Abstract
DNA methylation is one of the most important heritable epigenetic modifications of the genome and is involved in the regulation of many cellular processes. Aberrant DNA methylation has been frequently reported to influence gene expression and subsequently cause various human diseases, including cancer. Recent rapid advances in next-generation sequencing technologies have enabled investigators to profile genome methylation patterns at single-base resolution. Remarkably, more than 20 eukaryotic methylomes have been generated thus far, with a majority published since November 2009. Analysis of this vast amount of data has dramatically enriched our knowledge of biological function, conservation and divergence of DNA methylation in eukaryotes. Even so, many specific functions of DNA methylation and their underlying regulatory systems still remain unknown to us. Here, we briefly introduce current approaches for DNA methylation profiling and then systematically review the features of whole genome DNA methylation patterns in eight animals, six plants and five fungi. Our systematic comparison provides new insights into the conservation and divergence of DNA methylation in eukaryotes and their regulation of gene expression. This work aims to summarize the current state of available methylome data and features informatively.
Introduction
DNA methylation, which occurs in the genome of a vast array of bacteria, plants, fungi and animals, has been proven to play an important role in many cellular processes. These include embryonic development, transcription, chromatin structure, X chromosome inactivation, genomic imprinting and chromosome stability.Citation1–Citation6 Consistent with these important roles, a growing number of human diseases are now known to be associated with aberrant DNA methylation, often in CpG islands or promoter regions.Citation7 Consequently, regulation of DNA methylation is a vital part of normal development and when dysfunctional, may activate disease states such as cancer.Citation8–Citation10 For example, hypomethylation of the global genome or hypermethylation of specific tumor suppressor genes may contribute to human carcinogenesis.Citation11,Citation12 Therefore, the precise detection of both the extent and location of DNA methylation in specific genes and the genome in its entirety (often referred as the “methylome”) is urgently needed.Citation13 Recent rapid advances in sequencing technologies have enabled profiling of the genome methylation pattern at single-base resolution. So far more than 20 eukaryotic methylomes have been generated by next-generation sequencing technologies, as summarized in . Analysis of this vast amount of methylome data has dramatically enhanced our knowledge of the conservation, divergence and biological function of DNA methylation in eukaryotes.Citation14–Citation16 In this paper, we first briefly review the technologies of DNA methylation detection and then systematically compare the features of whole genome DNA methylation in eight animals, six plants and five fungi. This paper provides an overview of DNA methylation features and differences in these organisms, with data primarily generated since November 2009. This study also provides some new insights into the evolution and role of DNA methylation in eukaryotes.
Methods for Genome-Wide DNA Methylation Profiling
In recent years, many methods have been developed to detect the genome-wide cytosine methylation status.Citation17–Citation19 Widely used methods include restriction enzyme digestion of methylated DNA followed by hybridization to high-density oligo-nucleotide arrays or sequencing4,Citation20 and capture of methylated genomic DNA with antibodies that target 5-methylcytosine (methylated DNA immunoprecipitation, MeDIP) or methyl-binding domain (MBD) proteins, followed by array hybridization or sequencing.Citation21–Citation23 These approaches can determine genome-wide methylation levels and patterns but have major limitations in resolution, restriction enzyme bias, difficulty in characterizing genome regions that are rich in repeats and, most importantly, an inability to detect DNA methylation at a single-base resolution.
Currently, the “gold standard” method for the detection of DNA methylation at single-base resolution is an integration of sodium bisulfite (BS) conversion and next-generation sequencing. Sodium bisulfite converts unmethylated cytosine bases to uracils, whereas methylated cytosine (mC) bases remain unchanged. Thus, after PCR amplification, unmethylated Cs are converted to thymines (T) while methylated Cs remain unchanged. This method provides quantitative, contiguous and base-pair resolution of genome methylation map, including methylation on CpG sites and non-CpG sites.Citation19,Citation24 Recent rapid advances in sequencing technologies have enabled profiling more and more genome methylation patterns at the single-base resolution, including more than 20 eukaryotic methylomes ().
