Abstract
Chronic lymphocytic leukemia (CLL) exhibits a very variable clinical course. Altered DNA methylation of genes has shown promise as a source of novel prognostic makers in a number of cancers. Here we have studied the potential utility of a panel of methylation markers (CD38, HOXA4 and BTG4) in 118 CLL patients. Each of the three loci assessed exhibited frequent methylation, as determined by COBRA analysis, and individually correlated with either good (CD38, BTG4 methylation) or poor (HOXA4 methylation) prognosis. Using a combined approach to produce an overall methylation score, we found that methylation score was significantly associated with time to first treatment in CLL patients. Multivariate Cox regression analysis revealed that methylation score was the strongest predictor of time to first treatment, and was independent of IGHV gene mutational status and CD38 expression. This study provides proof of principle that a panel of methylation markers can be used for additional risk stratification of CLL patients.
Chronic lymphocytic leukemia (CLL) is the most commonly diagnosed form of leukemia in the UK and exhibits a highly variable clinical course.Citation1 While some patients rapidly succumb to the disease, others can survive for well over ten years, often without the need for treatment. This extreme variability in clinical course among patients presenting with early CLL emphasizes the need for good prognostic markers to help direct patient treatment.Citation1,Citation2 A number of molecular markers are already in frequent use, most notably IGHV mutational status.Citation1 In addition, other markers, such as protein expression of ZAP70 and CD38 and genomic abnormalities are also commonly used.Citation2 However, the group of patients defined as having a good prognosis through the use of current markers is still large (about 60% of CLL patientsCitation3) and these patients still exhibit a wide variety in outcome. Thus, additional markers are needed to aid clinical management in these cases.
Altered DNA methylation, especially at promoter associated CpG islands, is one of the hallmarks of cancer.Citation4 The high frequency of DNA methylation changes, associated with the relative ease and sensitivity with which DNA methylation can be detected, have led to increasing interest in the use of methylation based markers for diagnosis and prognosis.Citation5 A number of methylated genes have already been shown to correlate with clinical features of CLL. For example, TWIST2 has been reported to exhibit frequent hypermethylation in CLL with mutated IGHV genes, which have a good prognosis. It is, however, rarely methylated in unmutated CLL cases, which have a poorer prognosis.Citation6 Methylation of ZAP70 was strongly correlated with lack of ZAP70 expression and, similarly to TWIST2 methylation, was also strongly correlated with mutation of IGHV.Citation7 In contrast, we recently showed that the HOXA4 gene was a frequent target for hypermethylation in CLL but, in this case, methylation of the gene was associated with the poor prognosis IGHV unmutated cases.Citation8 While in many cases the functional role of the altered methylation still remains to be clearly demonstrated, the association between differential methylation and specific subsets of CLL patients raises the possibility that altered gene methylation could be used to identify patient populations with differing clinical outcome.
In this report we have more closely examined the relationship between methylation of specific candidate marker genes and patient outcome, using a panel of genes we have identified as frequently methylated in CLL. In addition to the previously described methylation of the HOXA4Citation8 gene, we have also found frequent methylation, as determined by COBRA analysis, of two further genes (CD38 and BTG4). All three genes independently exhibit a correlation with patient outcome. However, a combined approach using all three markers produced the strongest correlation with patient outcome. This combined marker was independently prognostic in multivariate analysis and identified a subset of patients with IGHV mutated genes (i.e., those regarded as good prognosis) who had a significantly greater likelihood of disease progression.
Results and Discussion
Expression of CD38 is a well established prognostic marker in CLL, with high levels of expression (either >30 or >7% cut-offs are generally used) being associated with poor outcome in CLL patients.Citation2 We analyzed a cohort of 118 patients to determine the potential importance of DNA methylation of the CD38 promoter region. As shown in , methylation of the CD38 promoter region was commonly observed in the CLL samples, with over half of the samples (69/118, ) exhibiting methylation by COBRA analysis. Methylation was not seen in normal peripheral blood samples. Methylation status was then compared to CD38 expression. Regardless of the cut-off used to define expression positive/negative samples, methylation status correlated very strongly with expression. Using a 30% cut-off, 66/77 (86%) of expression negative samples exhibited methylation and 3/40 (8%) of expression positive samples were methylated (p = 3.58 × 10−17, Fisher exact test). Using a 7% cut-off, 54/56 (96%) expression negative samples exhibited methylation, but only 15/61 (25%) of expression positive samples were methylated (p = 6.17 × 10−17, Fisher exact test).
