Abstract
This study was designed to determine the significance of DNA methyltransferases (DNMTs) in DNA hypermethylation in esophageal squamous cell carcinoma (ESCC) and to identify DNA methylation markers in serum for the early diagnosis of ESCC. A promoter methylation profile of 12 tumor-related genes was assessed using methylation-specific PCR in ESCC and paired non-tumor tissue samples from 47 patients. Expression levels of DNMTs were examined by real-time reverse transcription-PCR and immunohistochemistry. Using MethyLight, the methylation status of 5 genes was analyzed in serum samples from 45 patients and 15 healthy individuals. A total of 46 (97.9%) of 47 ESCC samples showed methylation in at least one of the examined genes, and methylation was most frequent for RAR-β (46.8%), DAPK (46.8%), p16 (44.7%), and CDH1 (42.6%). Methylation of RASSF1A was significantly correlated with the poorly differentiated tumors and the early pathologic tumor classification (P=0.035 and P=0.046, respectively). Tumoral DNMT3b mRNA up-regulation was significantly correlated with hypermethylation of multiple tumor-related genes (P=0.021). In addition, hypermethylation of cell-free serum DNA was common in ESCC patients, and diagnostic accuracy was increased when methylation of multiple genes (RAR-β, DAPK, CDH1, p16 and RASSF1A) were analyzed in combination (ROC AUC 0.911, 82.2% sensitivity and 100% specificity). The present study suggests that hypermethylation of multiple tumor-related genes may be involved in the pathogenesis of ESCC and mediated by the increase of DNMT3b expression. A cluster of multiple methylated genes in serum DNA has the potential as a novel biomarker for ESCC diagnosis.
Introduction
Esophageal cancer is the eighth most common cancer worldwide.Citation1 Esophageal squamous cell carcinoma (ESCC) constitutes 80% of esophageal cancer in the world and this tumor type is especially prevalent in East Asia, South Asia and South Africa. Despite significant advances in diagnosis and treatment of ESCC, the five year survival rate is still about 10% because of its asymptomatic progression in the early stage, the limited treatment options and the poor prognosis outcome. Therefore, further understanding of the molecular mechanism of this disease and translation of molecular findings into the clinical arena are clearly imperative.
Genetic factors, such as loss of heterozygosity at multiple chromosomal loci and mutation of p53 gene,Citation2,Citation3 are involved in ESCC progression. In addition, hypermethylation of normally unmethylated CpG islands in the promoter regions of tumor-related genes has often been observed in the development of human cancers.Citation4 It has been documented that hypermethylation is associated with loss of gene function that can provide a selective advantage to cancer cells. Numerous tumor-related genes involved in various carcinogenic pathways have been revealed to be directly silenced by DNA methylation in ESCC.Citation5 Furthermore, aberrant DNA methylation usually occurs somatically in cancers and can be released into blood circulation, so that it may serve as a novel marker for cancer.Citation6,Citation7 For example, p16 gene hypermethylation has been detected in serum of ESCC patients and can potentially be used for screening and monitoring this disease.Citation7 Despite these advances, in most cases, the methylation status has been investigated for just a single gene or a few genes in ESCC. Thus, the methylation profile of multiple genes in ESCC and its potential use as a valuable DNA marker for early diagnosis remain poorly understood.
To date, the molecular events leading to aberrant DNA methylation remain unclear. It has been reported that overexpression of DNA methyltransferases (DNMTs) is one potential mechanism for DNA hypermethylation in human cancers.Citation8–Citation11 However, studies regarding the relationship between the expression levels of DNMTs and the aberrant methylation of CpG islands have produced contradictory results in a variety of cancers.Citation12–Citation14 Currently, the relationship between expression of DNMTs and aberrant methylation of multiple tumor-related gene promoters is still unclear in ESCC.
In the current study, we analyzed the methylation profile of 12 tumor-related genes in Chinese ESCC, which are known to be involved in carcinogenesis and are frequently silenced by hypermethylation in ESCC or other malignancies. The genes chosen are involved in cell cycle regulation (p14; p16), repair of DNA damage (MGMT; GSTP1), apoptosis (DAPK), tumor cell invasion or tumor architecture (CDH1; APC; TIMP3; ADAM23) and growth factor response (RAR-β; RASSF1A; FHIT). We sought to determine whether a set of genes could serve as a novel marker for ESCC diagnosis. We also examined expression levels of DNMTs to determine whether high DNMTs expression was correlated with hypermethylation of the tumor-related genes.
