSF1 gene was co-regulated with TUSC2, ZMYND10 and NPRL2 genes. Furthermore, the promoter regions of these genes were unmethylated at the associated CpG islands in the untreated cells and showed low levels of trimethylation at lysine 9 of the H3 histone. These data suggest that other repressive chromatin modifications should mediate the epigenetic repression of neighboring genes in the 3p21.3 cluster.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| 5-Aza-dC | = | 5-Aza-2′-deoxycytidine |
| bp | = | base pair |
| CACNA2D2 | = | calcium channel, voltage-dependent, alpha2/delta subunit gene |
| cDNA | = | complementary DNA |
| ChIP | = | chromatin immunoprecipitation |
| chr3 | = | chromosome 3 |
| CpG | = | cytosine-guanine dinucleotide |
| CYB561D2 | = | cytochrome b-561 domain containing 2/101F6 gene |
| DMSO | = | dimethyl sulfoxide |
| DNA | = | deoxyribonucleic acid |
| EGF | = | epidermal growth factor |
| Egr-1 | = | early growth response |
| FBS | = | fetal bovine serum |
| H3K9ac | = | histone H3 lysine acetylation |
| H3K9me3 | = | histone H3 lysine trimethylation |
| HA | = | hyaluronic acid |
| HAase | = | hyaluronidase |
| HOX | = | homeobox gene |
| HRM | = | high resolution melting |
| HYAL1 | = | hyaluronidase type 1 gene |
| HYAL2 | = | hyaluronidase type 2 gene |
| HYAL3 | = | hyaluronidase type 3 gene |
| i.e. | = | id est |
| iRNA | = | interference RNA |
| Kb | = | kilo base |
| LOH | = | loss of heterozygosity |
| LRES | = | long range epigenetic silencing |
| MAGE | = | melanom antigen gene |
| MEBM | = | mammary epithelial basal medium |
| NEAA | = | non-essential amino acids |
| NPRL2 | = | nitrogen permease regulator-like2 gene |
| PHDC | = | protocadherin family genes |
| RASSF | = | ras-association domain family gene |
| RNA | = | ribonucleic acid |
| RT-PCR | = | reverse transcription polymerase chain reaction |
| SD | = | standard deviation |
| SEMA3B | = | semaphorin 3B gene |
| SP1 | = | Sp transcription factor |
| TMEM115 | = | transmembrane protein 115 gene |
| TSA | = | trichostatin A |
| TSG | = | tumor suppressor gene |
| TUSC2 | = | tumor suppressor candidate 2 gene |
| ZMYND10 | = | zinc finger, MYND-type containing 10 gene |
Figures and Tables
Figure 1 (A) Scheme of the genes and CpG islands organized according to their physical location on 3p21.3 (UCSC Genome Browser on Human, assembly GRCh37/hg19, http://genome.ucsc.edu). (B) Differential gene expression among breast cancer cell lines and non-tumorigenic breast cells. The HYAL2, TUSC2 and CACNA2D2 genes were differentially expressed in breast cancer compared with non-tumorigenic cell lines. Global RASSF1 transcripts showed a trend to overexpression in breast cancer cells lines compared with non-tumorigenic epithelial breast cells 184A1. All quantifications were performed by real-time PCR using specific hydrolysis probes for each target gene normalized to the endogenous control expression levels (GAPDH gene).
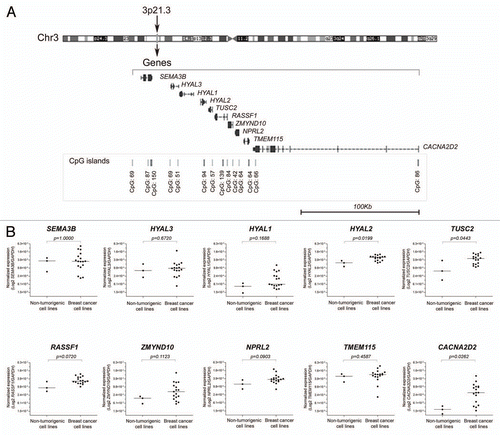
Figure 2 Drug induced expression levels of global RASSF1 transcripts and A isoform of RASSF1 gene in breast cancer cell lines. (A) Expression levels in untreated cell lines and after treatment with trichostatin A (TSA). Only two cell lines, Hs 578T and MDA-MB-415, expressed the A isoform of RASSF1 gene. As expected, the TSA treatment alone did not affect DNA methylation and as a result the expression levels of this isoform. (B) The treatment with 5-Aza-dC, isolated or combined with TSA, resulted in the reactivation of A isoform of RASSF1 gene. Gene expression levels were normalized to GAPDH gene. Error bars indicate standard deviation from duplicates.
