Abstract
Current data suggest that angiogenesis, smooth muscle cell migration, differentiation and proliferation may be epigenetically regulated. Prokaryotic DNA methyltransferases have been proposed as tools to modify mammalian DNA methylation. In order to assess the impact of DNA hypermethylation on smooth muscle pathophysiology, we expressed an HpaII site-specific methyltransferase transgene in smooth muscle cells in mice. The enzyme is expected to target only a subset (CCGG) of unmethylated CpG dinucleotides, thus avoiding possible deleterious effects of widespread hypermethylation. Transgenics of two independent lines were born at expected frequencies, showed no obvious abnormalities and were fertile. Nevertheless, ~30% of >1 year-old transgenics developed organomegaly and ~20% showed a range of tumors. Global DNA methylation was unchanged in transgenic tissue whether hyperplastic or normal, but tumor DNA showed a pronounced global hypermethylation. DNA hypermethylation was not indiscriminate, as five tested tumor suppressor genes showed promoter CpG and non-CpG hypermethylation and transcriptional down-regulation, whereas the methylation status of one intergenic CpG islands, repeated elements (n=2) and non-tumor suppressor gene promoters (n=3) was unchanged. Our work is the first report on the effects of HpaII methyltransferase on endogenous chromatin and in a whole animal. Furthermore, our data expand previous findings that imply that global DNA hypomethylation is not an obligate oncogenic pathway at least in the tumor types examined here.
Introduction
It has been long recognized that abnormal smooth muscle cell (SMC) migration, differentiation and proliferation are landmarks of various pathophysiological conditions including atherosclerosis, vascular aging and cancer.Citation1–Citation3 The latter diseases are associated with aberrant DNA methylation patterns.Citation4,Citation5 Epigenetic regulation of gene expression in SMC is capturing a significant interest, not only as a clue to novel molecular mechanisms of diseases, but also as a conceptual tool to explain how environmental and dietary factors affect disease predisposition.Citation6 A crucial issue in the field is whether DNA methylation pattern rearrangement is a consequence of other cellular abnormalities or whether it represents a primary, causal molecular event. For example, it is unclear whether the global DNA hypomethylation observed in atherosclerosis and cancer is secondary to increased cell proliferation or whether it can be actively imposed by an altered extracellular environment.Citation7,Citation8 Furthermore, DNA hypermethylation may well be a valuable cancer type- and stage marker, but genome-wide studies have shown a weak correlation between promoter methylation status and transcription (reviewed in ref. Citation9).
One approach to investigate these issues is to genetically impose de novo DNA methylation by overexpressing DNA methyltransferase (DNMT) enzymes in vivo. Relatively little data is available on overexpression of mammalian DNMTs. One study showed that exposure of the normally unmethylated Drosophila genome to the main mammalian DNMTs DNMT1 and DNMT3a resulted in embryonic lethality.Citation10 Recently, DNMT overexpression in mouse male germ line stem cells has been shown to cause defective spermatogenesis.Citation11 Taken together, these data support the predictable idea that endogenous DNMT overexpression causes widespread genome hypermethylation and impairment of cellular functions. One alternative strategy that has been followed is the expression of site-specific prokaryotic DNMTs. In principle, these enzymes target subsets of all methylable CpG dinucleotides, thus may selectively modify specific classes of regulatory sequences without imposing a widespread DNA hypermethylation. In particular, extensive work has been carried out on H. parainfluenzae HpaII methyltransferase (HpaIIM), a DNMT specific for the internal cytosine of the HpaII endonuclease restriction site 5′-CCGG-3′.Citation12 Efforts to reduce HpaIIM off-target activity and improve its specificity for promoter sequences, lead to the synthesis of zinc finger-HpaIIM fusions (FH).Citation13 These have been shown to hypermethylate zinc finger binding site- and HpaII site-containing reporter gene promoters in exogenous integrated constructs in cultured cells with high specificity.Citation14 Similar results have been obtained using zinc finger fusions of the mammalian DNMTs DNMT3a and DNMT3b in an extra-chromosomal reporter plasmid system.Citation15 Importantly, transient FH expression resulted in reporter gene silencing and permanent, inheritable promoter hypermethylation.Citation14 These important advances promise to pave the way towards obtaining completely gene- or gene family-specific DNMTs and provide insights into whether DNA methylation is a genuine upstream event in gene silencing. Nevertheless, one limitation of those approaches is the use of extra-chromosomal or exogenous integrated reporter constructs that do not necessarily mirror the complex regulation of endogenous promoter sequences. In addition, no information is available on the effects of HpaIIM expression in a whole animal. To address these latter issues and to explore the effects of artificially imposed de novo DNA methylation in SMC, we created transgenic mice expressing an HpaIIM transgene controlled by the mouse SMC ± actin promoter. We present the phenotype of such mutant mice and discuss our data in the context of current efforts to exogenously modify cellular DNA methylation patterns.
Result
Creation of Acta2HpaIIM transgenic mice.
We constructed the Acta2HpaIIM transgene, which included the coding sequence of HpaIIM fused to a C-terminal SV40 NLS to achieve nuclear targeting. Although we did not rigorously assay the biological activity of HpaIIM-NLS fusion in comparison with the unfused control, we observed a relative resistance to HpaII digestion in slow-growing E. coli transformed with one of the plasmids (pHSP0-1HpaIIMNLS) constructed during transgene assembling, presumably due to leaky expression (Sup. Fig. 1). We interpret this result as evidence that HpaIIM-NLS retains at least part of WT enzyme activity. The HpaIIM-NLS fusion was placed 5′ to the mouse Acta2 promoter, and the two elements were spaced by a rabbit β globin intron. A schematic Acta2HpaIIM transgene map is shown in . Pronuclear injections yielded a total of six transgenic mice, four of which transmitted the transgene to progeny. Transgene expression analysis by a semi-quantitative RT-PCR in the large intestine identified two highest-expressing lines. Heterozygous transgenics of these lines—referred to as S13 and Z—will be described in the present study.
Organ growth phenotype of Acta2HpaIIM mice.