To determine whether CD38 methylation was also able to predict patient outcome, CD38 methylation status was compared to time to first treatment (TFT). TFT was used as an endpoint as it is considered more clinically relevant than overall survival, as a significant number of CLL patients die of non-CLL related causes. As shown in , CD38 methylation status strongly correlated with TFT (p = 0.000008). The ability of CD38 methylation to predict TFT closely mirrored the results obtained using CD38 expression [with either a 30 or a7% cut-off, (p = 0.000001 and p = 0.0002, respectively)]. Furthermore each of these variables also retained their significance for prediction for TFT when the analysis was restricted to just stage A disease (p = 0.003 for methylation, p = 0.012 for expression at 7% and p = 0.001 for expression at 30%).
From our previous studies we have identified two additional genes in which methylation correlated with different subsets of CLL patients. These were the HOXA4 gene, which we have previously shown to be correlated with IGHV unmutated CLL patientsCitation8 and to be associated with poor prognosis patients in AML and CML.Citation10 Secondly, we have found that the BTG4 gene, which maps to the commonly deleted chromosome 11q23 region,Citation11 exhibited a correlation between methylation and the presence of the 11q23 deletion (unpublished results). Unexpectedly, the BTG4 gene was found to be more frequently methylated in patients lacking the 11q23 deletion, as opposed to acting as a second hit in patients with 11q23 deletions ().
The methylation status of these two genes was also assessed in the same set of CLL samples previously examined for CD38 methylation. Methylation of both genes, as determined by COBRA analysis, was found to be frequent, being identified in 38% (HOXA4) and 47% (BTG4) of the CLL patients (examples in ). BTG4 Methylation was absent in all normal peripheral blood samples, while HOXA4 methylation was very low/absent (examples in ). To determine whether methylation of these genes also correlated with outcome, the relationship between methylation and TFT was assessed. This analysis identified borderline correlations between methylation of each of the individual genes and outcome (). For HOXA4, a significant correlation, between methylation and reduced TFT, was seen when the analysis was restricted to stage A disease (), but this fell just short of significance when all patients were included. In contrast BTG4 exhibited a significant correlation, between methylation and increased TFT, when all patients were included (), but fell short of significance when the analysis was restricted to stage A disease. This suggests that both HOXA4 and BTG4 would only be weak predictors of outcome when assessed as single markers, with HOXA4 methylation weakly associated with poorer outcome and BTG4 methylation weakly associated with improved outcome.
The identification of multiple genes that independently correlated with patient outcome raised the possibility of producing a combined methylation marker. Furthermore, the methylation markers identified exhibited differential correlations with TFT, with methylation of CD38 and BTG4 being associated with improved TFT, while methylation of HOXA4 was associated with reduced TFT. This suggests that methylation of the individual markers is related to different aspects of the disease and not simply a reflection of the relative levels of CpG island methylation or the presence of a CpG island methylator phenotype.Citation12
We therefore assessed the utility of combining the three individual methylation markers that we had identified to produce an overall methylation score. As methylation of CD38 was individually a much stronger predictor of outcome, it was given increased weight in the analysis. The algorithm used to stratify patients into high risk and low risk methylation groups is shown in . Comparison of this combined methylation score to TFT in the patient population demonstrated an extremely strong correlation with patient outcome (p = 1.6 × 10−7, ). Combined methylation score was also strongly correlated with TFT when the analysis was restricted to stage A patients (p = 0.00028, ). Furthermore, multivariate Cox regression analysis demonstrated that combined methylation score was independent of other prognostic markers (IGHV gene mutational status, CD38 expression and del11q) in predicting TFT (). While IGHV gene mutational status and del11q were also independently predictive, methylation score was the most significant predictor of TFT in the patient population as a whole (). When only Binet stage A patients were included in the multivariate analysis both methylation score and del11q retained their independence and again methylation score was the most significant predictor of TFT ().