Results
Promoter hypermethylation profile of tumor-related genes in ESCC and its correlation with clinico-pathological parameters.
Using a panel of 12 gene promoter loci, the DNA methylation patterns were assessed in the tumor and paired adjacent non-tumor tissues from 47 ESCC patients. Examples of MSP analysis were shown in . The sequencing results of the MSP products confirmed that MSP reactions amplified the targeted genomic fragments and the CpG sites located inside the target fragments showed extensive methylation. The unmethylated form of all genes was detected in all the samples (data not show). MSP analysis revealed that at least one of these twelve genes had methylation in 97.9% (46/47) of the ESCC samples, whereas 74.5% (35/47) harbored two or more methylated genes. In ESCC samples, the methylation frequencies of 12 genes ranged from 0–46.8% (). Among these genes, DAPK and RAR-β stood out as the most frequently affected ones and the lowest methylation frequency was detected at GSTPI and ADAM23. By analyzing the paired non-tumor tissue samples, the frequencies of hypermethylated genes were lower than that in tumor samples (). The average number of methylated genes per sample was 2.5 for tumor samples and 0.8 for paired non-tumor samples, showing a significant increase (p < 0.0001).
We explored the relationships between the clinico-pathological parameters and DNA methylation patterns in the testing group cases. Methylation of RASSF1A was more frequently seen in female than in male (p = 0.029) and a clear relationship was observed between methylation of RAR-β and patient age (p = 0.042) (). Aberrant methylation of DAPK was more common in patients with a tumor < 5 cm than in patients with a tumor ≥ 5 cm (64 vs. 23.8%, p = 0.009) (). In this study, methylation of RASSF1A and MGMT were more frequent in the poorly or moderately differentiated tumors rather than in the well differentiated ones (p = 0.035 and p = 0.019, respectively) (). In addition, methylation of RASSF1A correlated significantly with the early pathologic tumor (pT) classification (pT1–pT2) (p = 0.046) (). No significant correlations were observed between methylation of any gene and location of tumor, lymph node metastasis (pN) or smoking and alcoholism.
DNMTs expression in ESCC and its correlation with hypermethylation of tumor-related genes.
To examine whether high level of DNMTs contributed to the promoter hypermethylation of tumor-related genes, we determined mRNA expression of DNMTs in the tumor and paired adjacent non-tumor tissues from 46 ESCC patients. Elevated mRNA expression of DNMT1, DNMT3a and DNMT3b were found in 22 (47.8%), 22 (47.8%) and 33 (71.7%) of 46 ESCC samples, respectively. The average expression level of DNMT1 and DNMT3a mRNA in ESCC tissues was not significantly different from that in the paired non-tumor tissues (), respectively. However, a significant increase in the mRNA level of DNMT3b was observed in ESCC tissues ( and p = 0.005). The statistical significance was still found after multiple testing (p < 0.017). The expression level of DNMT3b in cases with elevated mRNA expression was approximately 22 (range, 2.3–175.1) times higher than that of the paired non-tumor tissues. In addition, 29 (87.9%) of 33 patients with elevated mRNA expression of DNMT3b were in the high range (≥ 5-fold). Furthermore, mRNA levels for DNMT1, DNMT3a and DNMT3b appeared to be co-overexpressed in the same tumors ( and p < 0.001 for DNMT1/3a, p = 0.051 for DNMT1/3b and p = 0.008 for DNMT3a/3b).
DNMT3b protein expression was further evaluated in 44 cases of ESCC and paired adjacent non-tumor tissues because of tissue shortening. The immunohistochemical staining result of DNMT3b protein was shown in and . Thirty-two (72.7%) of 44 ESCC samples showed at least mild (1+) nuclear expression of DNMT3b protein and the expression was strong (3+ and 4+) in 17 (38.6%) ESCC samples, while only one (2.3%) of 44 paired non-tumor samples demonstrated strong nuclear expression of DNMT3b. DNMT3b protein was more frequently observed in ESCC tissues than in paired non-tumor tissues (p < 0.0001). However, there was no significant correlation between the mRNA expression and protein nuclear immunoreactivity of DNMT3b.