Mice of both lines were born at expected frequency, showed normal body weight and were macroscopically normal. An assessment of transgene expression and phenotype was performed in 2-month- and 1-year-old mice. Acta2HpaIIM was expressed in a variety of tissues in both lines, but not in peripheral blood cells, as expected from a vascular- and nonvascular SMC-specific promoter (), thus reproducing the expression pattern observed in transgenics bearing the same promoter and previously created by our group.Citation22 We inspected body weight, heart, lung and abdominal organ wet weight in transgenics and age- and litter-matched WT controls. A tendency to increased wet weight, although not statistically significant, was revealed in 2-month-old mice (n = 25 in each group; data not shown). To assess whether Acta2HpaIIM expression results in de novo DNA hypermethylation, we determined total 5mdC in the stomach, large intestine, colon, aorta and vesicular gland. Nonetheless, no significant difference in global DNA methylation was detected between transgenics and matched WT controls in either line at the age of 2 months (not shown).
By contrast, 1-year-old transgenics showed a number of abnormalities. The wet weight of the vesicular gland, large intestine and small intestine was significantly increased in both S13 and Z lines compared to WT controls, whereas all other organs remained unchanged (n = 50 in each group; ). In all cases, organomegaly was limited to either intestines or vesicular gland. Data point distribution showed a sub-group of transgenics with normal organ weight and another one with organomegaly (). Using a threshold value of OWav + 2SD (where OWav is the average organ weight), the latter sub-group amounted to ∼28–30% of transgenics depending on the organ and line considered. A portion (18% and 15% in the S13 and Z line, respectively) of transgenics with increased large intestine wet weight but none of WT controls examined, displayed rectal prolapse. An example of WT and transgenic hyperplastic vesicular gland is shown in and C. One curious observation was an asymmetric vesicular gland with a normal weight right horn and a ∼8-fold heavier hyperplastic left one (48.5 g and 418.0 g, respectively) in one Z line transgenic. Alimentary canal functionality was not grossly perturbed in transgenics with hyperplastic intestines. Similarly, a subset of transgenic males (n = 4, Z line) with hyperplastic vesicular glands that have been mated were reproductively normal. Transgene expression was detectable in both normal size and hyperplastic transgenic tissue in n = 10 mice examined for each group (). No significant difference in body weight was observed. Transgenics reached this age at the expected frequency.
Tumors in adult Acta2HpaIIM transgenic mice.
In addition, the sporadic detection of obvious subcutaneous tumors in transgenics prompted us to monitor animals (n = 40 in each transgenic line and controls) from the age of 1 year onwards. A proportion of transgenics developed spontaneous tumors. These were heterogeneous in type and were present in 25 and 20% of mice of the S13 and Z line, respectively (). Tumor presence was independent of transgene expression, since Acta2HpaIIM transcript was detectable in all tumor and adjacent tissue samples examined (). None of these tumor-bearing transgenic mice showed any concomitant organomegaly. Examples of histology of an intestinal carcinoma, a hemoangioma and a lymphoma are shown in –D.
Global DNA methylation in transgenic hyperplastic tissue and tumors.
Determination of global 5mdC in 1-year-old WT control, transgenic normal or hyperplastic tissue did not reveal any significant differences (). By contrast, a corresponding analysis of WT control, transgenic adjacent apparently normal tissue and tumor mass revealed a pronounced global hypermethylation of tumor DNA in four different tumor types (). Although we expected de novo DNA methylation as a result of HpaIIM expression, the marked global hypermethylation observed in transgenic tumors was slightly surprising given that global DNA hypomethylation is a landmark of many tumor types (reviewed in ref. Citation23). In particular, repeated elements such as LINE-1 retrotransposons are hypermethylated in normal cells but significantly contribute to global DNA hypomethylation in tumors.Citation24 Another example of normally hypermethylated repeated elements are MusD, which are transposition-autonomous members of the EtnII transposon family.Citation25,Citation26 To corroborate global DNA methylation data, we conducted a survey of the methylation status of LINE-1 and MusD by quantitative methylation-sensitive PCR in pooled intestinal carcinoma and hemoangioma samples. We chose to perform all further molecular studies in these two tumor types as they displayed the most extreme degrees of global DNA hypermethylation (). The data revealed that the bulk of tumor LINE-1 elements were hypermethylated as in WT control tissue and apparently normal transgenic tissue adjacent to tumors, in accordance with the absence of global DNA hypomethylation in the latter ( and C). Furthermore, LINE-1 elements showed tumor type-specific non-CpG hypermethylation involving the external cytosine of the CCGG HpaII site, as showed by resistance to MspI cleavage in intestinal carcinoma but not in hemoangioma ( and C). Interestingly, this pattern of relative insensitivity to MspI digestion was observed also for MusD ( and E). Furthermore, we observed a significant hypermethylation of MusD in intestinal carcinoma samples compared to WT control. 2−ΔCP values of HpaII-digested intestinal carcinoma samples and WT controls were 0.34 ± 0.32 and 0.022 ± 0.002, respectively (p < 0.04). Adjacent tissue samples had intermediate values (0.11 ± 0.09) and were not statistically different from tumor or WT samples. The same tendency was observed in hemoangioma samples, although differences were not significant.
Gene promoter hypermethylation in transgenic tumors.