IGHV gene mutational status is an extremely useful tool for identifying patients at high risk of disease progression. However, there is still considerable clinical variability in cases with mutated IGHV genes, which are defined as low risk. To assess the impact of methylation score in this group of patients the analysis was restricted to the 83 IGHV mutated patients. In this group, methylation score was still significantly associated with reduced TFT and significance was retained when the analysis was restricted to just stage A IGHV mutated patients ( and D, respectively). Similarly, methylation score identified patients with low CD38 expression and therefore, regarded as having good prognosis, who were at increased risk of disease progression ( and F). These results demonstrate the potential for methylation markers to identify high risk patients from patient sub-groups regarded as being at low risk for progression.
DNA methylation shows a great deal of promise as a rich source of biomarkers to aid in the management of cancer patients. In this report we have assessed the potential utility of a combination of three methylation based markers for the prediction of outcome in CLL patients. Initially we identified three loci (CD38, HOXA4 and BTG4) as frequent targets for hypermethylation in CLL patients. Each of these three markers was individually associated with TFT in the CLL patients. This included methylation events associated with both increased and decreased TFT, indicating that the identification of multiple markers was not simply due to an association between global methylation levels and patient outcome. Combining the three markers produced a stronger predictor of TFT than any of the three markers used individually.
Importantly, in the multivariate analysis the combined methylation score was independent of other prognostic factors and was the strongest predictor of outcome. Furthermore, restricting the analysis to populations regarded as having a good prognosis (either IGHV mutated or CD38 expression negative) demonstrated that the methylation score was able to identify a subset of these patients who were in fact at high risk of disease progression.
This study suggests that DNA methylation markers could be used to improve risk assessment in CLL patients and may be superior to currently used prognostic markers in this disease. In addition, it is likely that further improvements could be made to the methylation panel used in this study. Firstly, the genes included in the panel were chosen on the basis that we had recently identified them as novel methylated genes in CLL. More genome wide studies, aimed specifically at identifying methylation events associated with outcome, would very likely generate other methylation markers that could be used as well or instead of those included here to improve the potential clinical utility of the methylation panel. In addition, alternative methods for quantitatively assessing the methylation markers, such as pyrosequencing, could also result in further improvement of the approach. We have previously found that pyrosequencing and COBRA analysis produce very similar results at the HOXA4 locus in CLL patients;Citation8 however, this more sensitive technique may be especially valuable for loci such as BTG4 and CD38, where methylation levels are generally lower.
It is important to note that biomarkers need not necessarily be functionally relevant to be clinically useful for predicting the likely course of a patient's disease. Thus, the associations identified in this report do not necessarily imply an important function role for the identified methylation. The methylation seen at the BTG4 locus is nevertheless intriguing, especially in light of the recent report by Toyota et al.Citation13 identifying a role for methylation of this locus in colorectal cancer. This report also identified the BTG4 promoter as a bi-directional promoter that also drives expression of the p53-regulated microRNAs miR-34b/c. Thus the potential role of BTG4 and the microRNAs miR-34b/c in CLL merits further study. Of the three genes used in the panel of markers, methylation of HOXA4 appears to be the one most likely to be playing a functional role in CLL. We previously reported that HOXA4 was a frequent target for hypermethylation in CLL and that it was associated with IGHV gene mutational status.Citation8 In a separate report, we also demonstrated that HOXA4 hypermethylation was common in other types of leukemia and was associated with poor prognosis in patients with AML and CML.Citation10 Indeed, we have recently found that re-introduction of HOXA4 into AML cells resulted in upregulation of known anti-leukaemic genes and the induction of apoptosis (unpublished results). In contrast to the other two genes, the methylation levels at the HOXA4 gene are frequently very high (well above 50%), suggesting a direct role for methylation in gene suppression. In addition, treatment of methylated cell lines with the demethylating agent 2′deoxy-5-azacytidine results in re-expression of the gene.Citation10 Thus, in addition to its potential role as a prognostic marker, it is likely that methylation of the HOXA4 gene plays a functional role in CLL development.