The correlation between mRNA expression of DNMT3b and methylation of investigated genes was evaluated in 46 ESCC patients. Elevated DNMT3b mRNA expression was correlated with hypermethylation of p16 (p = 0.022) (), whereas there was no significant correlation between elevated mRNA expression of DNMT3b and hypermethylation of other genes. When high tumor-related genes alteration was defined as hypermethylation of two or greater genes, hypermethylation of multiple genes was correlated significantly with high expression level of DNMT3b mRNA (p = 0.021) ().
Hypermethylation in cell-free serum DNA and its correlation with clinico-pathological parameters.
DNA methylation patterns of five genes (p16, CDH1, RASSF1A, DAPK and RAR-β) most frequently hypermethylated in tumor tissues were assessed in cell-free serum DNA from 45 ESCC patients and 15 healthy individuals. The methylation patterns of five genes were shown in . In general, methylated DNA takes only a small proportion (mostly less than 5%) of total cell-free DNA. Methylation was detected in serum DNA at CDH1 (84.4%), DAPK (73.3%), RASSF1A (62.2%), RAR-β (26.7%) and p16 (6.7%). In healthy individuals, methylation was less common in serum DNA, including only 20.0% for CDH1, 13.3% for DAPK, 6.7% for RASSF1A, 13.3% for RAR-β and 0% for p16. The number of methylated genes per sample in ESCC patients was significantly higher than that in healthy individuals (2.5 vs. 0.5, p < 0.0001).
We analyzed whether serum DNA methylation was correlated with the clinico-pathological parameters. Hypermethylation of RAR-β was more common in female than in male (55.6% vs. 19.4%, p = 0.043) (Sup. Table 2). DAPK hypermethylation was correlated with the advanced tumor classification (pT3–pT4) (p = 0.016) (Sup. Table 2). When two or more methylated genes were considered, hypermethylation of multiple genes was also more frequent in the advanced tumor classification as compared to the early classification (90.9% vs. 54.5%, p = 0.016) (Sup. Table 2).
Evaluation of serum DNA methylation markers for ESCC diagnosis.
To assess the clinical usefulness of serum DNA methylation patterns as diagnostic biomarkers of ESCC, we calculated their sensitivity and specificity using ROC analysis. As shown in , methylation at CDH1, DAPK and RASSF1A genes provided a range of 62.2–84.4% in sensitivity and 80–93.3% in specificity for the diagnosis (AUC = 0.822, 0.8 and 0.778, respectively), while methylation of RAR-β and p16 genes was less informative with 26.7 and 6.7% sensitivity (AUC = 0.563 and 0.533), respectively. The overall diagnostic sensitivity of five genes for ESCC, when at least one of the five genes was positive, rose to 100%, but the overall specificity was relatively low (AUC = 0.733). With a combination of two or more methylated genes (cutoff value), the overall specificity rose to 100% and the overall diagnostic sensitivity was maintained at 82.2% (AUC = 0.911).
Discussion
Intensive studies have confirmed the importance of DNA methylation in human cancers and have focused on regions of the CpG islands that may have functional significance resulting from a loss of gene expression.Citation4,Citation5 Whereas most individual cancers have several, perhaps hundreds, of methylated genes, profiling studies have shown specific genes and their methylation frequencies for some tumor types.Citation18,Citation19 However, the profile of genes' hypermethylation in ESCC versus the corresponding non-tumor tissues and its clinical significance is still not well described. In the present study, the methylation profile of 12 tumor-related genes was firstly determined in Chinese ESCC patients. The results showed that methylation was detected in 10 genes with increasing percentage from 6.4% for FHIT to 46.8% for DAPK and RAR-β in ESCC tissues and 97.9% of ESCC samples showed at least one methylated gene. Our results suggest that CpG island hypermethylation is a common event in Chinese ESCC patients. In addition, promoter methylation of GSTP1 and ADAM23 did not exist in this study. Similar methylation frequency of GSPT1 also was reported in previous investigation in ESCC.Citation20 These data suggest that epigenetic alteration of ADAM23 and GSPT1 may be an uncommon molecular event in ESCC.