Another epigenetic landmark of tumors is hypermethylation of selected gene promoters. To assess the extent of promoter hypermethylation in transgenic tumors, we analyzed the corresponding regions of Apc, Cdh1, Cdkn2a, Mgmt and Mlh1, five tumor suppressor genes commonly hypermethylated in a variety of tumors.Citation27–Citation29 Promoter hypermethylation at HpaII- and non-HpaII CpG dinucleotides, and at the external cytosine of HpaII sites (resulting in mCCGG) were observed for all five tumor suppressor genes in the individual intestinal carcinoma and hemoangioma shown in , but not in WT control or transgenic adjacent tissue samples (). Promoter hypermethylation coincided with significant transcriptional downregulation of all five genes. Exceptions were Apc and Mgmt in the hemoangioma sample, where both genes remained largely unmethylated and expression unchanged (). The detection of mCCGG was unlikely to be due to incomplete bisulfite modification of DNA for the following two reasons. First, an HpaII site located 995 bp 3′ to Cdkn2a and surrounding sequences were found to be unmethylated in the tumors examined (). Second, simultaneous bisulfite treatment of human THP-1 cell and transgenic mouse intestinal carcinoma DNA revealed complete demethylation at a 206 bp fragment of a human MT2A gene CpG island that we had found consistently unmethylated in THP-1 macrophages in a concomitant independent study (data not shown) (). In order to assess whether DNA hypermethylation by HpaIIM was gene-specific or indiscriminate, we analyzed the methylation status of Cdh3, Lcn2 and Msln, three genes commonly found hypomethylated in tumors.Citation30,Citation31 These three genes were found variably methylated in WT tissue and their methylation status did not change in any tumor or adjacent tissue sample analyzed (). Expression analysis in intestinal carcinoma and control sample did not reveal any significant change for these three genes ().
Discussion
HpaIIM expression resulted in detectable phenotypic changes in a subset of animals and organs in the transgenic model presented here. One interpretation of this result is that functional access to chromatin by HpaIIM is generally constrained and takes place in a minority of expressing cells. The idea that access to chromatin by HpaIIM is intrinsically inefficient is consistent with previous observations that de novo DNA methylation by the enzyme does not exceed 40–50% of methylable residues even when interaction with chromatin is facilitated by fused zinc finger peptides.Citation13,Citation14 We speculate that the enzyme accesses chromatin during organ development, thus resulting in heritable DNA hypermethylation at a small number of loci including critical growth-controlling genes and organomegaly. The paucity of de novo methylated loci in hyperplastic tissue would explain why DNA hypermethylation was not detected by global 5mdC determination. Depending on the time point at which HpaIIM imposes the initial de novo DNA methylation event during organ development, the growth of part or the whole organ is affected, as shown by the a highly asymmetric vesicular gland detected in one transgenic. This hypothesis is consistent with previous data showing that HpaIIM can impose heritable transcriptional repression and DNA methylation patterns in a transient transfection assay.Citation14 Because of the SMC specificity of the promoter controlling the transgene, it is conceivable that organomegaly is driven by an abnormally high angiogenesis. The reason for the marked susceptibility of the intestine and the vesicular gland to organomegaly is unknown at this point. Noticeably, we could not detect any growth deficit in any transgenic examined, implying that HpaIIM targeted preferentially genes with a growth restriction function. This result could not be due to limited viability of a sub-population of transgenics with organ growth deficit or dwarfism that would have gone undetected, since transgenics reached the age of 1 year at the expected frequency. Furthermore, it is known that aberrant de novo DNA methylation often targets negative controllers of growth such as members of TGF β signaling.Citation32 In the case of tumors, DNA methylation by HpaIIM may act as a “second hit” favoring the expansion of sporadic, spontaneous lesions that are normally erased by the immune system. As for the origin of Acta2HpaIIM tumors, these were not typical SMC-derived lesions with the exception of hemoangiomas. It is therefore possible that ectopic expression of the Acta2 promoter caused by primary transcriptional deregulation events in sporadic pre-tumoral cells, leads to DNA hypermethylation acting as an additional hit for tumor progression. The observation that SMC actin is ectopically expressed in tumors supports this hypothesis.Citation33 Finally, our data suggest that HpaIIM targets an only partially overlapping gene set in tumors and hyperplastic tissue, as the latter was never coincident with the former.
One major concern in using HpaIIM to direct DNA methylation has been its relatively high off-target activity.Citation13,Citation14,Citation34 Our results expand the knowledge on DNA methylation patterns induced by HpaIIM expression, as we show that transgenic tumor DNA displays hypermethylation not only at the expected HpaIIM target, i.e., the internal cytosine in the CCGG sequence, but also at the external cytosine in CCGG (resulting in mCCGG) and non-CCGG CpG dinucleotides. Furthermore, the presence of a HpaII site was not required for de novo DNA methylation in HpaIIM transgenes, as hypermethylation was observed of HpaII site-containing (Cdh1, Cdkn2a, Mgmt) as well as HpaII site-devoid sequences (Mlh1). It is conceivable that HpaII sites in cis outside the sequences that were analyzed here dictate HpaIIM targeting, yet the HpaII site and flanking sequences of the Apc promoter were weakly hypermethylated in intestinal carcinoma samples. Taken together, our data suggest that mechanisms of DNA targeting by HpaIIM are complex. Whether these events are induced solely by HpaIIM or in concert with endogenous DNMTs cannot be concluded based on the present data, nevertheless the observed phenotype is clearly a downstream effect of transgene expression. Although in vitro experiments convincingly show that HpaIIM activity is highly specific in naked DNA, our results open the possibility that the enzyme behaves in a radically different fashion in endogenous chromatin. Accordingly, previous data do show that HpaIIM expression results in low but detectable non-CCGG CpG dinucleotide methylation.Citation14 De novo formation of mCCGG has not been addressed in the latter study and is therefore a novel finding. Future research will have to determine whether this form of non-CpG methylation is a serious issue that may undermine HpaIIM as a feasible methylome modifier in vivo. Methylation events at the cytosine of CNG trinucleotides are abundant in plants but are considered to be much less frequent than CpG methylation in mammals, although recent observations indicate that it is an epigenetic trait of human stem cells.Citation35 Interestingly, a study addressing the consequences of EcoRII methylase-induced CWG methylation showed that this modification does not detectably affect transcription or methylation of adjacent CpGs.Citation36 Provided that this result can be generalized, it may suggest that the functional consequences of mCCGG induced by HpaIIM expression are modest.
