Abstract
5-methyl-C (5mC) and 5-hydroxymethyl-C (5hmC) are epigenetic marks with well known and putative roles in gene regulation, respectively. These two DNA covalent modifications cannot be distinguished by bisulfite sequencing or restriction digestion, the standard methods of 5mC detection. The methylated CpG island recovery assay (MIRA), however, specifically detects 5mC but not 5hmC. We further developed MIRA for the analysis of allele-specific CpG methylation at differentially methylated regions (DMRs) of imprinted genes. MIRA specifically distinguished between the parental alleles by capturing the paternally methylated H19/Igf2 DMR and maternally methylated KvDMR1 in mouse embryo fibroblasts (MEFs) carrying paternal and maternal duplication of mouse distal Chr7, respectively. MIRA in combination with multiplex single nucleotide primer extension (SNuPE) assays specifically captured the methylated parental allele from normal cells at a set of maternally and paternally methylated DMRs. The assay correctly recognized aberrant biallelic methylation in a case of loss-of imprinting. The MIRA-SNuPE assays revealed that placenta exhibited less DNA methylation bias at DMRs compared to yolk sac, amnion, brain, heart, kidney, liver and muscle. This method should be useful for the analysis of allele-specific methylation events related to genomic imprinting, X chromosome inactivation and for verifying and screening haplotype-associated methylation differences in the human population.
Introduction
DNA CpG methylation is a major epigenetic mechanism implicated in gene regulation, normal development, differentiation, genomic imprinting, X-chromosome inactivation, transgenerational epigenetic inheritance and disease. It is important to correctly recognize 5-methylcytosine (5mC) in DNA. It is especially crucial to distinguish 5mC from another DNA covalent modification, 5-hydroxymethylcytosine (5hmC),Citation1,Citation2 that has recently come into the focus of attention being a potentially very important epigenetic mark.Citation3,Citation4 Significant amount of 5hmC exists in brain cells and ES cells but 5hmC is depleted in cancer cell lines. The Tet family proteins are responsible for catalyzing the oxidation of 5mC to 5hmC. The Tet proteins are expressed in a wide range of cell types.Citation5 It is therefore conceivable that 5hmC is present in several tissues. 5hmC may be an intermediate product in DNA demethylationCitation4,Citation5 and, as such, may be involved in diverse biological functions.
The standard method for detecting 5mC has been bisulfite sequencing and its derivatives, such as COBRA. Sodium bisulfite distinguishes between cytosine and 5mC, converting cytosine but not 5mC into uracil.Citation6,Citation7 However, the CpG sites that remain unconverted by sodium bisulfite can be either 5mC or 5hmC.Citation8,Citation9 DNA digestion by methylation-sensitive restriction enzymes and detection of the products by Southern blotting or PCR are alternative detection methods. However, methylation-sensitive restriction enzymes are equally blocked by 5mC or 5hmC.Citation8,Citation9 The methylated CpG island recovery assay (MIRA)Citation10,Citation11 is suitable for specific methyl-C detection, because unlike bisulfite sequencingCitation8,Citation9 or restriction digestion,Citation8,Citation9 it doesn't recognize 5hmC.Citation9 This method is based on affinity capture of methylated DNA by recombinant methyl-CpG binding protein 2b (MBD2b) and MBD3L1. We explored the potential of MIRA to measure allele-specific CpG methylation at imprinted regions ().
Imprinted genes exhibit parent-of-origin specific monoallelic expression. In somatic cells, the paternally and maternally inherited alleles of imprinted genes exist in opposite epigenetic states, characterized by allele-specific DNA methylation at DMRs (). Gamete-specific DNA methylation marks are established at paternally or maternally methylated germ line DMRsCitation12,Citation13 in the male and female germ lines, respectively, by the de novo DNA methyltransferases Dnmt3a and Dnmt3b together with Dnmt3LCitation14–Citation16 and are maintained by Dnmt1 during somatic cell divisions.Citation17 DNA methylation at DMRs is essential for the allele-specific expression of most imprinted genes.Citation17
DMRs that control the monoallelic expression of the associated genes in the respective domainsCitation18–Citation25 are considered imprinting control regions (ICRs). For example, in the H19/Igf2 imprinted region, a paternally methylated DMR, regulates parental allele-specific expression of the oppositely imprinted H19 and Igf2 genes.Citation21,Citation26,Citation27 CTCF insulator binds in the unmethylated maternal ICR allele and blocks communication between the Igf2 promoters and the shared downstream enhancers. CTCF binding is inhibited in the paternal ICR allele by DNA methylation, allowing Igf2 promoter access to the enhancers.Citation28–Citation32 The maternally methylated Kv differentially methylated region (KvDMR1)Citation19,Citation33,Citation34 controls the Cdkn1c/Kcnq1 imprinted domain. KvDMR1 is a CpG island in the intron of the Kcnq1 transcript.Citation35 The unmethylated paternal allele produces a non-coding RNA, Kcnq1 overlapping transcript 1 (Kcnq1ot1).Citation35 This RNA is required for repressing the paternal allele of an array of maternally expressed imprinted genes in the domain.Citation19,Citation36,Citation37
Methylation analysis at DMRs is usually performed by bisulfite conversion in combination with PCR and sequencing of the amplified and cloned fragments.Citation6,Citation7 Allele-specific methylation is observed when approximately half of the sequenced clones are fully methylated and the other half is fully unmethylated.Citation38 The parental allele can be ascertained when a single nucleotide polymorphism resides in the sequenced fragment. This analysis is labor intensive and not fully quantitative. Restriction digestion with methylation sensitive enzymes using Southern blotCitation39 is an alternative method, and can be allele-specific if the region contains a suitable RFLP, but requires large amounts (about 10 µg) of genomic DNA. PCR detection of the methylation-sensitive siteCitation40 requires less DNA but is not allele-specific. The drawback of these bisulfite- and restriction enzyme based methods is that they do not distinguish between 5mC and 5hmC.Citation8,Citation9
In this study we show that the MIRA method,Citation11,Citation41 which is specific to methyl-C,Citation9 can reliably measure allele-specific CpG methylation with great precision and sensitivity.
Results
We used two model systems () for detecting allele-specific CpG methylation with MIRA. In the first system, the maternal and paternal alleles were assessed separately in MEFs carrying maternal or paternal duplication of distal chromosome 7, MatDup.dist7 and PatDup.dist7, respectively ( and C). MatDup.dist7 embryos () are maternally duplicated for the hypomethylated Igf2/H19 ICRCitation21,Citation27,Citation42,Citation43 and hypermethylated KvDMR1 ICR.Citation19,Citation33,Citation34 PatDup.dist7 embryos are paternally duplicated for the hypermethylated H19/Igf2 ICR and hypomethylated KvDMR1 (). The maternally methylated Snrpn DMR lies outside the duplicated region and serves as an internal positive control in both MatDup.dist7 and PatDup.dist7 MEFs. In the second model system, SNPs between the mother and father allow allele-specific analysis and the two parental alleles are assessed in the same cell (). For this purpose we used MEFs and fetal organs from reciprocal mouse crosses between the Mus musculus musculus, 129S1 (129) and the Mus musculus molossinus, JF1 mice.