Despite these advances in BS treatment-based sequencing, there are several drawbacks to using this technique. For example, the sample preparation steps associated with BS treatment are costly and time-consuming. Additionally, the BS treatment reduces sequence complexity by converting all mCs to Ts, which complicates the alignment of short reads to reference genomes. Finally, BS treatment-based sequencing cannot discriminate mC and a newly identified epigenetic marker, 5-hydroxymethylcytosine (hmC). The presence of hmC was recently reported in the adult mouse brain and human embryonic stem (ES) cells;Citation25,Citation26 its functional significance has yet to be determined.
Several new technologies may bypass these problems by detecting DNA modification directly on single DNA molecules without BS treatment. The pioneering single-molecule sequencing approaches, including nanopore-based methods and single-molecule real-time (SMRT) DNA sequencing, with the capacity to discriminate native from modified bases, may enable methylome profiling for hundreds of thousands of contiguous bases, or even for entire chromosomes, as single long reads in which both primary sequence and methylation are simultaneously determined.Citation27–Citation29 Although accuracy of the data generated by these approaches at the current stage is not satisfactory, it is likely that it will replace the current BS-conversion based methods in the foreseeable future. Such methods may advance the frontier of DNA methylome study from only a small handful of studies towards the examination of the patterning and dynamics of cytosine methylation in diverse populations, tissues and disease states. These might include the examination of diet/nutrition, distinct cell types, mutants or diseases and large numbers of individuals from geographical populations (population methylomes).
Potential Function of Non-CG Methylation in Mammals
A surprising and provocative discovery in recent methylome studies was the prevalence of non-CG methylation in human ES cells.Citation30,Citation31 Nearly 25% of all methylated cytosines in the human ES cells did not occur exclusively at CG sites. The prevalence of non-CG methylation in another human ES cell line H9 revealed that the non-CG methylation sites are conserved, suggesting that such methylation may be a general feature of human ES cells. A previous study also found substantial non-CG methylation in mouse ES cells, although its prevalence and genomic location was not clearly defined.Citation32 Interestingly, non-CG methylation was found to be absent in fetal fibroblasts and mature peripheral blood mononuclear cells.Citation30,Citation31 The sharp contrast in non-CG methylation between ES cells and differentiated cells raises a critical question: is non-CG methylation functionally relevant and, if so, what is its primary importance in ES cells?
Examination of non-CG methylation patterns in human ES cells and comparison to DNA methylation among ES cells, differentiated cells and induced pluripotent stem cells (iPS) provided some insights into this question:Citation30,Citation31 (1) Non-CG methylation occurs more often in the gene bodies than in the promoter regions, where methylation is typically thought to suppress gene expression;Citation30,Citation31 (2) Non-CG methylation in gene bodies, however, is positively correlated with gene expression;Citation31 (3) Non-CG methylation is noticeably enriched on the antisense strand of gene bodies and a significantly more intronic transcription can be generated from non-CG methylation enriched genes;Citation30,Citation31 (4) Non-CG methylation is significantly enriched in genes encoding proteins involved in RNA processing, splicing and RNA metabolic processes;Citation31 (5) Although non-CG methylation is absent in fibroblasts, when differentiated cells were experimentally reprogrammed to an iPS, non-CG methylation was restored at the tested sites.Citation31,Citation33 This raises the possibility that non-CG methylation may play an important role in maintaining pluripotency and like DNA demethylation at specific pluripotency-related gene promoters, restoration of methylation at non-CG sites in gene bodies may be required for efficient reprogramming of induced pluripotent stem cells.Citation31 It is well accepted that evolutionarily conserved methylation patterns tend to be functionally important, so it is necessary to compare the methylomes of human and mouse ES cells to reveal the evolutionarily conserved sites of non-CG methylation, but such an effort is yet to be seen.