In addition to off-target activity, our study allows to assess gene-specific effects of HpaIIM expression. De novo methylation events in transgene samples are not indiscriminate, as they include substantial de novo methylation of at least four gene promoters and maintenance of retrotransposon methylation, but spare the four normally unmethylated sequences that were analyzed, i.e., three non-tumor suppressor gene promoters and one intergenic CpG island located 3′ to the Cdkn2a gene. Although a genome-wide analysis is needed to draw firm conclusions, the data suggest that HpaIIM expression may result in gene-specific de novo methylation. We propose that HpaIIM reinforces pre-determined blueprints of tumor-specific gene methylation patterns, rather determining novel ones. By contrast, our data suggest that the effects of HpaIIM expression on repeated elements may be widespread, as LINE-1 hypermethylation was retained and MusD elements were actually hypermethylated in selected tumor samples.
The observation that at least four different transgenic tumor types relevant for humans do not show any degree of global DNA hypomethylation was unexpected. It is widely accepted that most tumor types show a dual DNA methylation aberration. On the one hand, the genome undergoes a global DNA hypomethylation, whereas a subset of promoters becomes heavily hypermethylated (recently reviewed in ref. Citation37). The former is held responsible for genomic instability, transposon reactivation and oncogene upregulation, whereas the latter is thought to result in tumor suppressor gene silencing.Citation23,Citation37 Discerning the relative importance and functional relevance of those two events may hold important implications for cancer therapy, yet the issue remains unresolved. Perhaps the most convincing demonstration for a causal role of global DNA hypomethylation is the appearance of tumors in DNMT-null mutant mice.Citation38 This and other studies led to the hypothesis that global DNA hypomethylation has a broad pathogenic role extending to atherosclerosis and aging.Citation37,Citation39 On the other hand, DNA hypermethylation has been shown to precede hypomethylation in liver and prostate cancer, thus advocating a prominent role for the former epigenetic defect in tumor initiation.Citation40,Citation41 In at least thyroid cancer, selected promoter DNA hypermethylation occurs in the absence of widespread hypomethylation.Citation42 Furthermore, a significant global DNA hypermethylation was observed in hepatic carcinomas of mice that have genetically increase S-adenosylmethionine, the universal donor of methyl groups.Citation43 Our data expand the latter observations, by indicating that global DNA hypomethylation, whether or not causally relevant per se, is dispensable for the formation of a range of different tumors. Based on current literature and the present data, it is conceivable that any slight departure from the “normal” cell's DNA methylome, whether in the direction of global hypo- or hypermethylation, potentially leads to cellular transformation.
In summary, this work reports novel observations on the molecular and pathophysiological effects of HpaIIM in endogenous chromatin and in a whole animal. Growth abnormalities and tumors were noticeable traits of HpaIIM-expressing transgenic mice. This information should be useful to direct future efforts to manipulate cellular DNA methylation with exogenous DNMTs.
Materials and Methods
Transgene construction.
The SMC α actin promoter-HpaIIM transgene (Acta2HpaIIM) was constructed as follows. First, a C-terminal SV40 nuclear localization sequence (NLS)Citation16 was introduced into the H. parainfluenzae HpaII methyltransferase (HpaIIM, accession number X51322) coding sequence. The plasmid pHSP0-1HpaIIM containing the HpaIIM coding sequence was a generous gift from R.J. Roberts, New England Biolabs. The 3′ 561 bp portion of HpaIIM between the unique BclI and HindIII sites was mutated by PCR using the primers: 5′-GAT TGT AGT TGG AGG TAT GGG ACG TGA ACG-3′ (forward), ending 12 bp 5′ to the BclI site; and 5′-TCA GCC AAT AAC TTA AAG CTT GCG TAA GGC TCA CTC GAT ATC TCA AAC CTT CCT CTT CTT CTT AGG GTC ATA TAA ATT TCC TAA TTT TTC TAA AAT TTT CTT ACC TGT AGC TTG AAT AGC CGG-3′ (reverse), containing a SV40 NLS (underlined italics), a stop codon (italics), an EcoRV site (bold) and a HindIII site (underlined). The PCR product was digested with BclI and HindIII and inserted into the 3.36 kb pHSP0-1HpaIIM fragment obtained with the same two endonucleases to create the pHSP0-1HpaIIMNLS plasmid. Second, an SV40 polyadenylation cassette was introduced 3′ to HpaIIMNLS by ligating a 1,127 bp EcoRI-EcoRV fragment including its entire coding sequence into pcDNAI/Amp (Invitrogen) linearized with the same two enzymes, to obtain pcDNAHpaIIMNLS. Third, a triple ligation was performed between: (1) a 1.7 kb HindIII—BamHI fragment of pBSKCD11b, containing the human CD11b promoter (a kind gift from S. Jovinge, University of Lund, Sweden); (2) a 501 bp BamHI—EcoRI pBSKbovK5, containing a rabbit β globin intron (a kind gift from T. van Overeem Hansen, Rigshospitalet, Denmark); (3) pcDNAHpaIIMNLS linearized with HindIII and EcoRI, to obtain pCD11bHpaIIMTaseNLS. Finally, a 5.9 kb SpeI-BamHI fragment of pCD11bHpaIIM-TaseNLS including the whole CD11b promoter was replaced with the Acta2 promoter by insertion of a 3.6 kb SpeI-BamHI fragment of pSMP8 (a kind gift from J.A. Fagin, University of Cincinnati, OH, and A.R. Strauch, The Ohio State University, Ohio, described in Wang et al.Citation17). A SpeI site had been previously inserted into pSMP8 5′ to the Acta2 promoter as described in reference Citation18. All cloning steps were verified by sequencing. The 6.4 kb SpeI-KpnI transgene fragments was gel-purified and used in pronuclear injections.
Animal work.
Pronuclear injections were performed at the mouse genetic modification facility, University of Lund, Lund, Sweden. All animal work conducted in Sweden (2005–2006) was approved by the Ethical Committee of the University of Lund. All subsequent work conducted in Mexico (2007–2009) involved only animal culling with no live procedures and therefore did not need any ethical committee approval. Mice were genotyped by PCR (annealing at 50°C) with the primers 5′-CCC TCT GCT AAC CAT GTT CAT GCC-3′ and 5′-TTC AAA TAA ACT ATA TTG TGC AGC GGG-3′ using DNA from either tail tips or a single drop of blood obtained with a minimal incision of the tail. All samples were obtained under anesthesia. Transgene presence resulted in a single 216 bp band. Transgenic mice were maintained in the heterozygous state and were backcrossed into a C57BL/6 background for at least ten generations. For tissue wet weight measurement, fresh samples were rinsed in PBS, blotted with paper and weighed immediately. Intestine samples were cut into 2–3 cm fragments and the content was eliminated by gentle pressure with a spatula in PBS. Tumor and tissue samples were stored in RNAlater (Ambion) for molecular analysis. When appropriate a fragment was immediately fixed in formalin for histology.