In this study, DAPK, RAR-β, p16 and CDH1 showed high methylation frequencies, ranging from 42.6–46.8%. DAPK participates in a number of death signaling pathways, i.e., apoptosis triggered by interferon-γ, tumor necrosis factor-α and Fas. DAPK suppresses tumor growth and metastasis by increasing the occurrence of apoptosis in vivo. As disruption of processes involved in programmed cell death is a common feature of human cancers, it is significant that inactivation of DAPK by promoter methylation has been described in esophageal cancer.Citation20–Citation22 Retinoids regulate the growth, differentiation and apoptosis of premalignant and malignant cells during carcinogenesis. RAR-β, which acts as retinoic acid-dependent transcriptional activators, predominantly mediated the effects of retinoids. RAR-β methylation was found in 25–55% of ESCC.Citation20,Citation21,Citation23 p16 regulates the cell cycle through inhibiting the ability of CDK4 to interact with cyclin D1 and stimulating the passage through the G1 phase of cell cycle. p16 methylation was reported at a high frequency in ESCC.Citation20,Citation21 In addition, CDH1 encodes E-cadherin protein, a transmembrane glycoprotein that mediates calcium-dependent cell-cell adhesion. Loss of CDH1 enhances tumor progression and invasion by multiple mechanisms, including reduced cell-cell adhesion. CDH1 methylation was reported to be present in 31–43% of ESCC.Citation20,Citation21 Our results are in agreement with these reports since similar methylation frequencies of four genes were documented in ESCC. Our data suggest that the inactivation of these genes may play pivotal role in ESCC tumorigenesis.
We also investigated the correlation between promoter methylation and clinico-pathological parameters. A particular interest was addressed to RASSF1A, a novel tumor suppressor gene, frequently epigenetic inactivated in a number of epithelial cancers. RASSF1A regulates a proapoptotic pathway through heterodimerization with the Ras effector NORE1 and interacts with MST1, which mediates the apoptotic effect of Ras.Citation24 Also, RASSF1A protein interacts with Cdc20, an activator of the anaphase-promoting complex, resulting in the inhibition of activity of complexes and the prevention of mitotic progression.Citation25 Methylation of RASSF1A has been noticed in 7–52% of ESCC patients.Citation23,Citation26,Citation27 In this study, we found that 14.9% of patients exhibit methylation of RASSF1A. On the other hand, methylation of RASSF1A was also found in corresponding non-tumor tissues with a lesser frequency (4.3%). The present study also showed that methylation of RASSF1A was correlated with the early tumor classification. These data suggest that RASSF1A hypermethylation may be involved in the early step of ESCC progression. Methylation of RASSF1A or MGMT was more frequent in the poorly or moderately differentiated tumors rather than in the well differentiated ones. With regard to MGMT, aberrant methylation impairs the ability of the MGMT protein to remove alkyl groups from the O6-position of guanine and thereby increases the risk of cancer. MGMT methylation was also reported at a similar frequency in esophageal adenocarcinoma.Citation28 These data suggest that the loss of RASSF1A and MGMT protein due to the promoter hypermethylation may enhance malignant potentials in ESCC. Methylation of RASSF1A was seen more frequently in female, but the reason for this gender difference was unknown. In addition, hypermethylation of RAR-β was positively correlated with patient age. A recent study described that aging was associated with methylation of certain genes.Citation29 Therefore, the aging mechanism may be a possible explanation for detecting methylation in ESCC tissues.
Reports in literature had mentioned the lack of methylation in non-malignant tissues and considered methylation as a tumor-specific event. However, in this series, we found that 24 (51.1%) of 47 non-tumor tissues showed methylation in at least one gene. Methylation of p16, CDH1, DAPK and RAR-β was assessed at 21.3, 21.3, 12.8 and 12.8%, respectively. Similarly, Ishii et al. found that methylation of p16 and DAPK existed in 18 (32%) and 3 (5.4%) of 56 corresponding nonneoplastic esophageal epithelium in ESCC patients.Citation30 Kuroki et al. also showed high methylation frequency for RAR-β in non-tumor esophageal samples,Citation23 and a recent study indicated that smoking-damaged esophageal epithelium, even histologically normal, had suffered epigenetic changes.Citation27 Thus, our data suggest that the methylated alleles in the paired non-tumor tissues may represent premalignant changes and p16, CDH1, DAPK and RAR-β may be of the possible targets for predicting cancer risk and/or making an early cancer diagnosis.