Global DNA Methylation Patterns in Eukaryotes
New approaches combining BS conversion and next-generation sequencing allow us to measure the DNA methylation level more accurately and comprehensively. In addition to detecting whether a particular cytosine site is methylated, multiple reads covering each methylcytosine can be used as a readout of the fraction of the sequences within the sample that are methylated at that site.Citation34 For each strand of a DNA segment or the whole genome, there are many different measurements for the level of DNA methylation. Using different measurements to compare the diverse methylation patterns among species might help us to understand the regulation and function of DNA methylation from different angles. Here, we propose two novel measurements to estimate the methylation level of a DNA segment or the whole genome. One is methylation broadness, which represents the fraction of cytosine sites detected as methylated in a given DNA segment. It can be calculated as the proportion of methylated sites over the total sites in a sequence (i.e., #mCG sites/total CG sites). The other is methylation deepness, which represents the extent of methylation of the methylated cytosines from reads (i.e., #reads covering mCs/total reads covering the same sites). We obtained all the available methylome data and calculated their broadness and deepness. As shown in and , the methylation deepness of CG sites in plants is typically as strong as in vertebrates. However, the methylation broadness of CG sites is much weaker in plants than in vertebrates ( and ), indicating that the methylation at CG sites is conserved in different cell types in both plants and vertebrates and that CG methylation tends to occur in particular regions of plant genomes. We also found that the deepness of CHH methylation is higher than that of CHG methylation in most animals and fungi. Interestingly, the opposite pattern was observed in plant genomes, suggesting that the plant-specific machinery for establishing and maintaining CHG site methylation might be more efficient than in other organisms ().
The expansion of eukaryotic methylome data prompted a more comprehensive comparison of whole-genome methylation profiles across the plant and animal kingdoms, revealing both conserved and divergent features of DNA methylation in eukaryotes. Although DNA methylation appears to be a widespread epigenetic regulatory mechanism, genomes are methylated in different ways in diverse organisms. In animals, DNA methylation occurs mostly symmetrically (on both strands) at the cytosines of a CG dinucleotide. An intriguing exception is that a substantial portion of methylated cytosines in human ES cells were found in a CHG and CHH context.Citation30,Citation31 However, in higher plants, DNA methylation can occur at cytosines in both symmetric sequence contexts of CG and CHG (H = A, T or C), and also in an asymmetric CHH context, with the latter directed and maintained by small RNAs (see ).Citation35,Citation36 In the model plant Arabidopsis thaliana, methylation broadness at CG, CHG and CHH nucleotides are about 24.48, 14.47 and 6.47%, respectively.Citation37 Some fungi show substantial CpG site methylation and strong non-CpG methylation, corresponding to their Dnmt1-like and fungispecific Dim-2-type enzymes, respectively (). Despite the differing methylation sequence contexts, cytosine methylation is established and maintained by a family of conserved DNA methyltransferases. Citation36,Citation38 Not surprisingly, the absence of DNA methylation in some eukaryotes such as yeast, roundworm and fruit fly has been associated with the evolutionary loss of DNA methyltransferase homologs.Citation39
Recent methylome studies showed that cytosines are methylated not only in repetitive sequences and transposable elements (TEs) but also in promoters and gene bodies and that DNA methylation is highly correlated with transcription (). The methylation of gene's promoter regions has long been considered as a suppressor of gene expression. Consistent with DNA methylation's function of repressing transcription, gene expression correlates inversely with methylation level in the proximal region of the transcript start site (TSS) in all species with methylated promoters except invertebrates (). Considering that DNA methylation in invertebrate genomes occurs preferentially in gene bodies rather than promoter regions,Citation40–Citation42 the absence of correlation between promoter methylation and gene expression level might reflect background methylation signal in the promoter regions; thus, the role of suppression of gene expression via promoter methylation in invertebrates may be very weak. Furthermore, in the rice genome, the negative correlation was also observed at the 3′ end of genes. This suggests that the lack of methylation around the transcription termination site is important for gene expression.Citation24,Citation42
In plants and vertebrates, most methylated cytosines are found over repetitive elements and loss of this modification is associated with a transcriptional reactivation, as well as an increased mobilization of transposable elements. These observations likely reflect the ancestral role of cytosine methylation in the defense against invasive DNA. While DNA methylation in plants, fungi and vertebrates is concentrated in transposons, invertebrates showed an opposite pattern, with modifications occurring mainly in active genes (), suggesting that the use of DNA methylation to repress deleterious transposons in genomes may have evolved independently in plants and vertebrates, while this function was lost in the invertebrate lineage.Citation24,Citation42
