Abstract
Studies in humans and animal models indicate that profound immunosuppression is one of the chronic consequences of severe sepsis. This immune dysfunction encompasses deficiencies in activation of cells in both the myeloid and lymphoid cell lineages. As a result, survivors of severe sepsis are at risk of succumbing to infections perpetrated by opportunistic pathogens that are normally controlled by a fully functioning immune system. Recent studies have indicated that epigenetic mechanisms may be one driving force behind this immunosuppression, through suppression of proinflammatory gene production and subsequent immune cell activation, proliferation and effector function. A better understanding of epigenetics and post-septic immunosuppression can improve our diagnostic tools and may be an important potential source of novel molecular targets for new therapies. This review will discuss important pathways of immune cell activation affected by severe sepsis, and highlight pathways of epigenetic regulation that may be involved in post-septic immunosuppression.
Introduction
Sepsis represents an unchecked and uncontrolled immune response to exposure to microbes and microbial products, such as during a traumatic injury to the thoracic or peritoneal cavity. During the initial onset of sepsis, activation of immune responses expands beyond a restricted, local microenvironment and spreads to the systemic circulation. Activation of immune cells can then be amplified through the systemic spread of microbial products (such as through bacteremia) or through paracrine responses to the high levels of proinflammatory chemokines and cytokines produced during this initial phase. This unchecked immune response can result in hypotension, damage to the vasculature, unchecked coagulation events and ultimately cell and tissue injury. Together, these physiological responses are termed the systemic inflammatory response syndrome (SIRS).Citation1 The deleterious effects of SIRS, if left unchecked, can result in multi-organ dysfunction, septic shock and death if not treated early with proper medical interventions.Citation2,Citation3 Septic shock remains a significant human health concern, associated with high costs of medical treatment, along with both increased incidence and increased mortality rates.Citation4,Citation5
Similarly, the compensatory anti-inflammatory response syndrome (CARS) following SIRS presents its own challenges to human patients, and recent epidemiological studies indicate that the immunosuppression seen with CARS may last for extended periods of time (weeks, months and years) following the initial onset of SIRS.Citation6 CARS is thought to be a compensatory response by the immune system whereby anti-inflammatory chemokines and cytokines are globally upregulated in an attempt to restrict the harmful systemic TH1 immune responses driving tissue injury and organ failure.Citation7 CARS is a multifactorial response (as is SIRS) and can manifest itself in numerous phenomena. For example, leukocyte apoptosis is extensive during the transition from SIRS to CARS and can affect immune cell levels in both primary (e.g., bone marrow,Citation8 thymusCitation9,Citation10) and secondary (e.g., spleenCitation11,Citation12) lymphoid tissues. Additionally, global levels of anti-inflammatory mediators such as IL-10,Citation13 and TH1-opposing TH2 cytokines such as IL-4,Citation14 are increased during CARS. Finally, the increased activation threshold of leukocytes during CARS in response to pathogen-associated molecular patterns (PAMPs), such as bacterial cell wall components, results in reductions in proinflammatory chemokine and cytokine production by these cells.Citation15,Citation16 These anti-inflammatory mechanisms act in concert to counteract the harmful effects of SIRS; however, they leave the patient susceptible to opportunistic pathogens and otherwise innocuous infections, resulting in CARS-associated morbidity and mortality.
While SIRS is an acute phenomenon that manifests proximal to the initial causative insult, the pathophysiology of CARS can persist in patients for an extended period of time in both humans and animal models. This long-term immunosuppression manifests itself in patients as a significant reduction in 5- and 8-year survival for patients of severe sepsis as compared to the unaffected age-matched population, often due to the infection phenomena discussed previously.Citation17,Citation18 Additionally, mouse models of severe sepsis indicate that survivors of septic shock are susceptible to airway infection with innocuous pathogens (such as the fungus Aspergillus fumigatusCitation19,Citation20 and the Gram-negative bacteria Pseudomonas aeruginosaCitation21) long after sepsis-induced proinflammatory immune responses have resolved. These results indicate that CARS exists not only as an acute response to SIRS, but as a long-term phenomenon that may require its own diagnostic tools and therapies for the long-term survival of affected patients.
At the root of both SIRS and CARS pathology is the up or downregulation of immunomodulatory chemokines and cytokines that are responsible for the mobilization and/or suppression of immune responses. Regulation of protein expression in immune cells can occur both at the pre- and post-transcriptional level, both through regulation of gene expression and mRNA stability and through intracellular protein trafficking and proteasome degredation. In either scenario, mechanisms dictating modifications in protein expression must, by definition, be stable and transmittable from parent to daughter cell, especially for the maintenance of the CARS phenotype that persists in patients long-term. Therefore, mechanisms that can modify gene expression patterns are of particular interest to both researchers and clinicians as possible mediations of the immunosuppressive phenotype following severe sepsis.
Epigenetics is a general term encompassing any and all mechanisms that govern gene expression patterns without modifying the underlying DNA sequence of an organism.Citation22 Epigenetic mechanisms can include chemical modifications of DNA and/or associated histones that result in changing the physical accessibility of the DNA to transcription factors.Citation23 Additionally, post-transcriptional regulation of mRNA can be accomplished by complementary gene interference driven by micro-RNAs (miRNAs), resulting in the downregulation of protein expression through targeted degredation of specific mRNAs.Citation24 Of particular interest is the ability of epigenetic markings to be passed from parent to daughter cells; one classic example of this phenomenon is the changes in DNA methylation which occurs during development, restricting gene expression as cells progress from multipotent stem cells to committed cells of a specific lineage.Citation25 Epigenetic modification of gene expression is also a common phenomenon in the progression of cancer, as both activating and repressing histone methylation events are modulated by cancer cells to promote tumorigenesis.Citation26
Epigenetic regulation of gene products occurs through numerous mechanisms, but can be generalized as the regulated organization of gene loci into transcriptionally active or silent states. Transcriptionally active chromatin (euchromatin) is accessible to transcription factors and polymerases, while transcriptionally silent chromatin (heterochromatin) is sequestered from these factors.Citation27 Both euchromatin and heterochromatin are associated with histones, which are the proteins responsible for packaging and organizing the lengthy DNA strand(s) into organized chromatin. Modifications to the protein tails of histone core components, such as H2A, H2B, H3 and H4, can direct the winding or unwinding of the associated DNA through as yet unknown mechanisms. The activating or repressing nature of these histone tail modifications is dependent on both the chemical nature of the modification and the location of the modification on the histone tail. For example, acetylation of histone tails at specific amino acid residues is thought to act as an activation histone mark, regardless of its location on the histone tail.Citation28 In contrast, methylation of histone tails can be an activating or repressing transcriptional mark, depending on both the type and location of the modification on the histone tail.Citation29 Histone tails can be chemically modified in numerous ways, including acetylation, methylation, ubiquitination and phosphorylation, among others.Citation30 The coordinated modifications of a given histone can dictate the transcription state of the nearby DNA; this “histone code” (along with DNA methylation and other epigenetic mechanisms) can dictate the transcriptional status of a given gene locus, by exposing promoter regions in transcriptionally active euchromatin or sequestering them in transcriptionally silent heterochromatin.
Epigenetics has become an expanding field of study in immunology, as increasing studies indicate that epigenetic mechanisms can govern both productive immune responses and deleterious immunosuppression and autoimmunity. The importance of epigenetics to immune system development and effector function, as well as the apparent correlation between disease chronicity and epigenetic regulation of inflammatory genes, makes the role of epigenetic mechanisms in governing the pathophysiology of SIRS and CARS an important avenue of research. In the following review, the role of epigenetic modifications in driving post-septic immunosuppression will be summarized, with a focus on the currently available literature identifying specific events identified in both human patients and animal models. The purpose of this review is to provide a framework whereby further research into the pathophysiology of post-septic immunosuppression can be focused, with an emphasis on the identification of possible molecular markers of post-septic immunosuppression and novel therapeutic targets for treatment of individuals with long-term sepsis-induced immunosuppression.