Gene expression.
For transgene expression analysis, 1 µg total RNA extracted with the RNeasy system (Qiagen) from frozen ground tissue was reverse transcribed with a poly-T oligonucleotide. cDNA was amplified with the primers 5′-CAG AAC CCT GAG ACG CTG GAT CC-3′ (forward, spanning exon 2 of Acta2 promoter) and 5′-TTC AAA TAA ACT ATA TTG TGC AGC GGG-3′ (the same reverse primer used for genotyping, located in HpaIIM coding sequence) by conventional RT-PCR. Primers were located on either side of the rabbit β globin intron and were deduced by manual examination of transgene sequence. The spliced transgene RNA yielded a 190 bp band. Gapdh cDNA was co-amplified with commercial primers (R&D Systems, cat. no. RDP-106) as internal control. Cdh1, Cdkn2a and Mlh1 expression analysis was performed by quantitative RT-PCR. Briefly, one-tenth of cDNA obtained from 1 µg total RNA was amplified with the primers (5′ to 3′ sequences): GTG TTG CTT TCT GCC ACT CC and GCC AGC TTT TAC AGT CCC AG (Apc), CCA CTT TGA ATC GGG AGT CT and GAG GTC TAC ACC TTC CCG GT (Cdh1), AAA TCT TGG TGC CTC TGT CC and CCG CAT CTT AAG GAG ACG AA (Cdh3), TCG AAT CTG CAC CGT AGT TG and CGT GAA CAT GTT GTT GAG GC (Cdkn2a), ATT TCC CAG AGT GAA CTG GC and AAT GTC ACC TCC ATC CTG GT (Lcn2), AGA GGC TCT GGA ACT CCC TC and AGA TGG AGC TGT CTG GCT GT (Mgmt), TAG TGG ATG AGG AAG CCA CC and AGC ACA TTG AGA GCA AGC TG (Mlh1), CAG GTC CAG TGG AAG AGC AT and GGC TGG CTA TGG CTG TAA GA (Msln), by using the LightCycler Fast Start DNA MasterPLUS SYBR green I system (Roche) according to manufacturer's instructions in a LightCycler 1.5 (Roche). Primer sequences were obtained with qPrimerDepot (mouseprimerdepot.nci.nih.gov). Relative expression levels were calculated by subtracting the average Gapdh CP from the CP of the target gene obtained from the same cDNA and applying the formula 2−ΔCP.Citation19 CPs were calculated by the LightCycler 1.5 software. All samples were in triplicate.
DNA methylation.
DNA was extracted from RNAlater-stored tissue, paraffin embedded specimens or THP-1 cells (see below) with DNeasy (Qiagen). For global genome DNA methylation analysis, 5-methyldeoxycytidine was determined by HPLC as described in reference Citation20. Long interspersed nuclear element-1 (LINE-1) and MusD methylation status was assessed by quantitative methylation-sensitive PCR (QMS-PCR). Briefly, 10 ng DNA were digested with 1 u HpaII or MspI (New England Biolabs) or buffer alone (mock) for 3 h. Samples were amplified with the primers (5′ to 3′ sequences) AGA CTG CGG TAC ATA GGG AA and GGG TCT AGA TTT TCG GTT GTG (LINE-1), GCT TTA AGT GTT GGA GGT TT and AGC TGT CTT CTT TGA GTA AC (MusD) using LightCycler Fast Start DNA MasterPLUS SYBR green I (Roche) according to manufacturer's instructions in a Light Cycler 1.5 machine (Roche). For relative quantification, the average mock sample CP was subtracted from the CP of enzyme-treated triplicate samples and the formula 2−ΔCP was used. For analysis of bisulfite-treated DNA, 1 µg DNA was modified with the EZ DNA Methylation-Gold™ system (Zymo Research) according to manufacturer's instructions. One-fourth of modified DNA was amplified with the following primers (5′ to 3′ sequences): TTT TYG GAG TAA AGA TTA GAA GAG and CCA TCT TRT TRR ARR CRR RAA RC (Apc), GAA AAT AAA AAT ATA AGA AAT AAA AYG GAA and CCT ACA AAT AAC AAC CAA AAA AC (Cdh1), TTT GTT TTT TTG TYG GGT TTT AG and ACT CTT CTA AAA CTA CTT CT (Cdh3), GTT YGT TTT TTA GTA GTT AGT G and CCT ATT TTC TTC TTT TTA TAA AT (Cdkn2a), AGT GGA TAG GTA GTT TAG ATT T and CAA AAA ATA AAA AAA ACT CCC C (Lcn2), AGA AGA GGT TTG TTT TAG GAA and CAA AAT ACA ATT AAT TAC TAA AC (Mgmt), GTT TAA GGT TTT TTT TTA GTT TYG G and RTA TTC CCT CAA CTC TCA AA (Mlh1), AAA GGG ATT GAT TTA TGG AA and TAA AAA AAC CAA CCC TCC TAT AA (Msln), TTT TTA TTT TYG GAG GTA GAA GTG and ATC TTA CCT AAA CTA AAT TC (for an intergenic CpG island located 995 bp 3′ to Cdkn2a in chr4:88919313-88919637, mouse genome build 37.1). As unmethylated DNA control, we employed a human MT2A gene CpG island 206 bp fragment located in chr16:56642782-56642988 (human genome build 37.1) amplified from human THP-1 macrophages. THP-1 macrophages were cultured as previously reported.21 Human THP-1 and mouse tumor DNA were modified with bisulfite in the same tube. MT2A gene primers were 5′-ATG GTT TGT TTA AGT TTT TAG GAA T-3′ and 5′-AAA AAA AAT CCC ACT TAC TTT TCT TAC-3′. Primer sequences were obtained with BiSearch (bisearch.enzim.hu). Products were cloned in pCR4®-TOPO (Invitrogen) and sequenced. Ten clones were analyzed in each case, except that 2 clones were analyzed for human MT2A.