As shown in , methylation of the gene body is highly conserved and was likely present in the last common ancestor of eukaryotes ().Citation24,Citation42 Gene body methylation is not associated with gene expression level, thereby leaving its biological role an open question. Several methylome studies have revealed an interesting parabolic relationship between gene-body methylation and transcription levels.Citation23,Citation24,Citation41,Citation42 Whereas modestly expressed genes are more likely to be methylated, genes expressed at either the lowest level or the highest level are usually less methylated. This suggests the coexistence of two opposing targeting mechanisms that increase or decrease methylation with gene expression. It has been suggested that extreme transcription rates may affect the balance between chromatin disruption and polymerase association, both of which could prevent the generation of aberrant transcripts that might drive methylation via a small RNA dependent pathway.Citation23 Other interesting findings related to gene body methylation function are summarized as follows: (1) The gene body CHG and CHH methylation in human ES cells is correlated with gene expression level;Citation31 (2) The methylation level of 3′ gene region is negatively correlated with gene expression in rice;Citation42 (3) Exons tend to be more highly methylated than intronsCitation31 and (4) In gene bodies, methylated cytosines are more conserved than unmethylated cytosines and other nucleotides.Citation43 These observations imply general roles of DNA methylation in transcriptional elongation, termination and perhaps in alternative splicing. Consistent with these findings, a recent study found exons were densely methylated at CpGs and packaged into nucleosomes.Citation44 Exon enrichment of DNA methylation was specifically found in spliced exons and in exons with weak splice sites. Based on a high-resolution DNA methylation mapping of 24.7 million CpG sites in human brain, Maunakea et al.Citation45 found 34% of all intragenic CpG islands were methylated, compared with only 3% of CpG islands in the 5′ promoters. Interestingly, tissue-specific methylation was more common in intragenic CpG islands than in 5′ promoters. Intragenic CpG islands also overlapped with markers of transcription initiation and unmethylated CpG islands overlapped with trimethylated H3K4 (a histone mark enriched at promoters). Altogether, these findings suggest that gene body methylation might play an important role in regulating tissue-specific or cell-specific alternative promoters.Citation45 Although many assumptions have been proposed about the function of intragenic methylation, further genomewide comparison of DNA methylation patterns and experimental verification are warranted to illustrate the exact function of gene body methylation.
Perspectives
Although the latest studies have greatly improved our understanding of the evolutionary adaptations and conservation of DNA methylation, these studies nevertheless raised more questions than they could answer, as we are eager to further advance our knowledge in cellular systems. For example, the function of conserved gene body methylation is still unclear, although it has been proposed to suppress aberrant transcription from cryptic promoters inside the genes and regulate the alternative promoter.Citation31,Citation45 Furthermore, we still do not understand the mechanism by which DNA methylation regulates gene expression, especially with regard to the phenomenon of higher methylation of modestly transcribed genes than those expressed at the highest or lowest level. As the cost of sequencing drops rapidly and dramatically and single-molecule sequencing approaches evolve to be practical in the near future,Citation27,Citation29 we expect that much more methylome data will be generated from different samples and organisms. Subsequent comprehensive studies over a broad range of these methylomes will ultimately provide us a detailed view of the function and evolution of DNA methylation and ideally, a DNA methylation atlas.
Figures and Tables
Figure 1 Evolution of DNA methylation level of the available eukaryote methylomes. The phylogenetic tree was based on NCBI Taxonomy Browser (www.ncbi.nlm.nih.gov/taxonomy/taxonomyhome.html). Only the topology is shown and the branch lengths are not proportional to evolutionary divergence time. Green and red boxes indicate high methylation of gene body and transposon elements (TEs), respectively. On the right, a heatmap shows DNA methylation level (broadness and deepness for each kind of sequence context). The methylome data of human IMR90 fetal lung fibroblasts was used to represent the human.
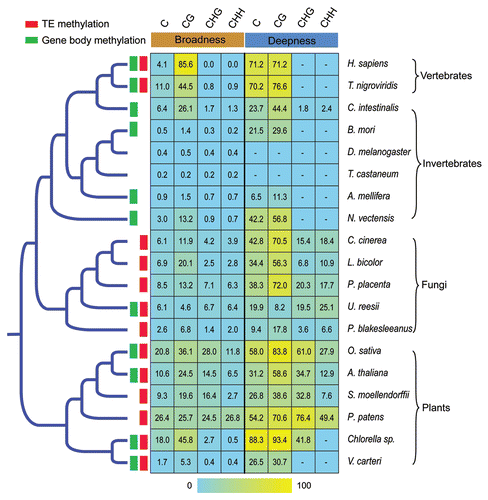
Table 1 Statistics and features of DNA methylation in vertebrates, invertebrates, plants and fungi
Table 2 Genome features and DNA methylation patterns of the available eukaryote methylomes
Acknowledgements
We would like to thank Jeffery Ewers for critical reading and improving the manuscript. This work was supported by a NIH grant (R03LM009598) from the National Library of Medicine, Vanderbilt's Specialized Program of Research Excellence in GI Cancer grant (P50CA95103) and the VICC Cancer Center Core grant (P30CA68485).