During ESCC tumorigenesis, overexpression of the three functional DNMTs, which catalyze cytosine methylation, may be of importance in dysregulating tumor-related genes expression. In this study, DNMT3b mRNA and protein expression were noticed to be remarkably upregulated in the tumor tissues. Our data point to a predominant role of DNMT3b in ESCC, relative to DNMT1 and DNMT3a. Our results also indicated that the mRNA expression of DNMT1, DNMT3a and DNMT3b was elevated in a coordinate manner in ESCC tissues. In patients with breast cancer, increased DNMTs mRNA expression reportedly was correlated with each other.Citation31 Our results support the conclusion that DNMTs expression may have a common regulatory pathway. The in vitro study of breast cell lines showed a significant correlation between the CpG island methylator phenotype and DNMT3b overexpression,Citation11 but some in vivo studies in various cancers failed to confirm this correlation.Citation12,Citation14 In our study, we demonstrated a strong correlation between the elevated expression of DNMT3b mRNA and hypermethylation of p16, and this result was consistent with the previous report in lung cancer.Citation16 If two or more methylated genes were considered, the increased expression of DNMT3b mRNA was correlated with hypermethylation of multiple genes. Our data suggest that DNMT3b upregulation may mainly contribute to aberrant methylation pattern in ESCC development.
Previous study clearly demonstrated the advantages of tumor-related genes methylation analysis in serum samples of several cancer types regarding diagnostic information.Citation6 To our knowledge, our study is the first study of CpG island hypermethylation of multiple genes in the serum of ESCC patients to date. Although most methylated DNA accounts for a relatively small proportion of cell-free serum DNA, the detection of tumor originated DNA is feasible in ESCC patients. The finding was in good agreement with the proportion of methylated DNA detected in the serum of patients with testicular cancer.Citation32 The methylation analysis of a single gene, including CDH1, DAPK or RASSF1A, allowed us to identify approximately 60–80% of ESCC patients, but the specificity was less than 95%. Some reports indicated that the diagnostic information could be increased if methylation of multiple genes of serum DNA were analyzed in combination.Citation17,Citation32 Indeed, we found that hypermethylation at two or more genes provided 82.2% in sensitivity with 100% in specificity. This finding was intriguing because currently available serum biomarkers such as CEA and CA199 were less informative for the early detection of ESCC.Citation33,Citation34 Thus, these data suggest that combinatorial methylation analysis of these genes in serum DNA has the potential of a valuable diagnostic marker by noninvasive testing. Interestingly, serum DNA hypermethylation at DAPK and two or more methylated genes was correlated with advanced tumor classification, respectively. These findings may be explained in two possibilities: (1) shedding of a large amount of DNA into blood indicates a more advanced stage of the maligany and (2) accumulation of a large proportion of methylated cells in tumor tissue indicates a more advanced stage of the malignancy.Citation35
In conclusion, our data indicate a comprehensive methylation profile in Chinese ESCC patients, suggesting the important role of epigenetic alteration in the pathogenesis of ESCC. The current results also suggest that hypermethylation of multiple genes may be mediated by the increase of DNMT3b expression. Further studies on the relationship between DNMT3b and other components of the DNA methylation machinery in ESCC tissue samples are necessary. Moreover, the detection of hypermethylated serum DNA is feasible in ESCC patients and combinatorial methylation analysis of CDH1, DAPK, RASSF1A, RAR-β and p16 may be used as a novel biomarker for early diagnosis of ESCC.
Materials and Methods
Clinical samples.
Tumors and the paired adjacent non-tumor tissue samples from 47 ESCC patients were collected from Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. Following surgery, tissue samples were promptly frozen in liquid nitrogen. The patients (37 males and ten females) ranged in age from 35–80 years, and their mean age was 58.9 ± 10.2 years. All of these patients had no history of receiving chemo or radiotherapy before the operation. The diagnosis was confirmed as squamous cell carcinoma by pathological examination. Tumor stages were evaluated according to the TNM classification of the International Union Against Cancer. Corresponding blood samples were collected from 45 patients before surgery, and serum was separated by centrifugation at 1,000x g and stored at −80°C. In addition, serum samples from 15 healthy volunteers selected by matching by age and sex were used as the negative controls. The healthy volunteers (eight males and seven females) ranged in age from 39–70 years, and their mean age was 53.7 ± 9.1 years. This study was approved by the Ethics Committee of the Chinese Academy of Medical Sciences and Peking Union Medical College, and informed consent was obtained from all participants.
DNA isolation and bisulfite treatment.
Genomic DNA was extracted from tissue samples using QIAmp DNA Mini Kit (Qiagen, Hilden, Germany). Cell-free serum DNA was isolated using QIAmp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and salmon testes DNA (Sigma, St. Louis, MO) as a carrier DNA. Extracted DNA was bisulfite-modified using EZ DNA methylation-gold kit (Zymo Research, Orange, CA).
Methylation-specific PCR and bisulfite sequencing.