Histology.
Formalin-fixed material was processed and stained with hematoxylin/eosin. All tumor samples were typed by a pathologist.
Statistics.
For growth parameters, sex-matched littermates were compared by a paired non-parametric test (Wilcoxon). The same test was used to compare within-gene DNA methylation profiles (average methylation frequency) after pairing cytosines at the same position. Genotype frequency was analyzed by a chi-square test. The Mann-Whitney test was used in all other cases.
Figures and Tables
Figure 1 Acta2HpaIIM transgene structure and expression. (A) Schematic map of the Acta2HpaIIM 6.4 kb transgene. Rectangles represent exons or coding sequences. (B) Semi-quantitative RT-PCR analysis of transgene expression in the two independent lines S13 and Z. St, stomach; SI, small intestine; Co, colon; Ao, aorta; VG, vesicular gland; Bl, whole blood cells.
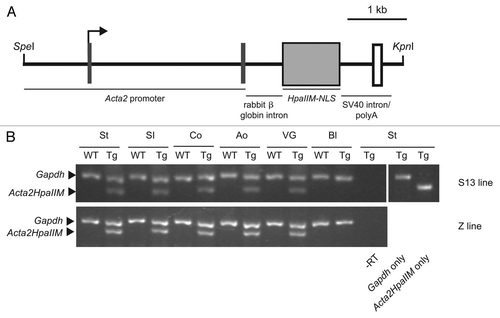
Figure 2 Organomegaly in a subset of Acta2HpaIIM mice. (A) Organ wet weight in WT controls and transgenics of the S13 and Z lines. Data points represent individual mice. Statistical differences are in comparisons with corresponding WT (*p < 0.05; **p < 0.01). Co, colon; SI, small intestine; VG, vesicular gland. (B and C) Examples of hyperplasia of the vesicular gland (arrows) in WT control and transgenic, respectively.
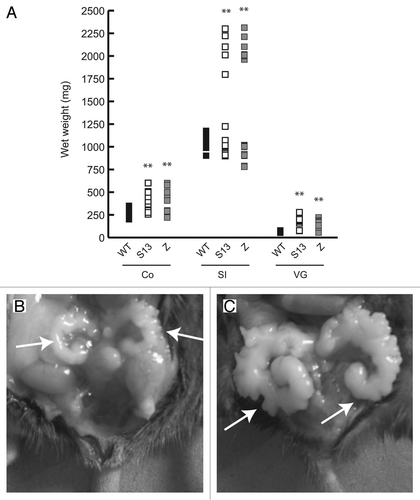
Figure 3 Acta2HpaIIM mice tumors. (A) Representative example of transgene expression in transgenic organs and tumors. VG, vesicular gland; SI, small intestine; N, normal size; H, hyperplastic; Adj, macroscopically normal tissue adjacent to a tumor; IC, intestinal carcinoma; He, hemoangioma. (B–D) Examples of transgenic tumor histology (hematoxylin/eosin). (B) Intestinal carcinoma. An area of well differentiated carcinoma is visible in the lower part, left of arrows (40× magnification). (C) Hemoangioma. The tumor mass surrounds blood vessels with abnormal architecture (arrow; 100×). (D) Lymphoma. Cells with enlarged nuclei (examples indicated by arrows, left) and phagocytes (arrowhead, right) are visible (200×).
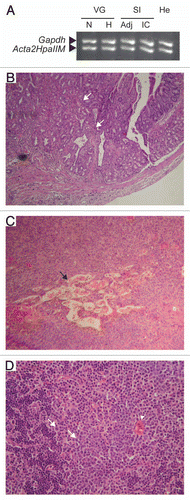
Figure 4 Global DNA hypermethylation in Acta2HpaIIM mice tumors. (A) Total 5mdC normalized as percent of dG in organs and tumors (average ± SD). Organs: white bars, WT controls; grey bars, transgenic normal size organs; black bars, transgenic hyperplastic organs (n = 5 in each group). Co, colon; SI, small intestine; VG, vesicular gland. Tumors: white and black bars, adjacent tissue and tumor mass, respectively. He, hemoangioma (n = 5); IC, intestinal carcinoma (n = 3); KA, kidney adenoma (n = 3); Ly, lymphoma (n = 5). Statistical significance: *p < 0.05; **p < 0.01. (B and C) quantitative methylation-sensitive PCR analysis of LINE-1 methylation status. HpaII endonuclease is sensitive to methylation of the underlined (internal and external) cytosine residues in CCGG. MspI is an isoschizomer of HpaII but is sensitive to methylation of the external cytosine residue (CCGG). Amplification indicates methylation at the corresponding endonuclease site residue(s). (D and E) as in (B and C) but for MusD elements. Adj., apparently normal tissue adjacent to tumor mass; SM, skeletal muscle; for other symbols see (A).