Innate Immunity: Epigenetic Regulation of Cytokine and Chemokine Responses
During the initial response to polymicrobial infection, innate immune activation leads to the overproduction of the proinflammatory chemokines and cytokines responsible for the initiation and amplification of SIRS. These include neutrophil and macrophage chemotactic factors such as CCL2 (MCP-1), CCL3 (MIP-1α), CCL6 (C10) and CXCL8 (IL-8).Citation31 Additionally, increased production of proinflammatory cytokines, such as TNFα, IL-1β, IL-6 and IFNγ, promotes the effector functions of neutrophils, macrophages and TH1 T cells, further exacerbating cellular immunity.Citation32 These responses work in context with complement activation during SIRS to promote cytotoxic immune responses, tissue damage and multiorgan dysfunction (MODS). As mentioned previously, the increase of anti-inflammatory cytokines (such as TGFβ and IL-10) and TH1-opposing TH2 cytokines (such as IL-13) during CARS serves to counteract the proinflammatory responses of SIRS; this anti-inflammatory response also leaves the patient susceptible to secondary and/or nosocomial infections that negatively impact survival. However, post-septic immunosuppression is not limited to the anti-inflammatory responses of CARS, but also includes the directed suppression of pro-inflammtory functions in innate immune cells.
Cells of the innate immune system arise from myeloid progenitors in the bone marrow, which give rise to distinct myeloid and granulocyte lineages in response to growth factor and differentiation stimuli.Citation33 During the acute phase of SIRS, myeloid progenitor cells give rise to mature innate immune cells responsible for the clearance of microorganisms, including macrophages and neutrophils. Clearance of infiltrating bacteria is dependent on both the phagocytic capacity of these cells and the ability of these cells to produce proinflammatory chemokines and cytokines. However, the unchecked production of these proinflammatory mediators leads to physiological stress, apoptosis and tissue damage. As mentioned previously, the CARS response is hypothesized to be a compensatory response aimed at countering the damaging effects of SIRS. Recent research has indicated that the CARS phenotype may be more than an increased production of anti-inflammatory mediators, but may also comprise directed suppression of proinflammatory mediators at the cellular level through epigenetic regulation of gene loci in mature immune cells.
In regards to bacterial-induced SIRS, responses to prokaryotic cell wall components, such as lipopolysaccharides (LPS) from the cell walls of gram-negative bacteria, are important mediators of host inflammatory processes. Extremely high levels of LPS in damaged tissues, blood or lymph can trigger the uncontrolled “cytokine storm” characteristic of SIRS by signaling through pattern-recognition receptors on the surface of innate immune cells. These include members of the Toll-like receptor (TLR) family, most notably TLR4.Citation34 TLR4 stimulation results in the production of pro-inflammatory chemokines and cytokines (as mentioned earlier) by macrophages, dendritic cells and neutrophils, among other myeloid lineage cells. Interestingly, many myeloid lineage cells lose their ability to respond to LPS following an initial high-dose response; this phenomenon has been termed “LPS/endotoxin tolerance,” especially in regards to the responsiveness of post-septic macrophages.Citation16 While it is possible that the anti-inflammatory (and/or TH2-biased) cytokine production during CARS may play a role in mediating this effect in vivo, recent studies indicate cell-intrinsic mechanisms for this LPS tolerance phenotype. In these models, epigenetic regulation of proinflammatory gene loci plays a prominent role.
During primary responses to gram-negative bacteria, production of IL-1β and TNFα by macrophages is critical for productive inflammation and clearance of the microorganism. However, following LPS tolerance induction, macrophages exhibit increased levels of repressive histone modifications at the promoter regions of both IL-1β and TNFα, mediated by dimethylation of histone 3 at lysine residue 9 (H3K9me2).Citation35,Citation36 DNA-associated transcription factors and transcriptional mediators, such as RelB and HMGB1, can direct this chromatin remodeling. Similar results were observed in both in vitro LPS tolerance experiments with human monocytic cell lines, as well as in peripheral blood monocytes (PBMCs) from septic patients. This increase in repressive histone modifications proximal to the promoter regions of IL-1β and TNFα limits the ability of the macrophage to respond to subsequent LPS challenges. In addition, the binding of the H3K9 histone methyltransferase G9a to the TNFα promoter during the induction of LPS tolerance can direct DNA methylation via the HP1-dependent recruitment of the DNA methylase Dmnt3a/b.Citation37 This further restricts the TNFα promoter in a transcriptionally inaccessible state, leading to decreased TNFα protein levels produced upon secondary challenge with LPS.
LPS stimulation of both human PBMCs and mouse macrophages has been shown in previous studies to direct chromatin remodeling essential for the production of pro-inflammatory chemokines and cytokines. For example, LPS-dependent expression of IL-8 and CCL2 (MCP-1) is partially dependent on histone acetylation, as treatment of LPS-stimulated PBMCs with histone deacetylase inhibitors results in decreased expression of both proinflammatory mediators in vitro.Citation38 The production of IL-8 and TNFα by human immune cell lines in response to LPS has also been shown to be dependent on histone acetylation events, as pretreatment of these cells with the broad-spectrum HDAC inhibitor trichostatin A (TSA) reduced the production of these proinflammatory mediators in vitro.Citation39 Interestingly, these studies also suggest that glucocorticoids may exert their immunosuppressive effects via modulation of histone acetylation, indicating one possible shared epigenetic mechanism between anti-inflammatory therapy and post-septic immunosuppression. Previously published reports suggest that bacteremia may promote endogenous glucocorticoid production; studies of LPS tolerance in mouse models indicate a time- and dose-dependent increase in plasma levels of adrenocorticotropic hormone (ACTH), a key upstream hormone responsible for stimulating the production of cortisol from the adrenal gland.Citation40 In addition, TLR4-dependent modulation of the calcium/calmodulin signal transduction pathway in macrophages is required for the chromatin remodeling function of the BAF complex, irrespective of BAF binding to target chromatin.Citation41 LPS has also been shown to directly increase the mRNA expression of numerous histone deacetylases (HDACs) in murine bone marrow-derived macrophages, and HDAC inhibition with TSA served to potentiate LPS-induced induction of numerous proinflammatory genes, most notably CXCL2 (MIP-2α).Citation42
In a similar fashion to the processes underlying developmental decisions in pluripotent stem cells during differentiation, immune cells rely on chromatin modifying enzymes to regulate gene expression directing maturation, lineage commitment and effector function. In macrophages, LPS stimulation has been shown to direct the increased expression of the histone demethylase KDM6B (JMJD3).Citation43 This increased expression of KDM6B can be directed by LPS through activation of NFκB, as shown in macrophage cell lines;Citation43 in fact, over 70% of LPS-inducible genes in murine bone marrow-derived macrophages appear to be controlled (at least in part) by the histone demethylase activity of KDM6B, indicating a direct link between polymicrobial stimulus and epigenetic gene regulation.Citation44 Surprisingly, KDM6B appears to have an important role in governing gene regulation in both LPS-stimulated (classically activated/M1) and in IL-4 stimulated (alternatively activated/M2) macrophages, as KDM6B is critical for the removal of the repressive histone methylation mark H3K27 at the promoter regions of M2-specific gene loci during IL-4 dependent maturation to M2.Citation45 Upregulation of KDM6B in bone marrow derived macrophages is governed by the IL-4 dependent activation of STAT6, and levels of KDM6B in IL-4 activated macrophages correlate with H3K27 levels at the promoters of M2 gene loci.Citation45
As KDM6B is responsible for removing repressive histone methylation marks, it may be counter-intuitive to suggest a role for this enzyme in post-septic immunosuppression, as it would seemingly prime gene products for transcription, as opposed to actively repressing genes as seen in previous examples of LPS tolerance. However, IL-4 has been shown to play a prominent role in the pathophysiology of CARS, as it is a potent TH2 cytokine that can counteract the pathogenic TH1 cytokines expressed during the “cytokine storm” of SIRS.Citation46 Additionally, IL-4 expression in serum is markedly increased in survivors of severe sepsis as compared to nonsurvivors; interestingly, this difference was not reflected in IL-4 expression levels between groups on the day of admission to the intensive care unit.Citation47 Multiplex analysis of cytokine expression in the serum of patients with severe sepsis indicates that IL-4 levels can serve as predictors of early mortality (<48 h), as elevated levels of serum IL-4 during the acute phase was observed in nonsurvivors.Citation48 It is interesting to postulate that the elevated IL-4 produced during CARS may promote the production of M2 macrophages through upregulation of KDM6B and removal of repressive H3K27 marks governing M2 macrophage genes. Supporting this concept are studies in BALB/c mice indicating that bone-marrow derived macrophages from mice subjected to severe sepsis via CLP exhibit an M2 phenotype.Citation49 In studies of M2 macrophages in helminth infections (which are potent inducers of the M2 phenotype), these M2 macrophages were shown to have significantly reduced bacterial killing capabilities, reminiscent of LPS-tolerant macrophages from septic animals.Citation50 The IL-4/KDM6B axis appears to be an important pathway for the epigenetic regulation of macrophage activation following SIRS and may provide a mechanistic pathway for epigenetic regulation of LPS tolerance in post-septic macrophages.