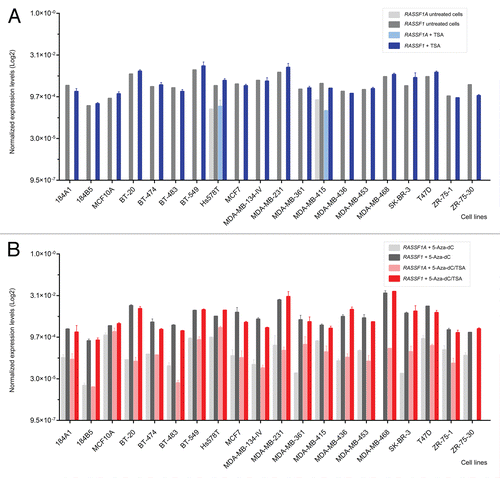
Figure 3 Expression profile summarized in a heat map. Effect of treatments with 5-Aza-dC and with 5-Aza-dC combined with TSA leading to the increase in the expression levels of the tumor suppressor cluster, except in the cell lines that were unmethylated at CpG island 1 of RASSF1A promoter (MDA-MB-415) or showing low levels of methylation (Hs 578T and BT-20).
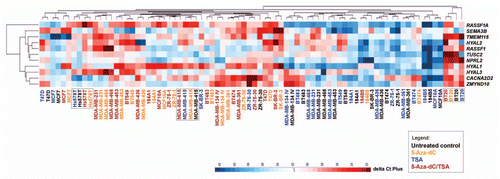
Figure 4 Effect of epigenetic drugs (5-Aza-dC and trichostatin A, TSA) on gene expression. (A and B) All genes were upregulated by treatment with 5-Aza-dC, isolated and combined with TSA. HYAL1 and CACNA2D2 genes showed the highest expression levels induced by these drugs. (C) Effects on gene transcript levels after isolated treatment with TSA. For each gene, the results are expressed as fold change relative to the reference (respective untreated control cell line) calculated using the ΔΔCt-method normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression levels and represent the mean of real-time PCR results of independent biological replicas.
Figure 5 Analysis of Chromatin Immunoprecipitation (ChIP) of genes close to RASSF1 in breast cancer and epithelial mammary breast cell lines. The amount of immunoprecipitated target (using antibodies against H3K9 acetylation and H3K9 trimethylation) was quantified by real time PCR and plotted as the ratio of immunoprecipitated DNA to input.
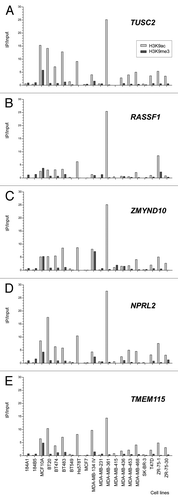
Table 1 Pair-wise correlation coefficient matrix of expression levels of contiguous genes mapped across 3p21.3 after treatments with 5-Aza-dC and 5-Aza-dC/TSA
Additional material
Download Zip (213.2 KB)Acknowledgments
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP grant# 07/59110-9) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Capes, Brazil. We are grateful to the Natállia Carrion Teodoro for her technical assistance.
The Tissue Culture Shared Resource (TCSR) of Georgetown University Lombardi Comprehensive Cancer Center is partially supported by NIH/NCI Cancer Center Support Grant (CCSG) Grant P30-CA51008, to the Lombardi Comprehensive Cancer Center.