MIRA assay correctly captures methylated DMRs.
To test whether the MIRA assay is suitable for detecting allele-specific CpG methylation at DMRs, we first subjected 500 ng of MatDup.dist7 and PatDup.dist7 MEF genomic DNA to MIRA assays and quantified the MIRA (5mC-enriched) fraction using real-time PCR at the H19/Igf2ICR and at the KvDMR1 (). MIRACitation11,Citation41 correctly captured the maternally methylated KvDMR1 allele in MatDup.dist7 MEFs but did not capture it in PatdDup.dist7 MEFs. The paternally methylated H19/Igf2 ICR was correctly captured from PatDup.dist7 MEFs but not from MatDup.dist.7 MEFs. The presumed maternal allele of the positive control Snrpn DMR was correctly captured from both MatDup.dist7 and PatDup.dist7 MEFs. The negative control, the unmethylated c-myc promoter, was correctly not captured in either MEF.
In the experiment above, MIRA proteins were presented with only the methylated or unmethylated allele of the H19/Igf2 ICR and KvDMR1 in the PatDup.dist7 or MatDup. dist7 MEFs. To test whether MIRA proteins can distinguish methylated and unmethylated alleles in the same reaction, we mixed DNA from MatDup.dist7 and PatDup.dist7 MEFs in different ratios and subjected these to MIRA assays (). Real-time PCR revealed that increasing amount of PatDup. dist7 DNA in the mix resulted in proportionally increased capture of the H19/Igf2 ICR and decreased capture of the KvDMR1. Therefore, the MIRA assay should be suitable for quantifying methylation level of two parental alleles in normal cells as well.
Testing the sensitivity of the MIRA assay at DMRs.
To test the limitations of the MIRA method we used different amounts of 129 X JF1 and JF1 X 129 MEF DNA in a series of MIRA assays (). The MIRA and the unbound fractions were quantified using real-time PCR at the H19/Igf2 ICR, KvDMR1, Snrpn DMR and c-myc promoter. The MIRA reactions captured a sufficient number of DNA molecules at the three DMRs, but not at the control c-myc promoter. The MIRA-captured fraction showed a linear response to the DNA amount. The reciprocal mouse crosses exhibited very similar levels of methylation, reflecting that each normal cell type has one copy of the methylated allele. Although expected to be equal, the unbound fraction was higher than the bound fraction at each region at 1,000 ng, 500 ng and 250 ng DNA, suggesting that the given amount (1 µg) of MBD protein is limiting for complete capture.
MIRA enrichment corresponds to the number of methylated alleles.
We performed MIRA with normal, MatDup. dist7 and PatDup.dist7 samples in the same experiment side-by-side () to test whether the MIRA enrichment follows a linear response with regard to the number of methyated alleles. 2 > 1 > 0 alleles of methylated KvDMR1 are present in MatDup.dist7 > wild type > PatDup.dist7 MEFs, and the levels of the normalized MIRA-captured fractions were consistent with these numbers. The same was true for 2 > 1 > 0 methylated alleles of the H19/Igf2 ICR in PatDup.dist7 > wild type > MatDup.dist7 MEFs. The MIRA-captured fractions had similar levels in the three types of cells consistent with one allele of methylated Snrpn DMR in each cell type. Only background level of MIRA enrichment was observed at the c-myc promoter, corresponding to zero methylated allele in each cell types. The level of MIRA capture, therefore, depended on the relative number of methylated DMR alleles in the different cell types.
Measuring allele-specific cpG methylation in normal cells.
Normal cells derived from JF1 mother X 129 father or from 129 mother X JF1 father mouse crosses () allow the assessment of allele-specific CpG methylation along the entire genome based on SNPs between the 129 and JF1 mouse lines. To measure allele-specific CpG methylation in normal cells at a set of maternally and paternally methylated germ line DMRs (), we used MIRA in combination with our recently developed 16-plex SNuPE assays.Citation44 The MIRA-SNuPE assays determine the ratio of maternal or paternal allele in the total captured fraction by measuring the incorporation of dideoxynucleotides at sites of SNPs between 129 and JF1 mouse genomic DNA ( and D). DNA methylation was measured within the maternally methylated DMRs (Zac1, Snrpn, Igf2r, Peg3, Peg1-Mest, U2af1 DMRs and the KvDMR1) and paternally methylated DMRs (H19/Igf2, Rasgrf1 DMRs and IG-DMR).Citation44 Alternative SNPs were included for the H19/Igf2 ICR (−3 kb and −2 kb from the TSS of H19). We found that each DMR, except for the U2af1 DMR, exhibited the expected parental-specific CpG methylation bias in the reciprocal crosses ( and not shown). The U2af1 SNP in the 16-plexCitation44 assay lies at the outside boundary of the U2af1 DMR, and although it is suitable to discern allele-specific chromatin features associated with the U2af1 DMR,Citation44 it does not map to an area of the actual differential CpG methylation. Other known SNPs that map to the DMR are embedded in repeat elements,Citation12 and cannot be used in SNuPE assays. The paternal allele was strongly overrepresented in the MIRA fraction at each paternally methylated DMR, the H19/Igf2 ICR, the Rasgrf1 DMR and the IG-DMR (). The maternal allele was overrepresented at each maternally methylated DMR, the Snrpn, Igf2r, Zac1 DMRs and at the KvDMR1 ().
To assess the sensitivity of allele-specific DNA methylation detection, MIRA fractions obtained with different amounts, 1,000, 500, 250, 125 and 62 ng of input DNA were subjected to Sequenom allelotyping (). We found that the allele-specific bias was similar at each of these conditions. We determined that the sensitivity of the allele-specific MIRA-SNuPE assays was sufficient at 62 ng of total input DNA.