Materials and Methods
For this study, DNA methylation at potential marker loci was analyzed in genomic DNA derived from a cohort of 118 peripheral blood samples from chronic lymphocytic leukemia patients, obtained from the Royal Bournemouth Hospital. Samples were derived from peripheral blood mononuclear cells obtained by Ficoll gradient centrifugation. This resulted in at least 80% (and usually >90%) malignant cells in all samples. The cohort consisted of 98 patients with Binet stage A disease, 14 with stage B and six with stage C. The median age of the patient population at diagnosis was 68 and the sex ratio was 73:45 (M:F). All patients had previously been assessed for Binet stage, IGHV mutational status and CD38 expression. For IGHV mutational status homologies of 98% or above were considered unmutated. For the majority of samples (110), HOXA4 methylation status had also previously been determined.Citation8 Peripheral blood samples (eight in total) were obtained from healthy volunteers. Ethical approval for all samples collected had been obtained.
DNA methylation analysis was performed using the COBRA assay,Citation9 largely as described before.Citation10 200 ng of genomic DNA was modified with sodium bisulfite using the Methylamp™ One-Step DNA Modification Kit (Epigentek) as per the manufacturer's instructions. All samples were resuspended in 15 µl of TE and 1 µl of this was used for subsequent PCR reactions. The samples were amplified in 25 µl volumes containing 1× manufacturer's buffer, 1 unit of Fast Start taq polymerase (Roche), 1.3–4 mM MgCl2, 10 mM dNTPs and 75 ng of each primer. PCR was performed with one cycle of 95°C for 6 min, 35 cycles of 95°C for 30 sec, 58–64°C for 30 sec and 72°C for 30 sec, followed by one cycle of 72°C for 5 min. Following amplification, the PCR products were digested with the appropriate restriction enzymes (New England Biolabs), specific for the methylated sequence after sodium bisulfite modification. For each locus two different restriction enzyme digests were used to increase the number of CpG sites analyzed. Digested PCR products were separated on a 2% agarose gels and visualized by ethidium bromide staining. In vitro methylated (IVM) DNA (Millipore) was diluted into DNA extracted from normal peripheral blood to produce standards (100, 66, 33 and 0%) of known methylation status for all COBRA assays.
Abbreviations
| CLL | = | chronic lymphocytic leukemia |
| TFT | = | time to first treatment |
| IVM | = | in vitro methylated DNA |
| COBRA | = | combined bisulfite and restriction analysis |
| AML | = | acute myeloid leukemia |
| CML | = | chronic myeloid leukemia |
| PBL | = | peripheral blood leukocytes |
| del11q | = | deletion of part/all chromosome 11q |
Figures and Tables
Figure 1 Methylation of the three marker loci in CLL samples. Examples of methylation analysis using COBRA assays for (A) CD38, (B) HOXA4 and (C) BTG4. Above each is a diagram of the locus analyzed indicating the position of the first exon and start of transcription (large arrow), the position of the CpG island (thick black line), the positions of the primers used (small horizontal arrows) and the positions of the restriction enzymes for the digest used in each example (small vertical arrows; H, HinFI and T, TaqI). The positions of bands representing methylated and unmethylated DNA are indicated by arrows and the controls used are indicated above their respective lanes [no DNA (water), 100, 66 and 33% (in vitro methylated DNA), diluted into PBL (peripheral blood leukocytes) DNA as required and PBL]. Under each sample lane an M or U indicates if the sample was considered methylated (M) or unmethylated (U) for the purposes of the subsequent analysis. In (A) samples were either high (>30%) or low (<30%) for CD38 expression. In (C) samples were either positive (11q23 del) or negative (no del) for the 11q23 deletion. Primers used were as previously described for HOXA4,Citation10 Forward 5′-GGT GTT AAG GTT AGT TGT TTT TGA AAG, Reverse 5′-ACT CTC TAA AAA AAC CCA ACT CTA TC for CD38 and Forward 5′-GTT TGG TAT TTT TGG GGG TTA TGG, Reverse 5′-CAA TAC AAT CAA CTA ATA ACA CTA CCT AC for BTG4.