Methylation-specific PCR (MSP) was used to examine the methylation status of 12 tumor-related genes, as described previously in reference Citation15. Primer sequences and conditions are available in Supplemental Table 1. SssI-treated normal lymphocyte DNA was used as positive control. Positive controls as well as negative controls (without DNA) were performed in each set of MSP and each MSP assay was repeated three times. The MSP products were separated electrophoretically on 2.5% agarose gels. The representative products were purified and cloned into the pMD19-T vector (Takara, Dalian, China). The cloned MSP fragments were sequenced by an ABI Prism 377 automated sequencer (Applied Biosystems, Foster City, CA).
RNA extraction and real-time reverse transcription-PCR.
Total RNA was isolated from the tissue samples by using TRIzol Reagent (Invitrogen, Carlsbad, CA). First strand cDNA was synthesized by using the Improm-II reverse transcriptase kit (Promega, Madison, WI). ACTB was used as an internal reference gene to normalize cDNA input. Real-time PCR was done in a 25 µL reaction volume containing 1x SYBR GreenER (Invitrogen), 300 nM each primer and 4 µl each diluted cDNA. Primer sequences and conditions are available in Supplemental Table 1. Each sample was performed in triplicate. The ratio (T/N) of target gene mRNA expression levels of ESCC tissues (T) and paired non-tumor tissues (N) was calculated. T/N ≥ 2 was considered as the elevated expression.
Immunohistochemistry.
After a routine dewaxing and endogenous peroxidase activity blockage, paraffin-embedded sections of tissue samples were subjected to the high-pressure antigen retrieval in 10 mM citric buffer (pH 6.0). Nonspecific binding was blocked by incubation with goat-nonimmune serum. Slides were then incubated sequentially with mouse monoclonal antibodies for DNMT3b (1:150, Imgenex, San Diego, CA) overnight at 4°C and peroxidase-labeled polymer secondary antibody (Dako, Glostrup, Denmark) for 30 min. After chromogen diaminobenzidine staining, sections were counterstained with hematoxylin. All the slides were inspected under double blindness and independently by two experienced pathologists. The positive cells were counted and classified into five grades according to previously described criteria:Citation16 score 0, no positive cell staining; score +, <10%; score ++, from 11–50%; score +++, from 51–70%; score ++++, >70% positive cells. In this study, scores +, ++, +++ and ++++ were considered to be a positive immunostaining result.
MethyLight analysis.
Real-time MSP was performed with a technique based on the principles of the MethyLight assay, as described previously in reference Citation17. A sequence of ACTB gene without CpG nucleotides served as the internal reference to normalize DNA input. The methylation status of five genes of interest was examined by real-time PCR with the primers and Taqman probes specific for fully methylated bisulfite-converted sequences. Primer and probe sequences are available in Supplemental Table 1. SssI-treated normal lymphocyte DNA served as a positive control and was used to construct a standard curve to quantify the amount of fully methylated alleles in each plate. Normal lymphocyte DNA and water served as negative and blank controls, respectively. Each sample was performed in triplicate. Relative levels of methylated DNA in each sample were described as the normalized index of methylation (NIM). The NIM was calculated by dividing the GENE:ACTB ratio of a sample by the GENE:ACTB ratio of SssI-treated lymphocyte DNA and multiplying by 100. A gene was deemed methylated if the NIM value was >0. A NIM scale from white (no methylation) to black (≥10% methylation) was designed with Microsoft Visual Basic (Microsoft Corporation, Seattle, WA).
Statistical analysis.
SPSS 16.0 software (SPSS Inc., Chicago, IL) was used for all the statistical analysis. Statistical analysis was performed using the Fisher exact test for differences between groups and the Student t test between means. ROC curve analysis was used to determine AUC, sensitivity and specificity of serum DNA methylation. p values <0.05 were considered statistically significant. All reported p values were based on 2-sided tests. Bonferroni tests were estimated to correct for multiple tests.