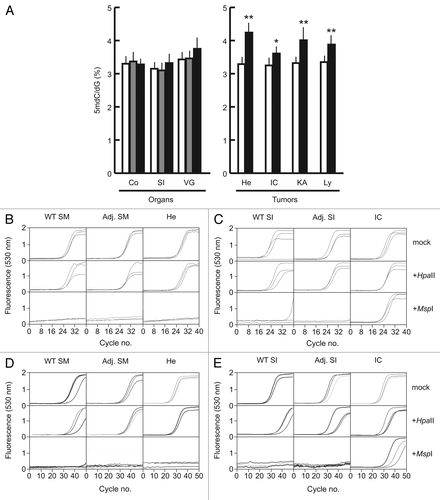
Figure 5 DNA methylation status of tumor suppressor genes and intragenic CpG islands in Acta2HpaIIM mice tumors. (A) Promoter bisulfite DNA methylation analysis and quantitative RT-PCR expression of selected genes in a hemoangioma and intestinal carcinoma. Gene symbols are on the left. In DNA methylation graphs, numbers on the vertical axis indicate the analyzed sequence position relative to the TSS (bp). Ticks represent CpG dinucleotides, arrows and arrowheads indicate HpaII site CpG (CCGG) and external cytosines of HpaII sites (CCGG), respectively. Horizontal bars represent the extent of methylation of the corresponding residues in the clones analyzed. In expression graphs, values are relative to Gapdh RNA levels (average ± SD). White bars, WT controls; grey bars, transgenic adjacent tissue; black bars, transgenic tumor mass. Adj., apparently normal transgenic tissue adjacent to tumors; He, hemoangioma; IC, intestinal carcinoma; SI, small intestine; SM, skeletal muscle. Statistical significance: *p < 0.05; **p < 0.01. (B) methylation status of a sequence located 3′ to Cdkn2a. Numbers on the vertical axis indicate the distance from the 3′ end of Cdkn2a. (C) methylation status of a negative control human MT2A CpG island. For graph symbols in (B and C) see (A).
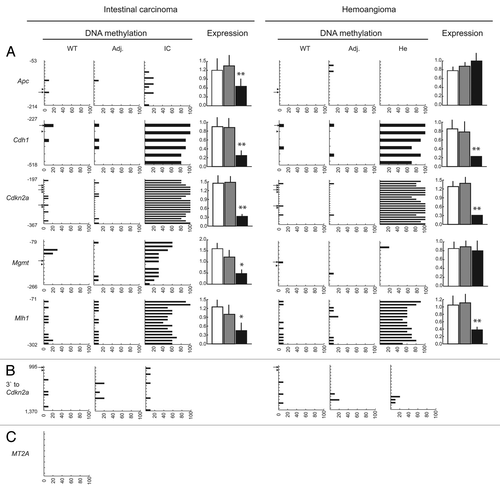
Figure 6 Promoter DNA methylation status of non-tumor suppressor genes in Acta2HpaIIM mice tumors. Symbols same as in legend of .
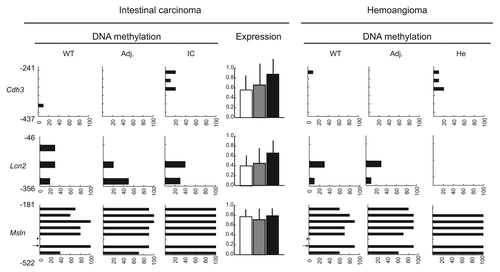
Table 1 Tumor type and frequency in transgenics from S13 and Z lines, and WT controls (n = 40 per group)
Additional material
Download Zip (372.1 KB)Acknowledgements
Work generously supported by Hjerteforeningen (Danish Heart Association, grant no. 04-10-B155-A236-22190), Lundbeck Foundation (grant no. 76/04), Novo Nordisk Foundation, Council for Science and Technology of the State of Guanajuato (CONCYTEG, grant no. 07-16-K662-095-A01) and the Initiative for the Improvement of the University Professorate (PROMEP, grant no. 103.5/08/1589) of the Mexican Ministry of Education (SEP). We thank Martha Olivia Solís-Martínez for technical assistance.
References
- Ross R, Glomset JA. Atherosclerosis and the arterial smooth muscle cell: Proliferation of smooth muscle is a key event in the genesis of the lesions of atherosclerosis. Science 1973; 180:1332 - 1339
- Stemerman MB, Weinstein R, Rowe JW, Maciag T, Fuhro R, Gardner R. Vascular smooth muscle cell growth kinetics in vivo in aged rats. Proc Natl Acad Sci USA 1982; 79:3863 - 3866
- Lagace R, Delage C, Boutet M. Light and electron microscopic study of cellular proliferation in carcinoid heart disease. Recent Adv Stud Cardiac Struct Metab 1975; 10:605 - 616
- Laukkanen MO, Mannermaa S, Hiltunen MO, Aittomäki S, Airenne K, Jänne J, et al. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler Thromb Vasc Biol 1999; 19:2171 - 2178
- Mays-Hoopes LL, Brown A, Huang RC. Methylation and rearrangement of mouse intracisternal a particle genes in development, aging and myeloma. Mol Cell Biol 1983; 3:1371 - 1380
- Gallou-Kabani C, Vigé A, Gross MS, Junien C. Nutri-epigenomics: lifelong remodelling of our epigenomes by nutritional and metabolic factors and beyond. Clin Chem Lab Med 2007; 45:321 - 327
- Zaina S, Lund G. Esteller M. Epigenetics and Cardiovascular Disease. Epigenetics in Biology and Medicine 2008; London, UK Taylor & Francis Group LLC 207 - 223
- Chawla RK, Watson WH, Jones DP. Effect of hypoxia on hepatic DNA methylation and tRNA methyltransferase in rat: similarities to effects of methyl-deficient diets. J Cell Biochem 1996; 61:72 - 80
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 2006; 38:1378 - 1385
- Lyko F, Ramsahoye BH, Kashevsky H, Tudor M, Mastrangelo MA, Orr-Weaver TL, et al. Mammalian (cytosine-5) methyltransferases cause genomic DNA methylation and lethality in Drosophila. Nat Genet 1999; 23:363 - 366
- Takashima S, Takehashi M, Lee J, Chuma S, Okano M, Hata K, et al. Abnormal DNA methyltransferase expression in mouse germline stem cells results in spermatogenic defects. Biol Reprod 2009; 81:155 - 164
- Card CO, Wilson GG, Weule K, Hasapes J, Kiss A, Roberts RJ. Cloning and characterization of the HpaII methylase gene. Nucleic Acids Res 1990; 18:1377 - 1383
- Smith AE, Ford KG. Specific targeting of cytosine methylation to DNA sequences in vivo. Nucleic Acids Res 2007; 35:740 - 754
- Smith AE, Hurd PJ, Bannister AJ, Kouzarides T, Ford KG. Heritable gene repression through the action of a directed DNA methyltransferase at a chromosomal locus. J Biol Chem 2008; 283:9878 - 9885
- Li F, Papworth M, Minczuk M, Rohde C, Zhang Y, Ragozin S, et al. Chimeric DNA methyltransferases target DNA methylation to specific DNA sequences and repress expression of target genes. Nucleic Acids Res 2007; 35:100 - 112