References
- Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet 2000; 9:2395 - 2402
- Brown SE, Fraga MF, Weaver IC, Berdasco M, Szyf M. Variations in DNA methylation patterns during the cell cycle of HeLa cells. Epigenetics 2007; 2:54 - 65
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69:915 - 926
- Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004; 430:471 - 476
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008; 133:523 - 536
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457 - 466
- Zhao Z, Han L. CpG islands: algorithms and applications in methylation studies. Biochem Biophys Res Commun 2009; 382:643 - 645
- Robertson KD. DNA methylation and human disease. Nat Rev Genet 2005; 6:597 - 610
- Szyf M. The role of DNA hypermethylation and demethylation in cancer and cancer therapy. Curr Oncol 2008; 15:72 - 75
- Szyf M, Pakneshan P, Rabbani SA. DNA methylation and breast cancer. Biochem Pharmacol 2004; 68:1187 - 1197
- De Smet C, Loriot A. DNA hypomethylation in cancer: Epigenetic scars of a neoplastic journey. Epigenetics 2010; 5:206 - 213
- Ting AH, McGarvey KM, Baylin SB. The cancer epigenome—components and functional correlates. Genes Dev 2006; 20:3215 - 3231
- Feinberg AP. Methylation meets genomics. Nat Genet 2001; 27:9 - 10
- Jeltsch A. Phylogeny of methylomes. Science 2010; 328:837 - 838
- Lee TF, Zhai J, Meyers BC. Conservation and divergence in eukaryotic DNA methylation. Proc Natl Acad Sci USA 2010; 107:9027 - 9028
- Yi SV, Goodisman MA. Computational approaches for understanding the evolution of DNA methylation in animals. Epigenetics 2009; 4:551 - 556
- Beck S, Rakyan VK. The methylome: approaches for global DNA methylation profiling. Trends Genet 2008; 24:231 - 237
- Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet 2010; 11:191 - 203
- Lister R, Ecker JR. Finding the fifth base: genomewide sequencing of cytosine methylation. Genome Res 2009; 19:959 - 966
- Vaughn MW, Tanurdzic M, Lippman Z, Jiang H, Carrasquillo R, Rabinowicz PD, et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol 2007; 5:e174
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853 - 862
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 2006; 126:1189 - 1201
- Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 2007; 39:61 - 69
- Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA 2010; 107:8689 - 8694
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009; 324:929 - 930
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324:930 - 935
- Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol 2009; 4:265 - 270
- Ecker JR. Zeroing in on DNA methylomes with no BS. Nat Methods 2010; 7:435 - 437
- Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 2010; 7:461 - 465
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res 2010; 20:320 - 331
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315 - 322
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase3a. Proc Natl Acad Sci USA 2000; 97:5237 - 5242
- Costello JF, Krzywinski M, Marra MA. A first look at entire human methylomes. Nat Biotechnol 2009; 27:1130 - 1132
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008; 452:215 - 219
- Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature 2007; 447:418 - 424
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 2010; 11:204 - 220
- Hsieh TF, Ibarra CA, Silva P, Zemach A, Eshed-Williams L, Fischer RL, et al. Genome-wide demethylation of Arabidopsis endosperm. Science 2009; 324:1451 - 1454
- Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 2009; 10:805 - 811
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem 2005; 74:481 - 514
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008; 9:465 - 476
- Xiang H, Zhu J, Chen Q, Dai F, Li X, Li M, et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat Biotechnol 2010; 28:516 - 520
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010; 328:916 - 919
- Widman N, Jacobsen SE, Pellegrini M. Determining the conservation of DNA methylation in Arabidopsis. Epigenetics 2009; 4:119 - 124
- Choi JK. Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol 2010; 11:70
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010; 466:253 - 257