Abbreviations
| ESCC | = | esophageal squamous cell carcinoma |
| DNMTs | = | DNA methyltransferases |
| RASSF1A | = | Ras association domain family 1A |
| MGMT | = | O6-methylguanine-DNA methyltransferase |
| GSTP1 | = | glutathione S-transferase P1 |
| DAPK | = | death-associated protein kinase |
| CDH1 | = | E-cadherin |
| APC | = | adenomatosis polyposis coli |
| TIMP3 | = | tissue inhibitor of metalloproteinase-3 |
| ADAM23 | = | a desintegrin and metalloprotease domain 23 |
| RAR-β | = | retinoic acid receptor β |
| FHIT | = | fragile histidine triad |
| MST1 | = | mammalian sterile 20-like 1 |
| ACTB | = | β-actin |
| MSP | = | methylation-specific polymerase chain reaction |
| CEA | = | carcinoembryonic antigen |
| CA199 | = | carbohydrate antigen 199 |
| WBCs | = | white blood cells |
| ROC | = | receiver operator characteristic curve |
| AUC | = | the area under ROC curve |
| NIM | = | the normalized index of methylation |
Figures and Tables
Figure 1 Representative results of MSP analysis of the methylated form of 12 tumor-related genes. M, markers; N, negative control (water blank); P, positive control (universal methylated DNA); T, tumor samples. Samples were scored as methylated when there was a clearly visible band with the methylated primers.
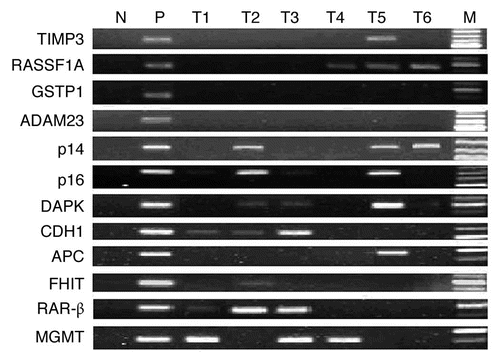
Figure 2 mRNA expression of DNMTs in patients with ESCC (n = 46). (A) Real-time reverse transcription-PCR results for DNMTs. The black bars correspond to non-tumor tissues (NT), and the white bars correspond to ESCC. The means of expression levels of DNMTs are depicted relative to ACTB housekeeping gene expression. Asterisk indicates statistical significance after multiple testing (p < 0.017). (B) DNMT1, 3a and 3b mRNA were expressed in a coordinate manner in the same tumor tissues. “+” indicates elevated mRNA expression as opposed to “−” which indicates a not elevated result. Numbers above the bars indicate the percentage in the total concordant group (+/+ and −/−) and non-concordant group (+/− and −/+). For the association between categories, p = 0.051 for DNMT1/3b, p < 0.001 for DNMT1/3a and p = 0.008 for DNMT3a/3b.
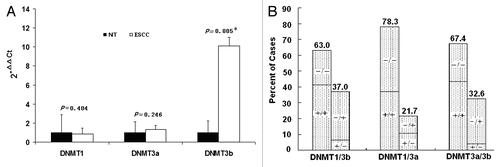
Figure 3 Representative immunohistochemical staining results for DNMT3b protein in two patients with ESCC. Strong nuclear expression was seen in the layer of epithelium in ESCC tissues (A and C). Negative staining was observed in paired non-tumor tissues (B and D). (Original magnification ×400 in A–D).
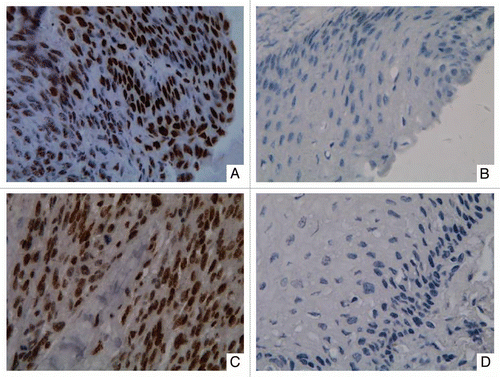
Figure 4 Hypermethylation of cell-free serum DNA at five gene promoter regions in ESCC patients (n = 45) and healthy individuals (n = 15). S, ESCC samples; HI, healthy individuals samples; WBC DNA isolated from the WBCs of healthy volunteers served as a negative control and SssI treated fully methylated DNA served as positive control. NIM was color-scaled between white (representing no methylation detected) and black (indicating that ≥10% of bisulfite-converted input copies were methylated). Asterisk indicates NIM values less than 0.5% but greater than 0.