In a similar fashion to high-dose LPS tolerance induction, macrophages from septic miceCitation13 and human patientsCitation51 exhibit refractory responses to ex vivo LPS re-challenge, as manifested by decreased pro-inflammatory cytokine production. Recent studies indicate that this reduced cytokine production may also have a prominent epigenetic component, as discussed previously with experimental LPS tolerance induction. For example, lung-resident macrophages from septic mice (24 h post-surgery/sepsis induction) exhibit significantly decreased expression of mRNA for TNF, IL-12p40 and iNOS when restimulated ex vivo with LPS as compared to sham surgery controls.Citation52 In addition, these post-septic lung macrophages exhibit decreased acetylation of histone 4 (H4Ac) and lysine 4 tri-methylation of histone 3 (H3K4me3), both of which are activating epigenetic marks.Citation52 These results suggest that the “cytokine storm” of sepsis may be directing the loss of specific activating epigenetic marks at promoters of proinflammatory genes in macrophages via some unknown mechanism. Interestingly, these epigenetic marks (and subsequent repression of pro-inflammatory gene expression) are reversed in cells lacking the TLR4 negative regulator Interleukin-1 receptorassociated kinase-M (IRAK-M).Citation52 The link between IRAK-M and histone modifications remains unclear; however, IRAK-M-/- macrophages exhibit modulated expression of histone deacetylase mRNA following sepsis as compared to wild-type macrophages, suggesting that IRAK-M may direct sepsis-induced chromatin modifications through modulations of the histone modifying machinery in the cell.Citation52
In addition to the deleterious effects on macrophages, the pathophysiological effects of SIRS also negatively impact the activation and effector function of dendritic cells (DCs). Professional antigen presenting cells such as DCs are a critical link between the innate and adaptive immune system, as these cells are responsible for activating T lymphocytes through presentation of cognate antigen in the context of MHC molecules.Citation53 In addition, dendritic cells are essential for modulating the adaptive immune response to infection via the production of proinflammatory chemokines and cytokines that guide the activation and lineage commitment of lymphocytes. For example, the production of IL-12 by DCs is critical for the development of TH1 T cells from immature CD4+ T cells during early responses to infection.Citation54 DCs are also intimately linked with the chemokine responses generated during infection that are critical for the trafficking of leukocytes, as well as their activation and effector function.Citation55 While chemokines are not as easily delineated into Th-type effector responses as are cytokines, certain DC-derived chemokine families are associated with TH1 or TH2 immune responses.Citation56 These differences in chemokine production are often associated with the origin of the DC under investigation; for example, peripheral DCs such as Langerhans cells exhibit different chemokine expression profiles as splenic DCs.Citation57 In the context of TH1-type stimulations such as IFNγ and LPS, DCs have been shown to produce CXCL9 (MIG), CXCL10 (IP-10) and CXCL11 (I-TAC).Citation57 In addition, IL-12-mediated interactions with CD8+ T cells has been shown to increase CCL1 and CCL17 (TARC) production by DCs,Citation58 and DCs have been shown to produce CX3CL1 (Fractalkine) in response to bacterial infections, most notably Legionella pneumophila.Citation59 Chemokine and cytokine production by DCs are essential for the proper orchestration of the adaptive immune response during immunity to infection.
DCs exert a significant effect on the pathophysiology of sepsis and in return the cellular stresses of SIRS reciprocally affect DC responses during the progression to CARS and post-septic immunosuppression; as such, DCs are important contributors to the pathogenesis of sepsis.Citation60 As bacterial products such as LPS readily activate DCs, DC-derived chemokines and cytokines contribute greatly to the immune responses necessary for clearance of bacteria. For example, DC functions are critically required for survival during the acute phase of sepsis, as evidenced by mouse models utilizing conditional genetic techniques for the in vivo deletion of DC subsets.Citation61 Additionally, loss of DC subsets in peripheral blood has been shown to predict poor outcomes in human patients with sepsis.Citation62 Sepsis-induced apoptosis of DCs has been noted in both spleensCitation63 and lymph nodesCitation64 of mice in experimental models of sepsis, indicating that the physiological stresses induced during SIRS have a negative effect on the viability of these cells. Interestingly, while DC functions during sepsis appear critical for the survival of both human patients and experimental animals, their activation and effector function is often blunted and diminished. For example, peripheral blood mononuclear cells (PBMCs) from septic patients exhibit an increased propensity to mature into DCs (kinetically); however, these ex vivo matured DCs have a decreased ability to stimulate T-cell proliferation in mixed-lymphocyte reactions (MLRs), indicating a deficiency in their antigen-presentation functions.Citation65
One of the hallmarks of CARS and post-septic immunosuppression is the susceptibility of survivors to opportunistic infections that are normally handled by a fully functioning immune system.Citation66 In animal models, this post-septic immunosuppression can be modeled utilizing airway challenges with the ubiquitous opportunistic fungi Aspergillus fumigatus. Immunocompetent humans and mice are efficient in clearing Aspergillus conidia from the airways through the co-ordinated microbicial activity of numerous granulocyte/myeloid lineage cells, including neutrophils and DCs.Citation67,Citation68 However, Aspergillus infection is a serious condition for immunocompromised individuals;Citation69 for example, invasive pulmonary aspergillosis is a leading cause of mortality in patients following bone marrow transplantation (BMT).Citation70 While functional immunity against Aspergillus infection is largely handled by the innate immune system, host immunity requires a productive TH1 type chemokine and cytokine response; for example, the production of IL-12 by lung-resident DCs is essential for clearance of Aspergillus conidia.Citation71,Citation72 In a similar fashion as BMT patients, post-septic mice are susceptible to Aspergillus infections, exhibiting up to 100% mortality after exposure at airway doses that are well-tolerated by sham surgery control mice.Citation20 This susceptibility appears to be directly related to the suppression of protective immunity mediated by DCs in post-septic mice; lung resident DCs from post-septic mice exhibit decreased IL-12 production following conidia antigen stimulation and intratracheal administration of wild-type DCs restores protective immunity in post-septic mice.Citation19 These data indicate that DCs are significantly affected by the stresses of septic shock, and while their presence is essential for survival during the acute phase of sepsis, the DCs that survive SIRS remain severely compromised in their function.