To assess the linearity of the allele-specific response, we mixed DNA from 129 X JF1 and JF1 X 129 MEFs in different ratios (0, 10, 25, 50, 75, 90 and 100% of 129 X JF1 DNA with 100, 90, 75, 50, 25, 10 and 0% of JF1 X 129 DNA) and 500 ng DNA from each of these mixes were subjected to MIRA-SNuPE assays (). These samples (a–h samples in ) resulted in a similar level of MIRA capture as assessed by real-time PCR. SNuPE assays were used to measure the 129 and JF1 component in the MIRA fraction at the H19/Igf2 ICR (−3 kb and −4 kb upstream of the H19 TSS), at the KvDMR1 and at the Snrpn DMR. At the paternally methylated H19/Igf2 ICR, the paternal JF1 allele increased with increasing amounts of the 129 X JF1 DNA. At the same time, the 129 paternal allele decreased with the decreasing amount of JF1 X 129 DNA. The maternally methylated KvDMR1 and Snrpn DMRs showed the reciprocal trend, as expected. We concluded that the MIRA-SNuPE is capable of detecting a wide range of allele-specific differences in CpG methylation and the measurements follow a linear response.
The amount of starting material can be limiting. DNA then has to be purified from small number of cells, separately sonicated, quantified and subjected to MIRA assays. To test the applicability of MIRA-SNuPE for analyzing small numbers of cells, we prepared DNA from aliquots of 100,000 cells from 129 X JF1 and JF1 X 129 MEFs and 500 ng from this DNA preparations were subjected to MIRA-SNuPE assays. MIRA-SNuPE assays correctly quantified the maternal and paternal allele-specific CpG methylation at each maternally and paternally methylated DMR (data not shown). We concluded that the MIRA-SNuPE can be used to sensitively detect allele-specific CpG methylation in small number of cells.
Measuring allele-specific CpG methylation at the H19/Igf2ICR in normal and CTCF site mutant fetuses.
We tested the applicability of the MIRA-SNuPE for detecting loss-of imprinting situations. We, and others have shown earlier that maternal allele-specific CTCF binding in the H19/Igf2 ICR is essential for parental-allele-specific expression of H19 and Igf2 and for protecting the ICR and the distantly located Igf2 DMRs from de novo methylation in somatic cells.Citation45–Citation47 We now performed MIRA-SNuPE analysis using kidney and liver DNA from 17.5 dpc 129 X CS normal and CTCFm X CS mutantCitation47 fetuses where the CTCF site mutations resided in the maternally inherited 129-type ICR allele. A previously validated 7-plex Sequenom assayCitation44 distinguished between 129 and CS alleles in the MIRA fraction. MIRA-SNuPE results () are in agreement with our previous bisulfite sequencing and Southern blot methylation analysesCitation47,Citation66 and confirmed that whereas CpG methylation is specific to the paternal allele in normal fetuses at the ICR and Igf2 DMRs, it is biallelic in fetuses carrying ICR CTCF site mutations. This result demonstrates that allele-specific methylation analysis using MIRA-SNuPE will be applicable for detecting loss of imprinting due to biallelic methylation, for example in tumors.
Measuring allele-specific methylation at 11 DMRs in normal mouse fetuses.
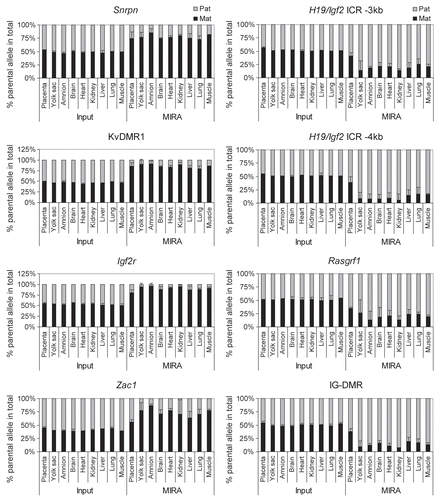
To test the applicability of the MIRA-SNuPE for allele-specific methylation analysis during fetal development, we measured the allele-specific DNA methylation in the placenta, yolk sac, amnion, brain, heart, kidney, liver, lung and muscle of 15.5 dpc JF1 X 129 normal fetuses (). DNA methylation was determined near single nucleotide polymorphisms (SNPs) within maternally methylated (Zac1, Snrpn, Igf2r, DMRs and at the KvDMR1) and paternally methylated DMRs (H19/Igf2 and Rasgrf1 DMRs and IG-DMR) by quantitative 16-plex assays.Citation44 Alternative SNPs were included for the H19/Igf2 ICR (−3 kb and −4 kb from the transcription start site of H19). MIRA was performed in three independent fetuses. We found that the MIRA fraction but not the input DNA in each fetal organ and the extraembryonic organs, amnion and yolk sac, exhibited strong bias of allele-specific CpG methylation. The allele-specific methylation, however, was less strict in the placenta. The placenta is a composite organ of cells originating from the zygote and from the mother and cells of maternal origin may mask the paternal bias at paternally methylated DMRs. The input DNA, however, was only very slightly biased towards the maternal allele, arguing that the contribution of maternal cells is not sufficient to mask the paternal methylation bias. Additionally, two maternally methylated DMRs, the Zac1 and the Igf2r DMRs, also exhibited less maternal bias in the placenta than in any other organs. In this case the methylation bias could not have been reduced because of maternal contamination. We also measured allele-specific methylation at DMRs in 13.5 dpc 129 X JF1 embryos (data not shown) and confirmed that placenta DNA exhibited weaker methylation bias than brain, liver, kidney, heart, lung, muscle, amnion and yolk sac. We determined that parental allele-specific DNA methylation is generally less biased at DMRs in the placenta than in the fetus, the amnion and the yolk sac.
Discussion
In this study we provided a novel sensitive and quantitative method for measuring allele-specific CpG methylation at DMRs of imprinted genes. We showed that the MIRA methodCitation11,Citation41 correctly captured the methylated alleles of known DMRs from MEFs carrying parental duplication of distal Chr7. The MIRA assay can be used for measuring DNA methylation not only in sequences with high but also with moderate CpG content. Whereas the KvDMR1 is a classical CpG island, the H19/Igf2 DMR is not. Additionally MIRA-SNuPE correctly detected the methylated parental alleles of a range of maternally and paternally methylated germ line DMRs in normal MEFs in a multiplex assay.