- Andreas S, Schwenk F, Küter-Luks B, Faust N, Kühn R. Enhanced efficiency through nuclear localization signal fusion on phage C31-integrase: activity comparison with Cre and FLPe recombinase in mammalian cells. Nucleic Acids Res 2002; 30:2299 - 2306
- Wang J, Niu W, Nikiforov Y, Naito S, Chernausek S, Witte D, et al. Targeted overexpression of IGF-I evokes distinct patterns of organ remodeling in smooth muscle cell tissue beds of transgenic mice. J Clin Invest 1997; 100:1425 - 1439
- Zaina S, Pettersson L, Ahrén B, Brånén L, Hassan AB, Lindholm M, et al. Insulin-like growth factor II plays a central role in atherosclerosis in a mouse model. J Biol Chem 2002; 277:4505 - 4511
- Pfaffl MW. Bustin SA. Quantification strategies in real-time PCR. A–Z of quantitative PCR 2004; La Jolla, CA International University Line (IUL) 87 - 112
- Alcázar Magaña A, Wrobel K, Alvarado Caudillo Y, Zaina S, Lund G, Wrobel K. High performance liquid chromatography determination of 5-methyl-2′-deoxycytidine (5mdC), 2′-deoxycytidine (dC) and other deoxynucleosides and nucleosides in DNA digests. Anal Biochem 2008; 374:378 - 385
- Lund G, Andersson L, Lauria M, Lindholm M, Fraga MF, Villar-Garea A, et al. DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem 2004; 279:29147 - 29154
- Zaina S, Pettersson L, Bloch Thomsen A, Thyberg J, Nilsson J. Shortened life span, bradycardia and hypotension in mice with targeted expression of an Igf2 transgene in smooth muscle cells. Endocrinology 2003; 144:2695 - 2703
- Pogribny IP, Beland FA. DNA hypomethylation in the origin and pathogenesis of human diseases. Cell Mol Life Sci 2009; 66:2249 - 2261
- Alves G, Tatro A, Fanning T. Differential methylation of human LINE-1 retrotransposons in malignant cells. Gene 1996; 176:39 - 44
- Baust C, Gagnier L, Baillie GJ, Harris MJ, Juriloff DM, Mager DL. Structure and expression of mobile ETnII retroelements and their coding-competent MusD relatives in the mouse. J Virol 2003; 77:11448 - 11458
- Maksakova IA, Zhang Y, Mager DL. Preferential epigenetic suppression of the autonomous MusD over the nonautonomous ETn mouse retrotransposons. Mol Cell Biol 2009; 29:2456 - 2468
- Eads CA, Lord RV, Kurumboor SK, Wickramasinghe K, Skinner ML, Long TI, et al. Fields of aberrant CpG island hypermethylation in Barrett's esophagus and associated adenocarcinoma. Cancer Res 2000; 60:5021 - 5026
- Otterson GA, Khleif SN, Chen W, Coxon AB, Kaye FJ. CDKN2 gene silencing in lung cancer by DNA hypermethylation and kinetics of p16INK4 protein induction by 5-aza 2′deoxycytidine. Oncogene 1995; 11:1211 - 1216
- Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998; 95:6870 - 6875
- Kim MA, Jung EJ, Lee HS, Lee HE, Yang HK, Oh DY, et al. P-cadherin expression in gastric carcinoma: its regulation mechanism and prognostic significance. Hum Pathol 2010; 41:877 - 885
- Sato N, Maitra A, Fukushima N, van Heek NT, Matsubayashi H, Iacobuzio-Donahue CA, et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res 2003; 63:4158 - 4166
- Kang SH, Bang YJ, Im YH, Yang HK, Lee DA, Lee HY, et al. Transcriptional repression of the transforming growth factor-beta type I receptor gene by DNA methylation results in the development of TGFbeta resistance in human gastric cancer. Oncogene 1999; 18:7280 - 7286
- Tsukamoto H, Mishima Y, Hayashibe K, Sasase A. Alpha-smooth muscle actin expression in tumor and stromal cells of benign and malignant human pigment cell tumors. J Invest Dermatol 1992; 98:116 - 120
- McNamara AR, Hurd PJ, Smith AE, Ford KG. Characterisation of site-biased DNA methyltransferases: specificity, affinity and subsite relationships. Nucleic Acids Res 2002; 30:3818 - 3830
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315 - 322
- Shevchuk T, Kretzner L, Munson K, Axume J, Clark J, Dyachenko OV, et al. Transgene-induced CCWGG methylation does not alter CG methylation patterning in human kidney cells. Nucleic Acids Res 2005; 33:6124 - 6136
- Kanai Y. Alterations of DNA methylation and clinicopathological diversity of human cancers. Pathol Int 2008; 58:544 - 558
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science 2003; 300:489 - 492
- Turunen MP, Aavik E, Ylä-Herttuala S. Epigenetics and atherosclerosis. Biochim Biophys Acta 2009; 1790:886 - 891
- Lee HS, Kim BH, Cho NY, Yoo EJ, Choi M, Shin SH, et al. Prognostic implications of and relationship between CpG island hypermethylation and repetitive DNA hypomethylation in hepatocellular carcinoma. Clin Cancer Res 2009; 15:812 - 820
- Yegnasubramanian S, Haffner MC, Zhang Y, Gurel B, Cornish TC, Wu Z, et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res 2008; 68:8954 - 8967
- Lee JJ, Geli J, Larsson C, Wallin G, Karimi M, Zedenius J, et al. Gene-specific promoter hypermethylation without global hypomethylation in follicular thyroid cancer. Int J Oncol 2008; 33:861 - 869
- Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, Martínez N, Varela M, Luka Z, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology 2008; 47:1191 - 1199