As with post-septic macrophages and LPS tolerance, post-septic DCs exhibit a reduced capacity to produce IL-12 in response to microbial stimulation, specifically in regards to TLR-dependent signal pathways.Citation73 IL-12 production via Il12 gene expression requires the active stabilization of the Il12 promoter into a transcriptionally active state. During chromatin remodeling events, the addition of activating methylation marks on lysine 4 of histone 3 (H3K4) and removal of repressive methylation marks on lysine 27 of histone 3 (H3K27) serve to stabilize gene loci for active transcription.Citation74,Citation75 Modulations in the balance of active vs. repressing histone methylation marks can have a dramatic effect on the level of gene transcription in a cell; for example, shifts towards increased H3K27 methylation can dramatically reduce specific gene transcription levels. Modulations in the relative levels of H3K4 and H3K27 can be used in other myeloid cells, namely macrophages, to delineate between gene families directly affected by LPS tolerance.Citation76 Therefore, concomitant increases in H3K27 and decreases in H3K4 in promoter regions can drastically decrease the resulting gene transcription of genes under the control of methylation-sensitive promoters.
Consistent with this model of histone methylation and gene regulation, studies utilizing lung-resident DCs from post-septic mice indicated that modulations in H3K4 and H3K27 methylation at the promoter regions of the Il12 gene correlate with decreased IL-12 protein production in response to TLR stimulus.Citation77 In addition, chromatin immunoprecipitation assays indicated that post-septic lung DCs exhibit increased recruitment of proteins mediating H3K27 methylation and concurrent decreased recruitment of H3K4 protein complexes, to the promoter regions of both the p35 and the p40 subunits of IL-12.Citation77 Of particular interest was the apparent stability of these methylation marks, as lung-resident DCs from post-septic mice continued to exhibit the imbalance towards repressive histone methylation at the Il12 promoter at six weeks following the experimental onset of acute sepsis.Citation77 These results are of particular importance when considering the persistent nature of post-septic immunosuppression; as these modulations in histone methylation appear to remain long after the acute phase of sepsis has resolved, it can be hypothesized that they play an important role in shaping the immune response at timepoints distal from recovery. As IL-12 is a critical early cytokine required for the development of a fully functioning TH1 immune response,Citation54 the epigenetic repression of IL-12 in DCs may also contribute to the TH2 bias observed in post-septic mice through suppression of TH1 responses.
Suppression of myeloid cell function following severe sepsis contributes greatly to the immunosuppression observed in both human patients and animal models. Both post-septic macrophages and dendritic cells exhibit decreased activation potential in response to pathogen stimuli, and their chemokine and cytokine production is reduced as a consequence. As the previous studies indicate, this reduced activation potential of myeloid cells correlates with modulations in the epigenetic signature of chemokine and cytokine genes, including changes in histone acetylation and methylation patterns (). These results suggest that the physiological stress imparted on the immune system during severe sepsis can have significant effects on long-term gene expression and that epigenetic regulatory “signatures” in myeloid cells can be modulated following uncontrolled immune responses. While critical for host defense, myeloid immune responses and innate immune responses in general, are only one part of a fully functioning immune response.
Competent host defense against pathogens often requires the activation of the adaptive immune system, mediated by cross-talk between myeloid cells and lymphoid cells. Clearly, the activation deficiencies mentioned previously can have a direct effect on lymphoid activation, especially in regards to the decreased cytokine and chemokine production by post-septic myeloid cells. However, recent studies have indicated that postseptic lymphocytes, and in particular CD4+ T-helper cells, exhibit cell-intrinsic defects in immune functions following sepsis. Additionally, as seen with myeloid cells, postseptic lymphocytes exhibit their own modulations in histone modifications, and these changes are correlated with cell-intrinsic defects in lineage commitment and effector function.
Adaptive Immunity: Epigenetic Regulation of Lymphocyte Activation and Effector Function
During the acute phase of sepsis, the bulk of the immune response is mediated by the innate immune system, and the “cytokine storm” of severe sepsis is largely a result of TLR-mediated stimulation of macrophages and dendritic cells, among others. However, the adaptive immune system is also involved in shaping the immune response during sepsis, and the physiological stresses of sepsis also exert their own effects on the lymphoid compartment. In this fashion, the adaptive immune response, and in particular the CD4+ T-helper response, is affected in two ways during the post-septic period—both through interactions with defective postseptic myeloid cells and through cell-intrinsic defects imparted to T cells following septic injury.
Lymphocyte apoptosis is a hallmark of severe sepsis; increased cell death of CD4+ T cells and T-cell precursors is observed in the thymus,Citation10 spleenCitation78 and bloodCitation79 of both humans and animals during the acute phase of sepsis. However, total numbers of CD4+ T cells appear to return to physiological levels following recovery, and the T-cell receptor repertoire of these cells appears unchanged from those found in control mice, suggesting that sepsis did not overtly prime the adaptive immune response to any given set of antigens.Citation80 These data indicate that the lymphocyte apoptosis observed during sepsis is a transient phenomenon and that some combination of re-seeding by the thymus and homeostatic proliferation of surviving T cells works in context to re-populate the CD4+ T-cell compartment following recovery. However, while these CD4+ T cells appear unchanged in number and distribution, they do exhibit numerous defects in activation and effector function that may contribute to the post-septic immunosuppression observed in both animal models and human patients.
One of the hallmarks of postseptic lymphocytes is a reduction in their proliferative capacity.Citation81,Citation82 This reduced proliferation is correlated with decreases in splenocyte production of IL-2, a potent T-cell growth factor.Citation83 Much of this reduction in proliferative capacity in vivo can be attributed to deficiencies in dendritic cell functions, as mentioned previously.Citation84 However, total numbers of lymphocytes (and CD4+ T cells in particular) return to normal levels in peripheral lymphoid tissues at later timepoints, suggesting that the reduction in proliferative capacity may be at some level a transient phenomenon. Interestingly, modulations in the quality of the T-helper response in post-septic patients and animals appear to remain for longer timepoints; in particular, the shift from TH1 immune responses towards TH2 responsesCitation14,Citation82 and repressive lymphocyte responses in general.Citation85 These data suggest that T lymphocytes retain cell-intrinsic defects that influence the quality of the T-helper response during future immune responses.
The development and maintenance of a helper T-cell immune response involves the stabilization of helper T-cell gene loci into transcriptionally active forms. As CD4+ T cells commit to a given effector lineage (e.g., TH1 vs. TH2), the associated lineage-specific genes (encoding transcription factors and cytokines, among others) undergo epigenetic modifications to activate the required gene products and silence unwanted gene products.Citation86 For example, hyperacetylation of histones 3 and 4 proximal to the Ifng promoter is associated with the differentiation of naïve T cells into TH1 cells.Citation87 Additonally, increases in transcription-activating modifications of histone 3 at various sites in the Il4-Il13 gene locus are associated with TH2 lineage commitment.Citation88 Importantly, epigenetic regulation of gene expression is essential not only for activation during lineage commitment; reciprocal silencing of TH1 and TH2 gene loci via sequestering into heterochromatin is characteristic of CD4+ T cells that have fully committed to a given effector lineage.Citation89 Therefore, proper CD4+ T-cell lineage commitment requires both epigenetic activation and silencing of genes and perturbations in the epigenetic landscape can have a deleterious effect on the ability of naïve T cells to commit to any specific effector lineage.