Assessing methylation using MIRA-SNuPE will have relevance in the fields of genomic imprinting and X-chromosome inactivation allowing the tissue-specific analysis of parental allele-specific 5mC patterns during embryo development. We provided quantitative information on the allele-specific CpG methylation at DMRs in fetal and extra-embryonic organs using MIRA-SNuPE assays. The MIRA will be very useful for example for analyzing cellular systems where imprinting is lost due to methylation defects such as hypomethylation or hypermethylation of DMRs, for example in cancer. Allele-specific analysis of hypermethylated DMRs is expected to reveal biallelic methylation. We demonstrated the feasibility of this approach using our previously developed mouse line containing CTCF site mutations in the H19/Igf2 ICR.Citation47 Other potential applications of the method can be to verify, quantify and screen at the population level, allele-specific haplotype-associated methylation differences resulting from deep sequencing after MeDIP, MIRA or bisulfite conversion,Citation48,Citation49 in genome-wide association studies.Citation50
SNuPE quantitation can potentially be similarly applied to methylated DNA immunoprecipitation (MeDIP)Citation51 and hydroxymethylated DNA immunoprecipitation. Unlike MeDIP, which is based on the enrichment of methyl-C using an anti-methyl-C antibody, MIRA does not require denaturation of the DNA. This is an advantage, because incomplete denaturation in MeDIP may contribute to bias against regions of high CpG content. The methylated and unmethylated alleles can be quantified after MIRA manually using traditional radioactive SNuPE assaysCitation52–Citation54 (Oates N and Szabó P, unpublished) when the Sequenom instrumentation is not available. The two alleles can also be potentially quantified using HPLC or by spotting on a membrane, similarly to MS-SNuPE.Citation55–Citation58 Multiplexing on the Sequenom platform, however, greatly reduces the work load and the amount of DNA required and allows CpG methylation studies using limiting amount of research material, such as small embryos and sorted cells.
There are multiple advantages of the current method over traditional bisulfite sequencing and restriction digestion-based methods. Restriction sites represent a very small fraction of a CpG island or a DMR. The MBD proteins bind to a large number of CpGs along a DNA fragment. Therefore, the MIRA assay, just like chromatin immunoprecipitation, is responsive to the epigenetic status of an entire DNA fragment and not only a single methylation sensitive restriction site. Bisulfite sequencing is labor intensive and not quantitative. A variation of the bisulfite method, MS-SNuPE can quantify the ratio of methylated and unmethylated alleles and can be used in a small multiplex. The SNuPE step in MS-SNuPE is primarily used to distinguish C/T resulting from converted C and unconverted 5mC.Citation55–Citation60 Proof of principle experiment showed that an additional SNP close to the CpG site can be utilized to distinguish parental alleles by molecular mass.Citation60 It is, however, easier to utilize the SNP in the MIRA-SNuPE, because the SNuPE step is exclusively used for discriminating between parental alleles after the methylated DNA fraction is collected. This allows a precise quantitation of allelic ratios. One important advantage of the MIRA-based allele-specific methylation detection is that whereas other commonly used methods, bisulfite genomic sequencing and restriction digestion using methylation sensitive enzymes do not distinguish between 5mC and 5hmC,Citation8,Citation9 the MIRA assay specifically detects 5mC.Citation9 It is important to distinguish 5mC from 5hmC, because 5hmC is emerging as the sixth nucleotide with potential relevance to gene regulation and development.Citation3,Citation4
DMR methylation is essential for the allele-specific expression of many imprinted genes,Citation17 but there are exceptions.Citation61–Citation63 Genes exhibiting imprinted expression specifically in the placenta often do not depend on DNA methylation. Imprinted gene expression in the placenta, at the same time is more sensitive to chromatin modifications, such as histone methylation.Citation62–Citation65 This is in agreement with our finding that placenta generally exhibited less allele-specific methylation at DMRs than the fetal organs and the other two extraembryonic layers, the yolk sac and the amnion. Using the MIRA-SNuPE, it will be interesting to address whether environmental insults, such as endocrine disruptor chemicals affected allele-specific CpG methylation at DMRs of imprinted genes, and if DNA in the placenta responded differently than in the embryo proper, yolk sac and amnion.
Materials and Methods
Cells and fetuses.
DNA for MIRA was prepared from fetal and extra-embryonic organs and MEFs by standard phenolchloroform extraction. Primary MEFs were derived from 13.5 dpc embryos.Citation66 Fetuses were derived from reciprocal matings of 129 mother X JF1 father and JF1 mother X 129 father at 13.5 and 15.5 dpc. To generate MatDup.dist7 and PatDup.dist7 MEFs,Citation43,Citation67–Citation69 intercrosses of mice heterozygous for a reciprocal T(7;15)9H translocation at the T9H breakpoint had been used.Citation68 PatDup.dist7 embryos die at 10.5 dpc due to failure in placental spongiotrophoblast development.Citation69 To obtain MEFs at 13.5 dpc, the PatDup.dist7 embryos had been rescued by an achaete-scute complex homolog 2, Ascl2 transgene.Citation70
MIRA.Citation10,Citation11
Genomic DNA was fragmented by sonication to an average fragment size of 500 bp. The amount of input DNA in the MIRA reaction was 500 ng if not stated otherwise in the Figures. The methylated fraction was captured using recombinant MBD3L1 and MBD2b proteins as described previously.Citation10,Citation11 The fragmented DNA was added to the pre-incubated mix containing one µg each of MBD2b and MBD3L1 proteins, and binding of the MBD2b/MBD3L1 complex to methylated CpGs was achieved after an overnight incubation. The MBD2b/MBD3L1/methylated DNA complex was magnetically captured using pre-washed MagneGST glutathione particles (Promega, Madison, WI), and purified according to the manufacturer's instructions. One condition differed from the standard MIRA protocol; the washing buffer contained 400 mM NaCl. This modification enhanced the enrichment of methylated DNA at non-CpG island sequences. The enriched MBD2b/MBD3L-bound methylated CpG fraction was further processed using the QIAquick PCR purification kit (Qiagen) to elute the methylated CpG fraction.
Real-time PCR.
Real-time PCR was performed to measure the region-specific overall MIRA enrichment levels at the H19-Igf2 ICR,Citation66 at the KvDMR1 and Snrpn DMRs and at the c-myc promoter,Citation44 as described. Three microliter aliquots out of the 100 µL MIRA elution or 3 ng input DNA were used to amplify targets with region-specific primers. A dilution series of genomic DNA was used for quantifying copy numbers from MIRA and input samples. PCR primers were previously described.Citation44
Allele-specific MIRA-SNuPE.
To measure allele-specific CpG methylation differences, we used the MALDI-TOF allelotyping analysis method from Sequenom.Citation71,Citation72 Relative enrichment of each allele in the total MIRA elution was quantified by mass spectrometry of the primer extension products at single nucleotide polymorphisms (SNPs) between parental alleles. A 16-plex assayCitation44 was used for 11 DMRs based on SNPs between inbred 129S1 (129) and JF1 mouse lines. A 7-plex assayCitation44 was used for the H19/Igf2 imprinted region based on SNPs between 129S1 and CAST/Ei lines as described.Citation66 Polymerase chain reaction and SNuPE primers for multiplex reactions were designed using MassArray Assay design software v3.1. Six microliter PCR reactions contained 1 pmole of each of the corresponding PCR primer pair, 3 ng genomic DNA or 10 µl of MIRA-captured DNA and Hot Start Reaction Mix (Qiagen). PCR conditions were as follows: 94°C for 15 min, followed by 40 cycles of 94°C (20 sec), 56°C (30 sec), 72°C (60 sec), and a final extension of 72°C for 3 min. Amplified samples were spotted onto a 384 SpectroCHIP Array using a Nanodispenser and analyzed in a MassArray Compact mass spectrometer (Sequenom). Automated spectra acquisition was performed using Spectroacquire (Sequenom). Samples were analyzed with the MassArray Typer v3.4. Allelotyping was performed by first generating an allele skew correction file using a heterozygote DNA sample to correct for any allelic imbalance that may be present in the allele mass products. All samples were then exported from Typer v3.4 and in the process were applied with skew correction. The final allelotype data report contained the ratio present of each allele product at that given SNP. Serial dilutions were included in every experiment for quality control.