![Figure 1 Methylation of the three marker loci in CLL samples. Examples of methylation analysis using COBRA assays for (A) CD38, (B) HOXA4 and (C) BTG4. Above each is a diagram of the locus analyzed indicating the position of the first exon and start of transcription (large arrow), the position of the CpG island (thick black line), the positions of the primers used (small horizontal arrows) and the positions of the restriction enzymes for the digest used in each example (small vertical arrows; H, HinFI and T, TaqI). The positions of bands representing methylated and unmethylated DNA are indicated by arrows and the controls used are indicated above their respective lanes [no DNA (water), 100, 66 and 33% (in vitro methylated DNA), diluted into PBL (peripheral blood leukocytes) DNA as required and PBL]. Under each sample lane an M or U indicates if the sample was considered methylated (M) or unmethylated (U) for the purposes of the subsequent analysis. In (A) samples were either high (>30%) or low (<30%) for CD38 expression. In (C) samples were either positive (11q23 del) or negative (no del) for the 11q23 deletion. Primers used were as previously described for HOXA4,Citation10 Forward 5′-GGT GTT AAG GTT AGT TGT TTT TGA AAG, Reverse 5′-ACT CTC TAA AAA AAC CCA ACT CTA TC for CD38 and Forward 5′-GTT TGG TAT TTT TGG GGG TTA TGG, Reverse 5′-CAA TAC AAT CAA CTA ATA ACA CTA CCT AC for BTG4.](/cms/asset/53fe72d3-aff2-4c92-ac2e-99d47cc4e35d/kepi_a_10914038_f0001.gif)
Figure 2 All three markers individually exhibit correlations with patient outcome. Kaplan-Meier graphs are shown to assess the relationship between methylation of (A) CD38, (B) HOXA4 and (C) BTG4 and TFT in CLL patients. In (A and C) all patients are included; however, in (B) only stage A patients are included. Methylation of CD38 and BTG4 were significantly correlated with longer TFT, whereas methylation of HOXA4 was associated with reduced TFT. Kaplan-Meier graphs were generated using the SPSS statistical software package (version 17) and p values were derived using the log rank test.
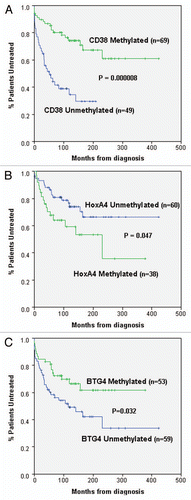
Figure 3 Use of methylation score identifies a subset of patients with reduced TFT from good prognostic groups. (A and B) Kaplan-Meier graphs showing the correlation between methylation score and TFT in all patients or Binet stage A only CLL patients, respectively. The predictive value of the combined score is greater than any of the single markers. (C and D) Kaplan-Meier graphs showing the correlation between methylation score and IGHV gene mutational status and TFT in all patients or stage A only CLL patients, respectively. Patients were stratified into four groups depending on both methylation score and IGHV gene mutational status. However only three groups are displayed as the fourth group [IGHV unmutated and methylation score low risk contained very few patients (3)]. (E and F) Kaplan-Meier graph showing the correlation between methylation score and CD38 expression and TFT in all patients or stage A only CLL patients, respectively. Patients were stratified into four groups depending on both methylation score and CD38 expression. However, again only three groups are displayed as the fourth group [CD38 expression positive and methylation score low risk contained very few patients (2)].