As mentioned previously, CD4+ T cells undergo apoptosis during the acute phase of severe sepsis. As post-septic mice and humans both exhibit a modulated TH-cytokine response (often skewed towards TH2), it has been hypothesized that the increased cell death may play a role in this phenomenon. Studies in humans indicate that post-septic TH1 CD4+ T cells may selectively undergo apoptosis following restimulation, suggesting one possible mechanism for apoptosis-induced TH2 skewing.Citation82 However, these results do not fully explain the deficiencies in effector function seen at distal timepoints after recovery from severe sepsis, when levels of CD4+ T cells have recovered to that of control populations. Recent studies utilizing the naïve CD4+ T-cell population in post-septic mice indicate that these cells exhibit difficulty in committing to either the TH1 or TH2 lineage in vitro when instructed to do so using polyclonal stimulus and exogenous cytokines.Citation90 This “difficulty” is manifested by the decreased production of IFNγ by post-septic CD4+ T cells instructed to become TH1 effectors and the production of both IFNγ and IL-4 in TH2 effector cultures, as compared to CD4+ T-cell cultures from sham surgery mice.Citation90 These results are especially striking as they indicate a CD4+ T-cell-intrinsic defect in lineage commitment, even in response to “optimized” in vitro differentiation cultures. Concurrent with this phenomenon, chromatin immunoprecipitation assays (ChIP) indicated an increase in repressive H3K27 methylation at the promoter regions of the TH1 cytokine gene Ifng, and the TH2 transcription factor gene Gata3.Citation90 Additionally, both Ifng and Gata3 mRNA were found to be decreased in abundance in post-septic naïve CD4+ T cells as compared to sham surgery cells when analyzed directly ex vivo, providing evidence that the increase in H3K27 methylation is involved with driving gene repression in these cells.Citation90
While the increase in H3K27 methylation in post-septic CD4+ T cells provides one possible mechanism for helper T-cell dysfunction seen in post-septic immunosuppression, it also creates its own questions. For example, if H3K27 methylation is increased at the Ifng promoter in post-septic CD4+ T cells, then why do these cells produce more IFNγ than sham surgery counterparts in TH2 cultures? Also, how does increased H3K27 methylation at the Gata3 promoter support the (general) shift towards TH2 responses seen in post-septic mice and humans? There are two possible explanations for this phenomenon. First, histone methylation is only one of the numerous epigenetics marks found on histone tails; gene regulation can be affected by histone acetylation, hosphorylation and ubiquitination, among others,Citation75 as well as direct methylation of the DNA itself.Citation22 Analysis of other chromatin remodeling events may clarify the cumulative effect of epigenetic modifications on post-septic CD4+ T-cell function. Secondly, recent high-throughput analysis of histone methylation in murine T cells indicates that many lineage-specific gene loci are “plastic,” in that they can contain both (or neither) H3K4 and H3K27 marks prior to lineage commitment.Citation91 Upsetting the balance between activating and repressive histone methylation marks could adversely affect the ability of the CD4+ T cell to stabilize the TH1/TH2 gene loci in response to cytokine stimuli, resulting in impaired decision making by naïve T cells in regards to T-helper lineage.
Epigenetic modification of gene loci in CD4+ T cells following sepsis not only limits certain lineage decisions, but can also guide decisions by priming certain gene loci for transcription prior to stimulation. Along with lymphocyte apoptosis, anergy and TH-lineage deficiencies, post-septic miceCitation85 and humansCitation92 also display a general increase in regulatory T cells (Treg). Regulatory T cells of the CD4+ lineage are cells capable of suppressing immune responses in a variety of contexts and can arise either directly from thymocytes (natural Tregs, nTreg) or from naïve T cells in response to the local cytokine milleu in the periphery (induced Tregs, iTreg).Citation93 Both populations of cells have suppressive activity in numerous in vivo and in vitro models; however, nTregs are committed to the suppressive lineage in the thymus, while iTregs arise from naïve T cells in the periphery.Citation94 In both cases functional Tregs require the expression of the transcription factor Foxp3 for the maintenance of their suppressive activity.Citation95 Expression of Foxp3 in CD4+ T cells requires coordinated permissive epigenetic modifications, including histone methylation and acetylation, as well as DNA demethylation at specific promoter and enhancer regions in the Foxp3 locus.Citation96 The pervasiveness of these epigenetic changes appear to correlate with the original source of the Treg in question; for example, DNA methylation levels in promoter and enhancer regions of Foxp3 can delineate nTregs versus TGFβ stimulated peripheral T cells (iTregs),Citation97 and the relative abundance of these DNA methylation marks may serve to stabilize Foxp3 transcription in fully differentiated Tregs.Citation98
Increases in Tregs may play a role in mediating post-septic immunosuppression through their ability to suppress inflammation during subsequent infections.Citation99 Consistent with this hypothesis, analysis of peripheral blood from human sepsis patients indicates that increases in circulating Tregs correlates with decreased proliferative capacity of lymphocytes in these patients.Citation100 Therefore, understanding the mechanisms governing the expansion of Tregs post-sepsis becomes essential for understanding the cellular basis of post-septic immunosuppression. Previous studies have indicated that the apparent increase in Tregs in post-septic patients may be due to the concomitant decrease in effector CD4+ T cells (defined as CD25−);Citation101 however, as mentioned previously the total CD4+ T-cell compartment recovers in total numbers in the long term following sepsis recovery. This suggests that there may be other mechanisms playing a role in maintaining Treg cell numbers in the periphery post-sepsis.
Interestingly, Foxp3 expression in T cells post-sepsis is increased not only in the putative Treg subset (expressing high and medium levels of CD25), but also in the conventional T-cell population characterized by absence of CD25 expression.Citation102 This suggests that the transcription factor necessary for Treg function may be turned on in post-septic effector T cells, which could skew their function towards suppressive activity, in other words these cells are converted in Tregs. Additionally, putative Tregs (CD4+ CD25+) from post-septic mice were shown to have increased suppressive activity as compared to control Tregs.Citation102 Interestingly, chromatin IP analysis of post-septic CD4+ CD25− T cells indicated a significant increase in acetylation of histone 3 at lysine 9 (H3K9ac), thought to be a permissive epigenetic mark.Citation102 In addition, these post-septic CD4+ CD25− T cells exhibited increases in expression of the mRNA encoding the histone acetyltransferase Kat2a, suggesting a mechanistic link between increased expression of this enzyme and increases in H3K9ac.Citation102 Based on these results, it can be hypothesized that the increase in permissive histone modifications in the Foxp3 locus serves to guide T-cell responses towards the Treg lineage, through an increased propensity to express Foxp3. These studies imply a mechanistic link between sepsis and epigenetic modification of the Foxp3 gene locus, leading to increased Treg lineage commitment in the post-septic T-cell pool and ultimately an increase in immune suppression.
Following severe sepsis, CD4+ T cells exhibit numerous functional deficiencies, including reduced proliferative capacity, activation potential and deficiencies in the directed production of TH-specific cytokines. In a similar vein as myeloid cells, these post-septic deficiencies correlate with epigenetic modifications in the promoter regions of genes essential for T-cell function. These include increases in repressive histone methylation proximal to genes important for effector TH cell function, as well as increases in activating histone acetylation proximal to genes important for driving regulatory T-cell function (). When coupled with the deficiencies in myeloid cell function, especially the decreases in cytokine production by dendritic cells, these modulations in gene expression provide a compelling case for epigenetics as a major driving factor in post-septic immunosuppression.
Perspectives: Sepsis, Epigenetics and Progenitor Cells
It has become clear that the “cytokine storm” of severe sepsis can drive the modulation of gene expression in numerous immune cell types. This modulation in gene expression can then affect the function of post-septic immune cells, by limiting their ability to mediate an effective response to secondary infections. A growing body of research in both animal models and human patients suggests that this modulation in gene expression can largely be ascribed to epigenetic mechanisms, including modifications in the histone code that both repress essential genes and activate unwanted or unwarranted gene products. While these studies provide new insight into the molecular mechanisms governing post-septic immunosuppression, questions remain as to the extent of epigenetic regulation as a driving factor in this phenomenon.