Conflict of Interest Statement
Under a licensing agreement between City of Hope and Active Motif (Carlsbad, CA) the methylated-CpG island recovery assay (MIRA) technique was licensed to Active Motif and the author G.P.P. is entitled to a share of the royalties received by City of Hope from sales of the licensed technology.
Abbreviations
| 5mC | = | 5-methyl-C |
| 5hmC | = | 5-hydroxymethyl-C |
| MIRA | = | methylated CpG island recovery assay |
| DMR | = | differentially methylated region |
| MEF | = | mouse embryo fibroblast |
| SNuPE | = | single nucleotide primer extension |
| MBD2b | = | methyl-CpG binding protein 2b |
| Dnmt | = | DNA methyltransferase |
| KvDMR1 | = | Kv differentially methylated region |
| ICR | = | imprinting control region |
| RFLP | = | restriction fragment length polymorphism |
| MatDup.dist7 | = | maternal duplication of distal chromosome 7 |
| PatDup.dist7 | = | paternal duplication of distal chromosome 7 |
| 129 | = | mouse inbred strain 129S1 |
| dpc | = | days post coitum |
| CTCFm | = | mouse line with CTCF binding site mutations in the H19/Igf2 imprinting control region |
| CS | = | distal Chr7 congenic mouse line for CAST/Ei in FVB background |
| SNP | = | single nucleotide polymorphism |
| MeDIP | = | methylated DNA immunoprecipitation |
| MS-SNuPE | = | methylation sensitive SNuPE |
| TSS | = | transcription start site |
Figures and Tables
Figure 1 MIRA assay for detecting allele-specific methylation. (A) DMRs exhibit parental allele-specific CpG methylation inherited either from the mother or the father. The latter case is shown with CpG-methylated (closed lollipops) paternal (PA T) allele and unmethylated (open lollipops) maternal (MAT) allele. The alleles can be distinguished by associated SNPs (G/T example is depicted). (B) In the MIRA assay, the MBD2b and MBD3L1 proteins are used to affinity-capture specifically the CpG-methylated allele of the DMR. The MIRA-captured alleles are proportionally PCR-amplified (horizontal arrows). (C) The ratio of the DMR alleles is quantified. In primer extension on the DMR templates ddCTP or ddATP nucleotides incorporate into the SNuPE primer, resulting in two different extension products. (D) The products are distinguished by their molecular masses using mass-spectrometry. The ratio of their peaks (indicated by vertical arrows) is proportional to the ratio of methylation between the MAT and PA T alleles of the DMR. The theoretical position of the unextended primer (UEP) is also indicated in the mass spectrum.
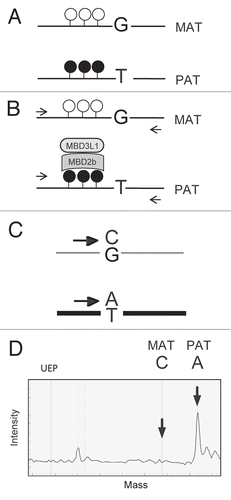
Figure 2 Model systems for allele-specific CpG methylation analysis. Maternally and paternally inherited alleles of Chr7 (red and blue) and Chr15 (pink and light blue) are shown. At least three known germ line DMRs can be investigated on Chr7. The Snrpn DMR and the KvDMR1 are methylated in the maternal allele and unmethylated in the paternal allele whereas the H19/Igf2 ICR is methylated in the paternal allele. (A) Normal embryos inherit one set of chromosomes from each parent. SNPs can be used to distinguish the parental alleles in 129 X JF1 or JF1 X 129 reciprocal mouse crosses (mother is written first). (B and C) Uniparental duplication of distal chromosome 7, telomeric to the T9H translocation breakpoint, allows the analysis of allele-specific methylation at the H19/Igf2 ICR and at the KvDMR1. The Snrpn DMR can be used as a control, because it is not in the duplicated region. (B) In MatDup. dist7 MEFs two copies of the distal Chr7 segment, are inherited from the mother. (C) In PatDup.dist7 MEFs two copies of distal Chr7 are inherited from the father.
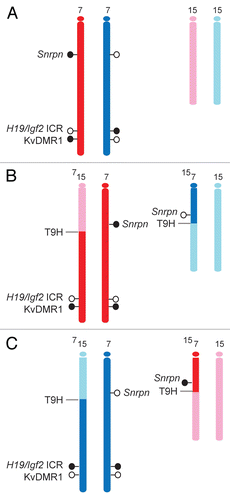
Figure 3 MIRA for detecting CpG methylation at DMRs. MatDup.dist7 (Mat) and PatDup.dist7 (Pat) MEF DNA was subjected to MIRA assays. Equal aliquots of the MIRA fraction were used for quantitation of the H19/Igf2 ICR, the KvDMR1 and the c-myc promoter regions. The copy numbers of methylated DNA molecules were determined by comparing the real-time amplification to a dilution set of genomic DNA. The assay captured the paternally methylated H19/Igf2 ICR from PatDup.dist7 cells and the maternally methylated KvDMR1 from MatDup.dist7 cells. The background was very low at the KvDMR1 in Pat MEFs and at the H19/Igf2 ICR in Mat MEFs and also at the unmethylated c-myc promoter in Mat and Pat MEFs. The positive control Snrpn DMR was captured in Pat and Mat MEFs. The MIRA assays and real-time PCR-s were done in duplicates. Average values and standard deviations were calculated from four measurements.
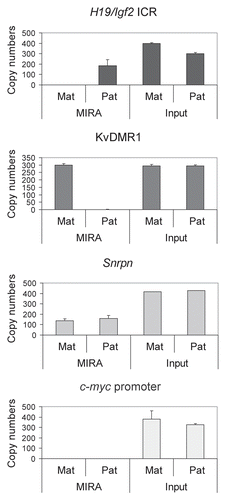
Figure 4 Mixing experiment. DNA from MatDup.dist7 and PatDup. dist7 MEF was mixed in the ratios indicated and was subjected to MIRA assays. The MIRA fraction was quantified using real-time PCR at the indicated regions. Increasing amounts of PatDup.dist7 (Pat) and decreasing amount of MatDup.dist7 (Mat) DNA in the mix resulted in increased capture of the H19/Igf2 ICR and decreasing capture of the KvDMR1. The linearity of the response was more obvious when we normalized the MIRA fraction of the H19/Igf2 ICR and the KvDMR1 separately to the MIRA fraction of the Snrpn DMR and expressed each DMR as a % in the total capture (last chart).