References
- Chari R, Coe BP, Vucic EA, Lockwood WW, Lam WL. An integrative multi-dimensional genetic and epigenetic strataegy to identify aberrant genes and pathways in cancer. BMC Biosystems Biology 2010; 4:67 - 80; http://dx.doi.org/10.1186/1752-0509-4-67
- Chari R, Thu KL, Wilson IM, Lockwood WW, Lonergan KM, Coe BP, et al. Integrating the multiple dimensions of genomic and epigenomic landscapes of cancer. Cancer Metastasis Rev 2010; 29:73 - 93; PMID: 20108112; http://dx.doi.org/10.1007/s10555-010-9199-2
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 692; PMID: 17320506; http://dx.doi.org/10.1016/j.cell.2007.01.029
- Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358:1148 - 1159; PMID: 18337604; http://dx.doi.org/10.1056/NEJMra072067
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis 2010; 31:27 - 36; PMID: 19752007; http://dx.doi.org/10.1093/carcin/bgp220
- Novak P, Jensen T, Oshiro MM, Wozniak RJ, Nouzova M, Watts GS, et al. Epigenetic inactivation of the HOXA gene cluster in breast cancer. Cancer Res 2006; 66:10664 - 10670; PMID: 17090521; http://dx.doi.org/10.1158/0008-5472.CAN-06-2761
- Novak P, Jensen T, Oshiro MM, Watts GS, Kim CJ, Futscher BW. Agglomerative epigenetic aberrations are a common event in human breast cancer. Cancer Res 2008; 68:8616 - 8625; PMID: 18922938; http://dx.doi.org/10.1158/0008-5472.CAN-08-1419
- Clark SJ. Action at a distance: epigenetic silencing of large chromosomal regions in carcinogenesis. Hum Mol Genet 2007; 16:88 - 95; PMID: 17613553; http://dx.doi.org/10.1093/hmg/ddm051
- Frigola J, Song J, Stirzaker C, Hinshelwood RA, Peinado MA, Clark SJ. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat Genet 2006; 38:540 - 549; PMID: 16642018; http://dx.doi.org/10.1038/ng1781
- Hitchins MP, Lin VA, Buckle A, Cheong K, Halani N, Ku S, et al. Epigenetic inactivation of a cluster of genes flanking MLH1 in microsatellite-unstable colorectal cancer. Cancer Res 2007; 67:9107 - 9116; PMID: 17909015; http://dx.doi.org/10.1158/0008-5472.CAN-07-0869
- Karpinski P, Ramsey D, Grzebleniak Z, Sasiadek MM, Blin N. The CpG isaland methylator phenotype correlates with long range epigenetic silencing in colorectal cancer. Mol Cancer Res 2008; 6:585 - 591; PMID: 18403637; http://dx.doi.org/10.1158/1541-7786.MCR-07-2158
- Mayor R, Casadomé L, Azuara D, Moreno V, Clark SJ, Capellà G, et al. Long-range epigenetic silencing at 2q14.2 affects most human colorectal cancers and may have application as a non-invasive biomarker of disease. Br J Cancer 2009; 100:1534 - 1539; PMID: 19384295; http://dx.doi.org/10.1038/sj.bjc.6605045
- Stransky N, Vallot C, Reyal F, Bernard-Pierrot I, de Medina SG, Segraves R, et al. Regional copy number-independent deregulation of transcription in cancer. Nat Genet 2006; 38:1386 - 1396; PMID: 17099711; http://dx.doi.org/10.1038/ng1923
- Devaney J, Stirzaker C, Qu W, Song JZ, Statham AL, Patterson KI, et al. Epigenetic deregulation across chromosome 2q14.2 differentiates normal from prostate cancer and provides a regional panel of novel DNA methylation cancer biomarkers. Cancer Epidemiol Biomarkers Prev 2011; 20:148 - 159; PMID: 21098650; http://dx.doi.org/10.1158/1055-9965.EPI-10-0719
- Coolen MW, Stirzaker C, Song JZ, Statham AL, Kassir Z, Moreno CS, et al. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity. Nat Cell Biol 2010; 12:235 - 246; PMID: 20173741
- Dallosso AR, Hancock AL, Szemes M, Moorwood K, Chilukamarri L, Tsai HH, et al. Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms' tumor. PLoS Genet 2009; 5:1000745; PMID: 19956686; http://dx.doi.org/10.1371/journal.pgen.1000745