As mentioned previously, epigenetics encompasses much more than DNA methylation, histone methylation and acetylation. Numerous other histone modifications can effect gene expression, including phosphorylation, ubiquitination and sumoylation, among others. Additionally, the sequestering of gene loci into transcriptionally silent heterochromatin or active euchromatin often relies on a wide range of co-ordinated histone modifications, rather than single methylation or acetylation events. Further studies aimed at elucidating the status of histone modifications in post-septic cells is required to get a clearer picture of gene regulation and how it drives immunosuppression. In addition, studies aimed at linking the up or downregulation of histone modifying proteins to the marks themselves in postseptic immune cells are essential, as these data could provide both a mechanistic explanation for the appearance of these marks, and can possibly serve as a molecular “fingerprint” for post-septic immunosuppression. Previous studies, such as the KDM6B/M2 macrophage and Kat2a/CD4+ T cell reports, show the possibility of linking changes in histone modifying protein expression with cell phenotype and function. Finally, the discussions in this review have omitted the roles of micro-RNA (miRNA) in modulating immune functions post-sepsis. miRNAs are small non-coding RNAs that can modulate the expression of multiple mRNAs through targeted degredation of messages, and they play an important role in epigenetic regulation of gene expression in immune cells, and in eukaryotes in general.Citation103 Recent studies have started to implicate miRNA species as potential biomarkers for sepsis severity,Citation104 as well as possible mediators of LPS tolerance.Citation105 The role of miRNAs in mediating post-septic immunosuppression remains an open field of study.
Of particular concern with post-septic immunosuppression is the persistence of the condition; both mouse models and human studies have indicated that the immune deficiencies discussed herein can persist for months, and perhaps years beyond recovery from acute symptoms. While the persistence of epigenetically “imprinted” leukocytes could in part explain the persistence of post-septic immunosuppression, it fails to explain why the immune system is not rescued by the generation of new immune cells by the bone marrow. One possible hypothesis is that the epigenetic imprints that occur on mature immune cells in the periphery could also imprint upon progenitor cells in the bone marrow (). In this scenario, the modulation in epigenetic signatures in progenitor cells may result in defects in hematopoiesis or activation defects in mature daughter leukocytes. Further, repressive histone modifications may be retained in the progenitor cells that survive SIRS, and their epigenetic signatures can be passed on to the daughter cells, perpetuating dysregulated gene expression. At present, these hypotheses are strictly speculative; however, the advent of next-generation technologies for epigenetic analysis have made it possible to analyze epigenetic signatures across the entire genome of a cell; for example, analyzing H3K4 or H3K27 methylation across numerous gene loci.Citation91 The application of these techniques to analyzing progenitor cells in animal models of sepsis and peripheral blood leukocytes in human patients, will allow for the quantification of the epigenome of post-septic immune cells. It will be of particular interest to determine if the epigenetic signatures apparent on mature immune cells can be traced back to similar modifications in progenitor cells following sepsis.
Sepsis poses a significant human health concern both in terms of the mortality associated with SIRS and the immunosuppression associated with CARS and long-term survivors of shock. Post-septic immunosuppression is characterized by multi-faceted deficiencies in activation and effector function by cells of both the innate and adaptive immune system. A growing body of experimental evidence suggests that epigenetic mechanisms, driven by as yet unknown signals generated during the acute phase of sepsis, play an important role in mediating post-septic immunosuppression. The study of these epigenetic changes in immune system functions may not only provide a better understanding of post-septic immunosuppression, but may also yield important biomarkers (histone modifying protein expression, histone modifications, etc.,) which can be used to assay the functionality of a post-septic patient's immune system.
Figures and Tables
Figure 1 Overview of histone modifications and gene regulation events observed in myeloid cells associated with endotoxin shock and sepsis. Phenotypic outcomes associated with the indicated modification events are listed below each cell subtype.
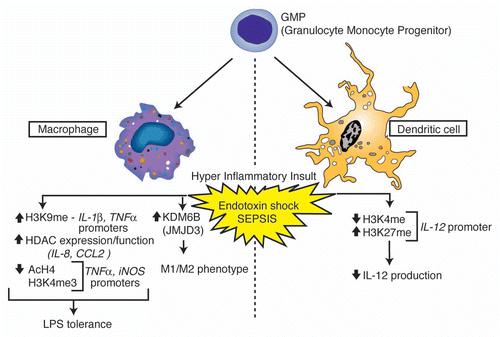
Figure 2 Overview of histone modifications and gene regulation events observed in CD4+ T lymphocyte subsets following severe sepsis. Phenotypic outcomes are listed below the indicated modification events.
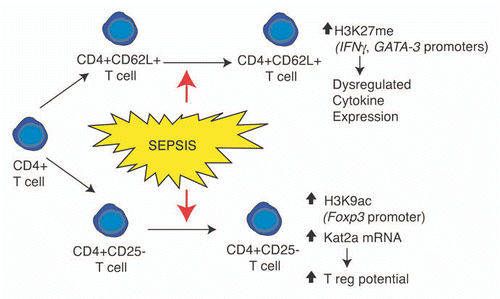
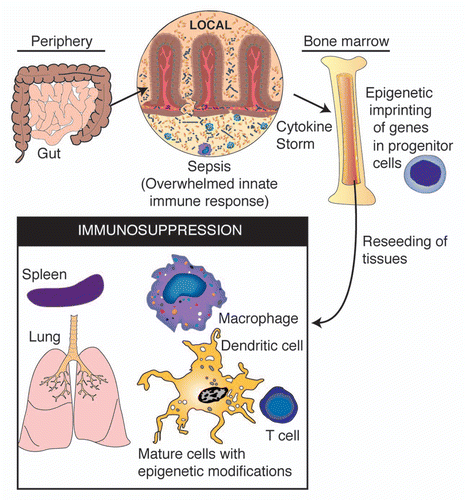
Figure 3 A schematic of the proposed feedback system between systemic inflammation and epigenetic imprinting of immune progenitor cells. In such a system, epigenetic modifications occur in progenitor cells during the acute phase of sepsis, resulting in re-seeding of tissues with daughter cells carrying similar histone modifications following recovery from sepsis.
Acknowledgements
The authors would like to thank R.G. Kunkel for the illustrations found in –. This work was supported in part by NIH grants HL31237 and HL089216.