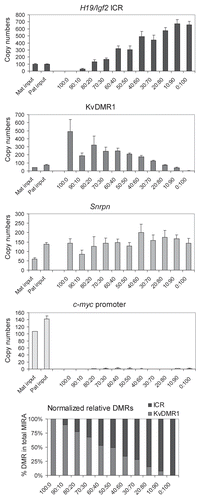
Figure 5 MIRA assays to quantify CpG methylation levels in normal cells. Different amounts (in ng, as indicated) of 129 X JF1 and JF1 X 129 MEF DNA were used in MIRA assays. The MIRA and the unbound fractions were quantified using real-time PCR. The reciprocal crosses exhibited very similar levels of methylation, reflecting that each cell type had one copy of the methylated allele. The unbound fraction was very high at the 1,000 ng data point for each region, suggesting that the DNA amount is saturating at the given MBD protein concentration. To the right, 500 ng total DNA from different mixes were used for MIRA (0, 10, 25, 50, 75, 90 and 100% 129 X JF1 DNA was combined with 100, 90, 75, 50, 25, 10 and 0% JF1 X 129, respectively in samples a–g). These mixes correctly exhibited similar MIRA enrichment levels, reflecting that the total amount of the methylated DMR was similar.
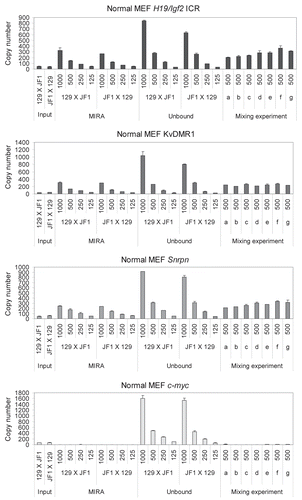
Figure 6 The number of MIRA-captured DNA molecules depends on the number of methylated DMR alleles per the cell. MIRA enrichment was quantified using real-time PCR and was normalized for corresponding input values in MatDup.dist7, wild type and PatDup.dist7 MEF cell DNA. Average values and standard deviations were calculated from four independent MIRA experiments. Statistically significant differences, determined using student's t-test, are indicated by asterisks. The relative levels of MIRA fractions corresponded to 2 > 1 > 0 methylated KvDMR1 alleles and 0 < 1 < 2 methylated H19/Igf2 ICR alleles in MatDup.dist7 > wild type > PatDup.dist7 MEFs. The MIRA enrichment at the Snrpn DMR and c-myc promoter was similar between the three cell types, consistently with the constant number of methylated alleles (1 and 0, respectively).
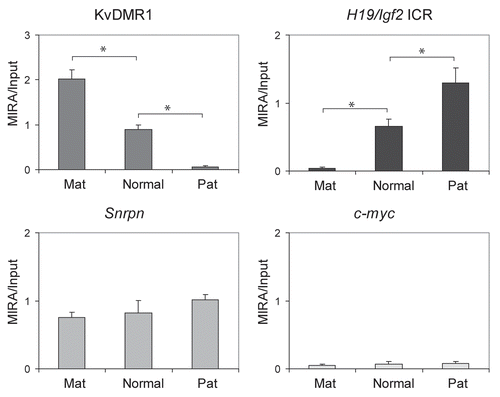
Figure 7 Measuring the ratio of CpG methylated and CpG-unmethylated alleles at maternally and paternally methylated DMRs. Allele-specific DNA methylation was measured by quantitative MIRA-SNuPE assays in normal MEFs using Sequenom 16-plex assays.Citation44 MIRA was performed from different amounts of genomic DNA as indicated under the bars in nanograms from JF1 mother X 129 father or from 129 mother X JF1 father crosses (maternal allele comes first). The percent allele-specific DNA methylation is shown at the maternally methylated (to the left) and paternally methylated (to the right) DMRs. Alternative SNPs were included for the H19/Igf2 ICR (-3 kb and -4 kb from the transcription start site of H19). The ratio of allele-specific CpG methylation at a specific region was expressed as percent of 129 (black bars) or JF1 (grey bars) alleles in the total (129 + JF1 or 100%) of MIRA-enriched DNA. Standard deviations are indicated as error bars. The MIRA-SNuPE is quantitative at 62 ng of DNA.
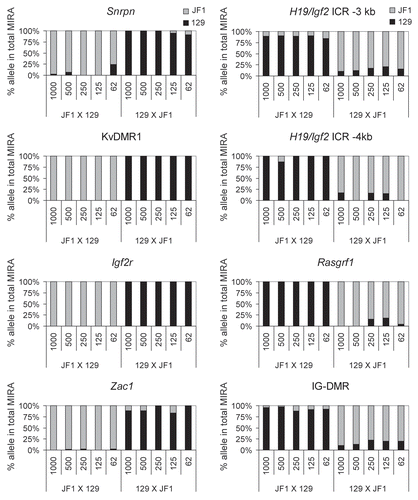
Figure 8 Mixing experiment. DNA from 129 X JF1 and JF1 X 129 MEFs was mixed in different ratios: (0, 10, 25, 50, 75, 90 and 100% 129 X JF1 with 100, 90, 75, 50, 25, 10 and 0% JF1 X 129) and was subjected to MIRA (a–h samples in ). SNuPE assays were used to measure the 129 and JF1 alleles in the total MIRA fraction. At the paternally methylated H19/Igf2 ICR (−3 kb and −4 kb upstream of the H19 TSS ), the paternally inherited JF1 allele increased with increasing amounts of the 129 X JF1 DNA. At the same time with the decreasing amount of JF1 X 129 DNA, the paternally inherited 129 allele decreased. The maternally methylated KvDMR1 and Snrpn DMRs showed the reciprocal trend, as expected.