- Martinez A, Walker RA, Shaw JA, Dearing SJ, Maher ER, Latif F. Chromosome 3p allele loss in early invasive breast cancer: detailed mapping and association with clinicopathological features. Mol Pathol 2001; 54:300 - 306; PMID: 11577171; http://dx.doi.org/10.1136/mp.54.5.300
- Maitra A, Wistuba II, Washington C, Virmani AK, Ashfaq R, Milchgrub S, et al. High-resolution chromosome 3p allelotyping of breast carcinomas and precursor lesions demonstrates frequent loss of heterozygosity and a discontinuous pattern of allele loss. Am J Pathol 2001; 159:119 - 130; PMID: 11438460; http://dx.doi.org/10.1016/S0002-9440(10)61679-3
- van der Weyden L, Adams DJ. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim Biophys Acta 2007; 1776:58 - 85; PMID: 17692468; http://dx.doi.org/10.1016/j.bbcan.2007.06.003
- Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer: from epigenetic silencing to functional characterization. Biochim Biophys Acta 2009; 1796:114 - 128; PMID: 19344752; http://dx.doi.org/10.1016/j.bbcan.2009.03.004
- Hesson LB, Cooper WN, Latif F. Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene 2007; 26:7283 - 7301; PMID: 17533367; http://dx.doi.org/10.1038/sj.onc.1210547
- Ji L, Minna JD, Roth JA. 3p21.3 tumor suppressor cluster: prospects for translational applications. Future Oncol 2005; 1:79 - 92; PMID: 16555978; http://dx.doi.org/10.1517/14796694.1.1.79
- Sekido Y, Ahmadian M, Wistuba I, Latif F, Bader S, Wei MH, et al. Cloning of a breast cancer homozygous deletion junction narrows the region of search for a 3p21.3 tumor suppressor gene. Oncogene 1998; 16:3151 - 3157; PMID: 9671394; http://dx.doi.org/10.1038/sj.onc.1201858
- Lerman MI, Minna JD. The 630 Kb lung cancer homozygous deletion region on chromosome 3p21.3: identification and evaluation of the resident candidate tumor suppressor genes. Cancer Res 2000; 60:6116 - 6133; PMID: 11085536
- Hsu PY, Hsu HK, Singer GAC, Yan PS, Rodriguez BAT, Liu JC, et al. Estrogen-mediated epigenetic repression of large chromosomal regions through DNA looping. Genome Res 2010; 20:733 - 744; PMID: 20442245; http://dx.doi.org/10.1101/gr.101923.109
- Hesson L, Bièche I, Krex D, Criniere E, Hoang-Xuan K, Maher ER, et al. Frequent epigenetic inactivation of RASSF1A and BLU genes located within the critical 3p21.3 region in gliomas. Oncogene 2004; 23:2408 - 2419; PMID: 14743209; http://dx.doi.org/10.1038/sj.onc.1207407
- Ito M, Ito G, Kondo M, Uchiyama M, Fukui T, Mori S, et al. Frequent inactivation of RASSF1A, BLU and SEMA3B on3p21.3 by promoter hypermethylation and allele loss in non-small cell lung cancer. Cancer Lett 2005; 225:131 - 139; PMID: 15922865; http://dx.doi.org/10.1016/j.canlet.2004.10.041
- Senchenko VN, Anedchenko EA, Kondratieva TT, Krasnov GS, Dmitriev AA, Zabarovska VI, et al. Simultaneous downregulation of tumor suppressor genes RBSP3/CTDSPL, NPRL2/G21 and RASSF1A in primary non-small cell lung cancer. BMC Cancer 2010; 1:10 75; PMID: 20193080; http://dx.doi.org/10.1186/1471-2407-10-75
- Lai HC, Lin YW, Chang CC, Wang HC, Chu TW, Yu MH, et al. Hypermethylation of two consecutive tumor suppressor genes, BLU and RASSF1A, located at 3p21.3 in cervical neoplasias. Gynecol Oncol 2007; 104:629 - 635; PMID: 17097722; http://dx.doi.org/10.1016/j.ygyno.2006.10.003
- Lai HC, Lin YW, Chang CC, Wang HC, Chu TW, Yu MH, et al. Hypermethylation of two consecutive tumor suppressor genes, BLU and RASSF1A, located at 3p21.3 in cervical neoplasias. Gynecol Oncol 2007; 104:629 - 635; PMID: 17097722; http://dx.doi.org/10.1016/j.ygyno.2006.10.003