References
- Remick DG. Pathophysiology of sepsis. Am J Pathol 2007; 170:1435 - 1444
- Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, Cohen J, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004; 32:858 - 873
- Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368 - 1377
- Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: A trend analysis from 1993 to 2003. Crit Care Med 2007; 35:1244 - 1250
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome and associated costs of care. Crit Care Med 2001; 29:1303 - 1310
- Reddy RC, Chen GH, Tekchandani PK, Standiford TJ. Sepsis-induced immunosuppression: From bad to worse. Immunol Res 2001; 24:273 - 287
- Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med 2008; 29:617 - 625
- Barthlen W, Zantl N, Pfeffer K, Heidecke CD, Holzmann B, Stadler J. Impact of experimental peritonitis on bone marrow cell function. Surgery 1999; 126:41 - 47
- Rocha PN, Plumb TJ, Robinson LA, Spurney R, Pisetsky D, Koller BH, et al. Role of thromboxane A2 in the induction of apoptosis of immature thymocytes by lipopolysaccharide. Clin Diagn Lab Immunol 2005; 12:896 - 903
- Wang SD, Huang KJ, Lin YS, Lei HY. Sepsis-induced apoptosis of the thymocytes in mice. J Immunol 1994; 152:5014 - 5021
- McDunn JE, Turnbull IR, Polpitiya AD, Tong A, MacMillan SK, Osborne DF, et al. Splenic CD4+ T cells have a distinct transcriptional response six hours after the onset of sepsis. J Am Coll Surg 2006; 203:365 - 375
- Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE Jr, Hui JJ, Chang KC, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol 2001; 166:6952 - 6963
- Reddy RC, Chen GH, Newstead MW, Moore T, Zeng X, Tateda K, et al. Alveolar macrophage deactivation in murine septic peritonitis: role of interleukin 10. Infect Immun 2001; 69:1394 - 1401
- O'Sullivan ST, Lederer JA, Horgan AF, Chin DH, Mannick JA, Rodrick ML. Major injury leads to predominance of the T helper-2 lymphocyte phenotype and diminished interleukin-12 production associated with decreased resistance to infection. Ann Surg 1995; 222:482 - 490
- de Vos AF, Pater JM, van den Pangaart PS, de Kruif MD, van 't Veer C, van der Poll T. In vivo lipopolysaccharide exposure of human blood leukocytes induces cross-tolerance to multiple TLR ligands. J Immunol 2009; 183:533 - 542
- Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care 2006; 10:233
- Quartin AA, Schein RM, Kett DH, Peduzzi PN. Magnitude and duration of the effect of sepsis on survival, Department of Veterans Affairs Systemic Sepsis Cooperative Studies Group. JAMA 1997; 277:1058 - 1063
- Perl TM, Dvorak L, Hwang T, Wenzel RP. Long-term survival and function after suspected gram-negative sepsis. JAMA 1995; 274:338 - 345
- Benjamim CF, Lundy SK, Lukacs NW, Hogaboam CM, Kunkel SL. Reversal of long-term sepsis-induced immunosuppression by dendritic cells. Blood 2005; 105:3588 - 3595
- Benjamim CF, Hogaboam CM, Lukacs NW, Kunkel SL. Septic mice are susceptible to pulmonary aspergillosis. Am J Pathol 2003; 163:2605 - 2617
- Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, et al. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest 2006; 116:2532 - 2542
- Delcuve GP, Rastegar M, Davie JR. Epigenetic control. J Cell Physiol 2009; 219:243 - 250
- Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol 2009; 10:192 - 206
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight?. Nat Rev Genet 2008; 9:102 - 114
- Nafee TM, Farrell WE, Carroll WD, Fryer AA, Ismail KM. Epigenetic control of fetal gene expression. BJOG 2008; 115:158 - 168
- Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther 2009; 8:1409 - 1420
- Fedorova E, Zink D. Nuclear architecture and gene regulation. Biochim Biophys Acta 2008; 1783:2174 - 2184
- Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep 2002; 3:224 - 229
- Hublitz P, Albert M, Peters AH. Mechanisms of transcriptional repression by histone lysine methylation. Int J Dev Biol 2009; 53:335 - 354
- Zhu Q, Wani AA. Histone modifications: crucial elements for damage response and chromatin restoration. J Cell Physiol 2010; 223:283 - 288
- Coelho AL, Hogaboam CM, Kunkel SL. Chemokines provide the sustained inflammatory bridge between innate and acquired immunity. Cytokine Growth Factor Rev 2005; 16:553 - 560
- Netea MG, van der Meer JW, van Deuren M, Kullberg BJ. Proinflammatory cytokines and sepsis syndrome: not enough or too much of a good thing?. Trends Immunol 2003; 24:254 - 258
- Friedman AD. Transcriptional regulation of granulocyte and monocyte development. Oncogene 2002; 21:3377 - 3390
- Kaczorowski DJ, Mollen KP, Edmonds R, Billiar TR. Early events in the recognition of danger signals after tissue injury. J Leukoc Biol 2008; 83:546 - 552
- Chan C, Li L, McCall CE, Yoza BK. Endotoxin tolerance disrupts chromatin remodeling and NFkappaB transactivation at the IL-1beta promoter. J Immunol 2005; 175:461 - 468
- El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol Cell Biol 2009; 29:1959 - 1971
- El Gazzar M, Yoza BK, Chen X, Hu J, Hawkins GA, McCall CE. G9a and HP1 couple histone and DNA methylation to TNFalpha transcription silencing during endotoxin tolerance. J Biol Chem 2008; 283:32198 - 32208
- Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D, et al. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood 2007; 109:1123 - 1130
- Tsaprouni LG, Ito K, Adcock IM, Punchard N. Suppression of lipopolysaccharide- and tumour necrosis factoralpha-induced interleukin (IL)-8 expression by glucocorticoids involves changes in IL-8 promoter acetylation. Clin Exp Immunol 2007; 150:151 - 157
- Beishuizen A, Thijs LG. Endotoxin and the hypothalamo-pituitary-adrenal (HPA) axis. J Endotoxin Res 2003; 9:3 - 24
- Lai D, Wan M, Wu J, Preston-Hurlburt P, Kushwaha R, Grundstrom T, et al. Induction of TLR4-target genes entails calcium/calmodulin-dependent regulation of chromatin remodeling. Proc Natl Acad Sci USA 2009; 106:1169 - 1174
- Aung HT, Schroder K, Himes SR, Brion K, van Zuylen W, Trieu A, et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J 2006; 20:1315 - 1327
- De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007; 130:1083 - 1094
- De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J 2009; 28:3341 - 3352
- Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009; 114:3244 - 3254
- Miller AC, Rashid RM, Elamin EM. The “T” in trauma: the helper T-cell response and the role of immunomodulation in trauma and burn patients. J Trauma 2007; 63:1407 - 1417
- Wu HP, Wu CL, Chen CK, Chung K, Tseng JC, Liu YC, et al. The interleukin-4 expression in patients with severe sepsis. J Crit Care 2008; 23:519 - 524
- Bozza FA, Salluh JI, Japiassu AM, Soares M, Assis EF, Gomes RN, et al. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit Care 2007; 11:49
- Takahashi H, Tsuda Y, Takeuchi D, Kobayashi M, Herndon DN, Suzuki F. Influence of systemic inflammatory response syndrome on host resistance against bacterial infections. Crit Care Med 2004; 32:1879 - 1885
- Weng M, Huntley D, Huang IF, Foye-Jackson O, Wang L, Sarkissian A, et al. Alternatively activated macrophages in intestinal helminth infection: effects on concurrent bacterial colitis. J Immunol 2007; 179:4721 - 4731
- Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest 1991; 88:1747 - 1754
- Lyn-Kew K, Rich E, Zeng X, Wen H, Kunkel SL, Newstead MW, et al. IRAK-M regulates chromatin remodeling in lung macrophages during experimental sepsis. PLoS ONE 2010; 5:11145
- Coquerelle C, Moser M. DC subsets in positive and negative regulation of immunity. Immunol Rev 234:317 - 334
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003; 3:133 - 146
- McColl SR. Chemokines and dendritic cells: a crucial alliance. Immunol Cell Biol 2002; 80:489 - 496
- Lebre MC, Burwell T, Vieira PL, Lora J, Coyle AJ, Kapsenberg ML, et al. Differential expression of inflammatory chemokines by Th1- and Th2-cell promoting dendritic cells: a role for different mature dendritic cell populations in attracting appropriate effector cells to peripheral sites of inflammation. Immunol Cell Biol 2005; 83:525 - 535