![Figure 3 Use of methylation score identifies a subset of patients with reduced TFT from good prognostic groups. (A and B) Kaplan-Meier graphs showing the correlation between methylation score and TFT in all patients or Binet stage A only CLL patients, respectively. The predictive value of the combined score is greater than any of the single markers. (C and D) Kaplan-Meier graphs showing the correlation between methylation score and IGHV gene mutational status and TFT in all patients or stage A only CLL patients, respectively. Patients were stratified into four groups depending on both methylation score and IGHV gene mutational status. However only three groups are displayed as the fourth group [IGHV unmutated and methylation score low risk contained very few patients (3)]. (E and F) Kaplan-Meier graph showing the correlation between methylation score and CD38 expression and TFT in all patients or stage A only CLL patients, respectively. Patients were stratified into four groups depending on both methylation score and CD38 expression. However, again only three groups are displayed as the fourth group [CD38 expression positive and methylation score low risk contained very few patients (2)].](/cms/asset/47d0fe32-0747-49ad-bd0d-ff6cce45f595/kepi_a_10914038_f0003.gif)
Figure 4 Algorithm used to define high and low risk based on methylation of the three marker genes. Information from the three marker genes was combined using an algorithm as shown. This was designed to give greater weight to CD38 methylation status as this correlated most strongly with outcome when used in isolation. Patients were defined as methylation low risk if their samples exhibited methylation at CD38 and the low risk variant at least one of the other two genes (i.e., either HOXA4 unmethylated and/or BTG4 methylated). Patients lacking CD38 methylation or with CD38 methylated but high risk variants at both of the other two genes (i.e., HOXA4 methylated and BTG4 unmethylated) were defined as methylation high risk.
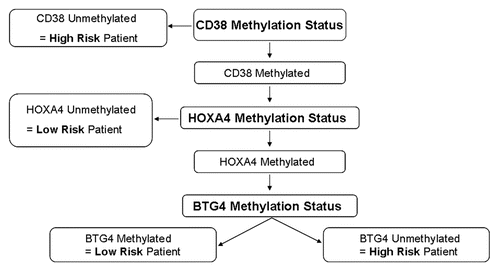
Table 1 Methylation frequencies in CLL samples
Table 2 Multivariate Cox regression analysis
Acknowledgements
The authors would like to thank all UK hematologists who have contributed to the collections of samples used for the analysis.
References
- Montillo M, Hamblin T, Hallek M, Montserrat E, Morra E. Chronic lymphocytic leukemia: Novel prognostic factors and their relevance for risk-adapted therapeutic strategies. Haematologica 2005; 90:391 - 399
- Moreno C, Montserrat E. New prognostic markers in chronic lymphocytic leukemia. Blood Rev 2008; 22:211 - 219
- Oscier DG, Gardiner AC, Mould SJ, Glide S, Davis ZA, Ibbotson RE, et al. Multivariate analysis of prognostic factors in CLL: Clinical stage, IGVH gene mutational status, and loss or mutation of the p53 gene are independent prognostic factors. Blood 2002; 100:1177 - 1184
- Costello JF, Plass C. Methylation matters. J Med Genet 2001; 38:285 - 303
- Miyamoto K, Ushijima T. Diagnostic and therapeutic applications of epigenetics. Japanese J Clin Oncol 2005; 35:293 - 301
- Raval A, Lucas DM, Matkovic JJ, Bennett KL, Liyanarachchi S, Young DC, et al. TWIST2 demonstrates differential methylation in immunoglobulin variable heavy chain mutated and unmutated chronic lymphocytic leukemia. J Clin Oncol 2005; 23:3877 - 3885
- Corcoran M, Parker A, Orchard J, Davis Z, Wirtz M, Schmitz OJ, et al. ZAP-70 methylation status is associated with ZAP-70 expression status in chronic lymphocytic leukemia. Haematologica 2005; 90:1078 - 1088
- Strathdee G, Sim A, Parker A, Oscier D, Brown R. Promoter hypermethylation silences expression of the HoxA4 gene and correlates with IgVh mutational status in CLL. Leukemia 2006; 20:1326 - 1329
- Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nuc Acid Res 1997; 25:2532 - 2534
- Strathdee G, Holyoake TL, Sim A, Parker A, Oscier DG, Melo JV, et al. Inactivation of HOXA genes by hypermethylation in myeloid and lymphoid malignancy is frequent and associated with poor prognosis. Clin Cancer Res 2007; 13:5048 - 5055
- Auer RL, Starczynski J, McElwaine S, Bertoni F, Newland AC, Fegan CD, et al. Identification of a potential role for POU2AF1 and BTG4 in the deletion of 11q23 in chronic lymphocytic leukemia. Gene Chrom Cancer 2005; 43:1 - 10
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999; 96:8681 - 8686
- Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, et al. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 2008; 68:4123 - 4132