- Dworkin AM, Huang TH, Toland AE. Epigenetic alterations in the breast: Implications for breast cancer detection, prognosis and treatment. Semin Cancer Biol 2009; 19:165 - 171; PMID: 19429480; http://dx.doi.org/10.1016/j.semcancer.2009.02.007
- Reeves ME, Baldwin SW, Baldwin ML, Chen ST, Moretz JM, Aragon RJ, et al. Ras-association domain family 1C protein promotes breast cancer cell migration and attenuates apoptosis. BMC Cancer 2010; 10:562; PMID: 20955597; http://dx.doi.org/10.1186/1471-2407-10-562
- Estrabaud E, Lassot I, Blot G, Le Rouzic E, Tanchou V, Quemeneur E, et al. RASSF1C, an isoform of the tumor suppressor RASSF1A, promotes the accumulation of beta-catenin by interacting with betaTrCP. Cancer Res 2007; 67:1054 - 1061; PMID: 17283138; http://dx.doi.org/10.1158/0008-5472.CAN-06-2530
- Udabage L, Brownlee GR, Nilsson SK, Brown TJ. The overexpression of HAS2, Hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Exp Cell Res 2005; 310:205 - 217; PMID: 16125700; http://dx.doi.org/10.1016/j.yexcr.2005.07.026
- Bertrand P, Girard N, Duval C, d'Anjou J, Chauzy C, Menard JF, et al. Increased hyaluronidase levels in breast tumor metastases. Int J Cancer 1997; 73:327 - 331; PMID: 9359477; http://dx.doi.org/10.1002/(SICI)1097-0215(19971104)73:3<327::AID-IJC4>3.0.CO;2-1
- Tan JX, Wang XY, Li HY, Su XL, Wang L, Ran L, et al. HYAL1 overexpression is correlated with the malignant behavior of human breast cancer. Int J Cancer 2011; 128:1303 - 1315; PMID: 20473947; http://dx.doi.org/10.1002/ijc.25460
- Lokeshwar VB, Gomez P, Kramer M, Knapp J, McCornack MA, Lopez LE, et al. Epigenetic regulation of HYAL-1 hyaluronidase expression. identification of HYAL-1 promoter. J Biol Chem 2008; 283:29215 - 29227; PMID: 18718911; http://dx.doi.org/10.1074/jbc.M801101200
- Lokeshwar VB, Selzer MG. Hyalurondiase: both a tumor promoter and suppressor. Semin Cancer Biol 2008; 18:281 - 287; PMID: 18448355; http://dx.doi.org/10.1016/j.semcancer.2008.03.008
- Chow LS, Lo KW, Kwong J, To KF, Tsang KS, Lam CW, et al. RASSF1A is a target tumor suppressor from 3p21.3 in nasopharyngeal carcinoma. Int J Cancer 2004; 109:839 - 847; PMID: 15027117; http://dx.doi.org/10.1002/ijc.20079
- Hesson L, Bièche I, Krex D, Criniere E, Hoang-Xuan K, Maher ER, et al. Frequent epigenetic inactivation of RASSF1A and BLU genes located within critical 3p21.3 region in gliomas. Oncogene 2004; 23:2408 - 2419; PMID: 14743209; http://dx.doi.org/10.1038/sj.onc.1207407
- Carboni GL, Gao B, Nishizaki M, Xu K, Minna JD, Roth JA, et al. CACNA2D2-mediated apoptosis in NSCLC cells is associated with alterations of the intracellular calcium signaling and disruption of mitochondria membrane integrity. Oncogene 2003; 22:615 - 626; PMID: 12555074; http://dx.doi.org/10.1038/sj.onc.1206134
- Komashko VM, Farnham PJ. 5-azacytidine treatment reorganizes genomic histone modification patterns. Epigenetics 2010; 5:229 - 240; PMID: 20305384; http://dx.doi.org/10.4161/epi.5.3.11409
- Nguyen CT, Weisenberger DJ, Velicescu M, Gonzales FA, Lin JC, Liang G, et al. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res 2002; 62:6456 - 6461; PMID: 12438235
- Kondo Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Med J 2009; 50:455 - 463; PMID: 19718392; http://dx.doi.org/10.3349/ymj.2009.50.4.455
- Mayor R, Munõz M, Coolen MW, Custodio J, Esteller M, Clark SJ, et al. Dynamics of bivalent chromatin domains upon drug induced reactivation and resilencing in cancer cells. Epigenetics 2011; 6:1138 - 1148; PMID: 21852760