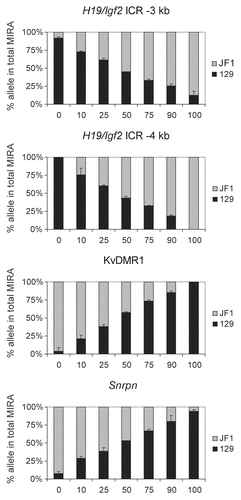
Figure 9 Measuring allele-specific CpG methylation in case of loss of imprinting. DNA from livers and kidneys of normal (Wt) and mutant (Mut) fetuses, carrying CTCF site mutations in the maternal H19/Igf2 ICR allele,Citation47 was subjected to MIRA-SNuPE assays using previously validated 7-plex Sequenom assays.Citation44 DNA methylation was paternal allele-specific in normal, but biallelic in mutant organs at the H19/Igf2 ICR (at −4 and −3 kb from the TSS ) and at the Igf2 DMR2, confirming that the CTCF sites maintain DNA hypomethylation in the maternal allele.Citation47,Citation66 The control Igf2 P2 promoter is not differentially methylated in normal or mutant organs. The ratio of allele-specific CpG methylation at a specific region was expressed as a percent of maternal (black bars) or paternal (grey bars) in the total of MIRA-enriched DNA. The ratio of the parental alleles in the control input DNA was close to 50%. Average values from two independent fetuses are shown with standard deviations.
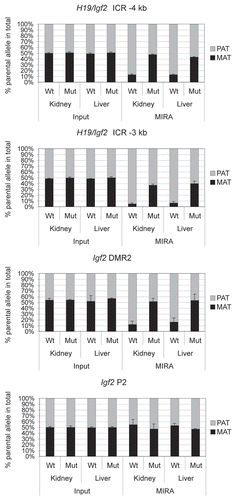
Figure 10 Allele-specific methylation at 7 DMRs in developing normal mouse fetuses. DNA methylation was assessed by quantitative MIRA-SNuPE assays in the placenta, yolk sac, amnion, brain, heart, kidney, liver, lung and muscle of 15.5 dpc JF1 X 129 type normal fetuses. Previously validated 16-plex DMR Sequenom assaysCitation44 were used at SNPs within maternally methylated (to the left) and paternally methylated (to the right) DMRs. The ratio of allele-specific CpG methylation at a specific region was expressed as a percent of maternal or paternal allele in the total of MIRA-enriched DNA. The ratio of the parental alleles in the control input DNA was close to 50%. Average values from three independent fetuses are shown with standard deviations.
Acknowledgements
We thank Jeff Mann for providing the MatDup.dist7 and PatDup.dist7 MEFs. This work was supported by the National Institutes of Health [ES015185 and GM064378 to P.E.S. and AG036041 to G.P.P.].
References
- Wu R, Geiduschek EP. The role of replication proteins in the regulation of bacteriophage T4 transcription. I. Gene 45 and hydroxymethyl-C-containing DNA. J Mol Biol 1975; 96:513 - 538
- Wyatt GR, Cohen SS. The bases of the nucleic acids of some bacterial and animal viruses: the occurrence of 5-hydroxymethylcytosine. Biochem J 1953; 55:774 - 782
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009; 324:929 - 930
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324:930 - 935
- Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010; 466:1129 - 1133
- Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res 1994; 22:2990 - 2997
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA 1992; 89:1827 - 1831
- Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One 5:8888
- Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res 2010; 38:e125
- Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res 2006; 66:7939 - 7947
- Rauch TA, Pfeifer GP. The MIRA method for DNA methylation analysis. Methods Mol Biol 2009; 507:65 - 75
- Kobayashi H, Suda C, Abe T, Kohara Y, Ikemura T, Sasaki H. Bisulfite sequencing and dinucleotide content analysis of 15 imprinted mouse differentially methylated regions (DMRs): paternally methylated DMRs contain less CpGs than maternally methylated DMRs. Cytogenet Genome Res 2006; 113:130 - 137
- Thorvaldsen JL, Bartolomei MS. SnapShot: imprinted gene clusters. Cell 2007; 130:958
- Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science 2001; 294:2536 - 2539
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 2004; 429:900 - 903
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99:247 - 257
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature 1993; 366:362 - 365
- Bielinska B, Blaydes SM, Buiting K, Yang T, Krajewska-Walasek M, Horsthemke B, et al. De novo deletions of SNRPN exon 1 in early human and mouse embryos result in a paternal to maternal imprint switch. Nat Genet 2000; 25:74 - 78
- Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet 2002; 32:426 - 431
- Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, et al. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet 2003; 35:97 - 102
- Thorvaldsen JL, Duran KL, Bartolomei MS. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev 1998; 12:3693 - 3702
- Williamson CM, Ball ST, Nottingham WT, Skinner JA, Plagge A, Turner MD, et al. A cis-acting control region is required exclusively for the tissue-specific imprinting of Gnas. Nat Genet 2004; 36:894 - 899
- Williamson CM, Turner MD, Ball ST, Nottingham WT, Glenister P, Fray M, et al. Identification of an imprinting control region affecting the expression of all transcripts in the Gnas cluster. Nat Genet 2006; 38:350 - 355
- Wutz A, Smrzka OW, Schweifer N, Schellander K, Wagner EF, Barlow DP. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature 1997; 389:745 - 749
- Yoon BJ, Herman H, Sikora A, Smith LT, Plass C, Soloway PD. Regulation of DNA methylation of Rasgrf1. Nat Genet 2002; 30:92 - 96
- Tremblay KD, Duran KL, Bartolomei MS. A 5′ 2-kilobase-pair region of the imprinted mouse H19 gene exhibits exclusive paternal methylation throughout development. Mol Cell Biol 1997; 17:4322 - 4329
- Tremblay KD, Saam JR, Ingram RS, Tilghman SM, Bartolomei MS. A paternal-specific methylation imprint marks the alleles of the mouse H19 gene. Nat Genet 1995; 9:407 - 413
- Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000; 405:482 - 485
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylationsensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000; 405:486 - 489
- Kaffer CR, Srivastava M, Park KY, Ives E, Hsieh S, Batlle J, et al. A transcriptional insulator at the imprinted H19/Igf2 locus. Genes Dev 2000; 14:1908 - 1919
- Kanduri C, Pant V, Loukinov D, Pugacheva E, Qi CF, Wolffe A, et al. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr Biol 2000; 10:853 - 856
- Szabó P, Tang SH, Rentsendorj A, Pfeifer GP, Mann JR. Maternal-specific footprints at putative CTCF sites in the H19 imprinting control region give evidence for insulator function. Curr Biol 2000; 10:607 - 610
- Engemann S, Strodicke M, Paulsen M, Franck O, Reinhardt R, Lane N, et al. Sequence and functional comparison in the Beckwith-Wiedemann region: implications for a novel imprinting centre and extended imprinting. Hum Mol Genet 2000; 9:2691 - 2706