- Fujita H, Asahina A, Sugaya M, Nakamura K, Gao P, Fujiwara H, et al. Differential production of Th1- and Th2-type chemokines by mouse Langerhans cells and splenic dendritic cells. J Invest Dermatol 2005; 124:343 - 350
- Henry CJ, Ornelles DA, Mitchell LM, Brzoza-Lewis KL, Hiltbold EM. IL-12 produced by dendritic cells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J Immunol 2008; 181:8576 - 8584
- Kikuchi T, Andarini S, Xin H, Gomi K, Tokue Y, Saijo Y, et al. Involvement of fractalkine/CX3CL1 expression by dendritic cells in the enhancement of host immunity against Legionella pneumophila. Infect Immun 2005; 73:5350 - 5357
- Efron P, Moldawer LL. Sepsis and the dendritic cell. Shock 2003; 20:386 - 401
- Scumpia PO, McAuliffe PF, O'Malley KA, Ungaro R, Uchida T, Matsumoto T, et al. CD11c+ dendritic cells are required for survival in murine polymicrobial sepsis. J Immunol 2005; 175:3282 - 3286
- Guisset O, Dilhuydy MS, Thiebaut R, Lefevre J, Camou F, Sarrat A, et al. Decrease in circulating dendritic cells predicts fatal outcome in septic shock. Intensive Care Med 2007; 33:148 - 152
- Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, et al. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol 2003; 171:909 - 914
- Efron PA, Martins A, Minnich D, Tinsley K, Ungaro R, Bahjat FR, et al. Characterization of the systemic loss of dendritic cells in murine lymph nodes during polymicrobial sepsis. J Immunol 2004; 173:3035 - 3043
- Faivre V, Lukaszewicz AC, Alves A, Charron D, Payen D, Haziot A. Accelerated in vitro differentiation of blood monocytes into dendritic cells in human sepsis. Clin Exp Immunol 2007; 147:426 - 439
- Benjamim CF, Hogaboam CM, Kunkel SL. The chronic consequences of severe sepsis. J Leukoc Biol 2004; 75:408 - 412
- Romani L. Immunity to fungal infections. Nat Rev Immunol 2004; 4:1 - 23
- Walsh TJ, Roilides E, Cortez K, Kottilil S, Bailey J, Lyman CA. Control, immunoregulation and expression of innate pulmonary host defenses against Aspergillus fumigatus. Med Mycol 2005; 43:165 - 172
- Thompson GR 3rd, Patterson TF. Pulmonary aspergillosis. Semin Respir Crit Care Med 2008; 29:103 - 110
- Garcia-Vidal C, Upton A, Kirby KA, Marr KA. Epidemiology of invasive mold infections in allogeneic stem cell transplant recipients: biological risk factors for infection according to time after transplantation. Clin Infect Dis 2008; 47:1041 - 1050
- Bozza S, Perruccio K, Montagnoli C, Gaziano R, Bellocchio S, Burchielli E, et al. A dendritic cell vaccine against invasive aspergillosis in allogeneic hematopoietic transplantation. Blood 2003; 102:3807 - 3814
- Gafa V, Lande R, Gagliardi MC, Severa M, Giacomini E, Remoli ME, et al. Human dendritic cells following Aspergillus fumigatus infection express the CCR7 receptor and a differential pattern of interleukin-12 (IL-12), IL-23 and IL-27 cytokines, which lead to a Th1 response. Infect Immun 2006; 74:1480 - 1489
- Wen H, Hogaboam CM, Gauldie J, Kunkel SL. Severe sepsis exacerbates cell-mediated immunity in the lung due to an altered dendritic cell cytokine profile. Am J Pathol 2006; 168:1940 - 1950
- Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol 2007; 8:307 - 318
- Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693 - 705
- Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007; 447:972 - 978
- Wen H, Dou Y, Hogaboam CM, Kunkel SL. Epigenetic regulation of dendritic cell-derived interleukin-12 facilitates immunosuppression after a severe innate immune response. Blood 2008; 111:1797 - 1804
- Ayala A, Chung CS, Xu YX, Evans TA, Redmond KM, Chaudry IH. Increased inducible apoptosis in CD4+ T lymphocytes during polymicrobial sepsis is mediated by Fas ligand and not endotoxin. Immunology 1999; 97:45 - 55
- Le Tulzo Y, Pangault C, Gacouin A, Guilloux V, Tribut O, Amiot L, et al. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock 2002; 18:487 - 494
- Unsinger J, Kazama H, McDonough JS, Hotchkiss RS, Ferguson TA. Differential lymphopenia-induced homeostatic proliferation for CD4+ and CD8+ T cells following septic injury. J Leukoc Biol 2009; 85:382 - 390
- Napolitano LM, Campbell C. Polymicrobial sepsis following trauma inhibits interleukin-10 secretion and lymphocyte proliferation. J Trauma 1995; 39:104 - 110
- Roth G, Moser B, Krenn C, Brunner M, Haisjackl M, Almer G, et al. Susceptibility to programmed cell death in T-lymphocytes from septic patients: a mechanism for lymphopenia and Th2 predominance. Biochem Biophys Res Commun 2003; 308:840 - 846
- Ayala A, Deol ZK, Lehman DL, Herdon CD, Chaudry IH. Polymicrobial sepsis but not low-dose endotoxin infusion causes decreased splenocyte IL-2/IFNgamma release while increasing IL-4/IL-10 production. J Surg Res 1994; 56:579 - 585
- Flohe SB, Agrawal H, Schmitz D, Gertz M, Flohe S, Schade FU. Dendritic cells during polymicrobial sepsis rapidly mature but fail to initiate a protective Th1-type immune response. J Leukoc Biol 2006; 79:473 - 481
- Scumpia PO, Delano MJ, Kelly KM, O'Malley KA, Efron PA, McAuliffe PF, et al. Increased natural CD4+CD25+ regulatory T cells and their suppressor activity do not contribute to mortality in murine polymicrobial sepsis. J Immunol 2006; 177:7943 - 7949
- Ansel KM, Lee DU, Rao A. An epigenetic view of helper T cell differentiation. Nat Immunol 2003; 4:616 - 623
- Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat Immunol 2002; 3:643 - 651
- Baguet A, Bix M. Chromatin landscape dynamics of the Il4-Il13 locus during T helper 1 and 2 development. Proc Natl Acad Sci USA 2004; 101:11410 - 11415
- Grogan JL, Mohrs M, Harmon B, Lacy DA, Sedat JW, Locksley RM. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity 2001; 14:205 - 215
- Carson WF IV, Cavassani KA, Ito T, Schaller M, Ishii M, Dou Y, et al. Impaired CD4+ T-cell proliferation and effector function correlates with repressive histone methylation events in a mouse model of severe sepsis. Eur J Immunol 2010; 40:998 - 1010
- Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009; 30:155 - 167
- Monneret G, Debard AL, Venet F, Bohe J, Hequet O, Bienvenu J, et al. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit Care Med 2003; 31:2068 - 2071
- Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol 2003; 3:253 - 257
- Apostolou I, Verginis P, Kretschmer K, Polansky J, Huhn J, von Boehmer H. Peripherally induced Treg: mode, stability and role in specific tolerance. J Clin Immunol 2008; 28:619 - 624
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005; 22:329 - 341
- Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood 2009; 114:3727 - 3735
- Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol 2009; 182:259 - 273
- Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 2007; 5:38
- Venet F, Chung CS, Monneret G, Huang X, Horner B, Garber M, et al. Regulatory T cell populations in sepsis and trauma. J Leukoc Biol 2008; 83:523 - 535
- Venet F, Chung CS, Kherouf H, Geeraert A, Malcus C, Poitevin F, et al. Increased circulating regulatory T cells (CD4(+)CD25 (+)CD127 (-)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med 2009; 35:678 - 686
- Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, et al. Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25− lymphocytes. Crit Care Med 2004; 32:2329 - 2331
- Cavassani KA, Carson Wt, Moreira AP, Wen H, Schaller MA, Ishii M, et al. The post sepsis-induced expansion and enhanced function of regulatory T cells creates an environment to potentiate tumor growth. Blood 2010; 115:4403 - 4411
- O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 2010; 10:111 - 122
- Wang JF, Yu ML, Yu G, Bian JJ, Deng XM, Wan XJ, et al. Serum miR-146a and miR-223 as potential new biomarkers for sepsis. Biochem Biophys Res Commun 2010; 394:184 - 188
- El Gazzar MA, McCall CE. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J Biol Chem 2010; 285:20940 - 20951