- Yatsuki H, Joh K, Higashimoto K, Soejima H, Arai Y, Wang Y, et al. Domain regulation of imprinting cluster in Kip2/Lit1 subdomain on mouse chromosome 7F4/F5: large-scale DNA methylation analysis reveals that DMR-Lit1 is a putative imprinting control region. Genome Res 2002; 12:1860 - 1870
- Smilinich NJ, Day CD, Fitzpatrick GV, Caldwell GM, Lossie AC, Cooper PR, et al. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Natl Acad Sci USA 1999; 96:8064 - 8069
- Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev 2006; 20:1268 - 1282
- Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 2008; 32:232 - 246
- Warnecke PM, Mann JR, Frommer M, Clark SJ. Bisulfite sequencing in preimplantation embryos: DNA methylation profile of the upstream region of the mouse imprinted H19 gene. Genomics 1998; 51:182 - 190
- Driscoll DJ, Waters MF, Williams CA, Zori RT, Glenn CC, Avidano KM, et al. A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics 1992; 13:917 - 924
- Singer-Sam J, LeBon JM, Tanguay RL, Riggs AD. A quantitative HpaII-PCR assay to measure methylation of DNA from a small number of cells. Nucleic Acids Res 1990; 18:687
- Rauch T, Pfeifer GP. Methylated-CpG island recovery assay: a new technique for the rapid detection of methylated-CpG islands in cancer. Lab Invest 2005; 85:1172 - 1180
- Olek A, Walter J. The pre-implantation ontogeny of the H19 methylation imprint. Nat Genet 1997; 17:275 - 276
- Ferguson-Smith AC, Sasaki H, Cattanach BM, Surani MA. Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature 1993; 362:751 - 755
- Singh P, Han L, Rivas GE, Lee DH, Nicholson TB, Larson GP, et al. Allele-specific H3K79 Di-versus trimethylation distinguishes opposite parental alleles at imprinted regions. Mol Cell Biol 2010; 30:2693 - 2707
- Pant V, Mariano P, Kanduri C, Mattsson A, Lobanenkov V, Heuchel R, et al. The nucleotides responsible for the direct physical contact between the chromatin insulator protein CTCF and the H19 imprinting control region manifest parent of origin-specific long-distance insulation and methylation-free domains. Genes Dev 2003; 17:586 - 590
- Schoenherr CJ, Levorse JM, Tilghman SM. CTCF maintains differential methylation at the Igf2/H19 locus. Nat Genet 2003; 33:66 - 69
- Szabó PE, Tang SH, Silva FJ, Tsark WM, Mann JR. Role of CTCF binding sites in the Igf2/H19 imprinting control region. Mol Cell Biol 2004; 24:4791 - 4800
- Zuo T, Tycko B, Liu TM, Lin HJ, Huang TH. Methods in DNA methylation profiling. Epigenomics 2009; 1:331 - 345
- Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res 20:883 - 889
- Tycko B. Mapping allele-specific DNA methylation: a new tool for maximizing information from GWAS. Am J Hum Genet 2010; 86:109 - 112
- Wilson IM, Davies JJ, Weber M, Brown CJ, Alvarez CE, MacAulay C, et al. Epigenomics: mapping the methylome. Cell Cycle 2006; 5:155 - 158
- Singer-Sam J. Quantitation of specific transcripts by RT-PCR SNuPE assay. PCR Methods Appl 1994; 3:48 - 50
- Szabó PE, Mann JR. Allele-specific expression and total expression levels of imprinted genes during early mouse development: implications for imprinting mechanisms. Genes Dev 1995; 9:3097 - 3108
- Szabó PE, Mann JR. Biallelic expression of imprinted genes in the mouse germ line: implications for erasure, establishment and mechanisms of genomic imprinting. Genes Dev 1995; 9:1857 - 1868
- El-Maarri O. SIRPH analysis: SNuPE with IP-RP-HPLC for quantitative measurements of DNA methylation at specific CpG sites. Methods Mol Biol 2004; 287:195 - 205
- El-Maarri O, Herbiniaux U, Walter J, Oldenburg J. A rapid, quantitative, non-radioactive bisulfite-SNuPE-IP RP HPLC assay for methylation analysis at specific CpG sites. Nucleic Acids Res 2002; 30:25
- Gonzalgo ML, Jones PA. Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE). Nucleic Acids Res 1997; 25:2529 - 2531
- Gonzalgo ML, Liang G. Methylation-sensitive single-nucleotide primer extension (Ms-SNuPE) for quantitative measurement of DNA methylation. Nat Protoc 2007; 2:1931 - 1936
- Kaminsky Z, Petronis A. Methylation SNaPshot: a method for the quantification of site-specific DNA methylation levels. Methods Mol Biol 2009; 507:241 - 255
- Coolen MW, Statham AL, Gardiner-Garden M, Clark SJ. Genomic profiling of CpG methylation and allelic specificity using quantitative high-throughput mass spectrometry: critical evaluation and improvements. Nucleic Acids Res 2007; 35:119
- Tanaka M, Puchyr M, Gertsenstein M, Harpal K, Jaenisch R, Rossant J, et al. Parental origin-specific expression of Mash2 is established at the time of implantation with its imprinting mechanism highly resistant to genome-wide demethylation. Mech Dev 1999; 87:129 - 142
- Lewis A, Mitsuya K, Umlauf D, Smith P, Dean W, Walter J, et al. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet 2004; 36:1291 - 1295
- Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, et al. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 2004; 36:1296 - 1300
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008; 322:1717 - 1720
- Wagschal A, Sutherland HG, Woodfine K, Henckel A, Chebli K, Schulz R, et al. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol Cell Biol 2008; 28:1104 - 1113
- Han L, Lee DH, Szabó PE. CTCF is the master organizer of domain-wide allele-specific chromatin at the H19/Igf2 imprinted region. Mol Cell Biol 2008; 28:1124 - 1135
- Cattanach BM, Barr JA, Beechey CV, Martin J, Noebels J, Jones J. A candidate model for Angelman syndrome in the mouse. Mamm Genome 1997; 8:472 - 478
- Cattanach BM, Barr JA, Evans EP, Burtenshaw M, Beechey CV, Leff SE, et al. A candidate mouse model for Prader-Willi syndrome which shows an absence of Snrpn expression. Nat Genet 1992; 2:270 - 274
- McLaughlin KJ, Szabó P, Haegel H, Mann JR. Mouse embryos with paternal duplication of an imprinted chromosome 7 region die at midgestation and lack placental spongiotrophoblast. Development 1996; 122:265 - 270
- Rentsendorj A, Mohan S, Szabó PE, Mann JR. A genomic imprinting fefect in mice traced to a single gene. Genetics 2010; 186:917 - 927
- Haun WJ, Springer NM. Maternal and paternal alleles exhibit differential histone methylation and acetylation at maize imprinted genes. Plant J 2008; 56:903 - 912
- Jurinke C, Denissenko MF, Oeth P, Ehrich M, van den Boom D, Cantor CR. A single nucleotide polymorphism based approach for the identification and characterization of gene expression modulation using MassARRAY. Mutat Res 2005; 573:83 - 95