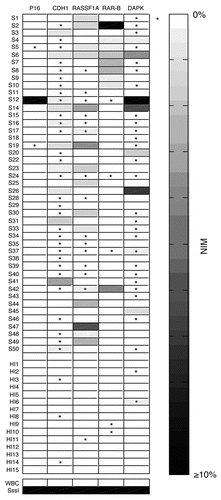
Table 1 Frequency of promoter methylation for individual gene in ESCC patients (n = 47)
Table 2 Correlation between DNA hypermethylation and clinico-pathological parameters of ESCC patients (n = 47)
Table 3 Nuclear immunoreactivity of DNMT3b in ESCC patients (n = 44)
Table 4 Correlation between DNA hypermethylation and mRNA expression of DNMT3b in ESCC (n = 46)
Table 5 Diagnostic information of serum DNA methylation at five genes
Additional material
Download Zip (117.9 KB)Acknowledgements
We thank Professor Ming-Peng She and Dr. Xiao-Bing Zhang for the revision of the article and Dr. Li-Bin Deng for advice on the design of . This study was supported by National Natural Science Foundation of China (30393130, 30470651), National Basic Research Program (2006CB504103), Key Projects in the National Science and Technology Pillar Program during the Eleventh Five-year Plan Period (2006BAI119B07).
References
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics 2002. CA Cancer J Clin 2005; 55:74 - 108
- Roth MJ, Hu N, Emmert-Buck MR, Wang QH, Dawsey SM, Li G, et al. Genetic progression and heterogeneity associated with the development of esophageal squamous cell carcinoma. Cancer Res 2001; 61:4098 - 4104
- Gao H, Wang LD, Zhou Q, Hong JY, Huang TY, Yang CS. p53 tumor suppressor gene mutation in early esophageal precancerous lesions and carcinoma among high-risk populations in Henan, China. Cancer Res 1994; 54:4342 - 4346
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415 - 428
- Sato F, Meltzer SJ. CpG island hypermethylation in progression of esophageal and gastric cancer. Cancer 2006; 106:483 - 493
- Duffy MJ, Napieralski R, Martens JW, Span PN, Spyratos F, Sweep FC, et al. Methylated genes as new cancer biomarkers. Eur J Cancer 2009; 45:335 - 346
- Hibi K, Taguchi M, Nakayama H, Takase T, Kasai Y, Ito K, et al. Molecular detection of p16 promoter methylation in the serum of patients with esophageal squamous cell carcinoma. Clin Cancer Res 2001; 7:3135 - 3138
- Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002; 416:552 - 556
- Peng DF, Kanai Y, Sawada M, Ushijima S, Hiraoka N, Kitazawa S, et al. DNA methylation of multiple tumor-related genes in association with overexpression of DNA methyltransferase 1 (DNMT1) during multistage carcinogenesis of the pancreas. Carcinogenesis 2006; 27:1160 - 1168
- Linhart HG, Lin H, Yamada Y, Moran E, Steine EJ, Gokhale S, et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev 2007; 21:3110 - 3122
- Roll JD, Rivenbark AG, Jones WD, Coleman WB. DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol Cancer 2008; 7:15
- Eads CA, Danenberg KD, Kawakami K, Saltz LB, Danenberg PV, Laird PW. CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase overexpression. Cancer Res 1999; 59:2302 - 2306
- Sato M, Horio Y, Sekido Y, Minna JD, Shimokata K, Hasegawa Y. The expression of DNA methyltransferases and methyl-CpG-binding proteins is not associated with the methylation status of p14(ARF), p16(INK4a) and RASSF1A in human lung cancer cell lines. Oncogene 2002; 21:4822 - 4829
- Park HJ, Yu E, Shim YH. DNA methyltransferase expression and DNA hypermethylation in human hepatocellular carcinoma. Cancer Lett 2006; 233:271 - 278
- Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93:9821 - 9826
- Lin TS, Lee H, Chen RA, Ho ML, Lin CY, Chen YH, et al. An association of DNMT3b protein expression with p16INK4a promoter hypermethylation in non-smoking female lung cancer with human papillomavirus infection. Cancer Lett 2005; 226:77 - 84
- Hoque MO, Feng Q, Toure P, Dem A, Critchlow CW, Hawes SE, et al. Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J Clin Oncol 2006; 24:4262 - 4269
- Costello JF, Frühwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet 2000; 24:132 - 138
- Houshdaran S, Hawley S, Palmer C, Campan M, Olsen MN, Ventura AP, et al. DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS One 2010; 5:9359
- Lee EJ, Lee BB, Han J, Cho EY, Shim YM, Park J, et al. CpG island hypermethylation of E-cadherin (CDH1) and integrin alpha4 is associated with recurrence of early stage esophageal squamous cell carcinoma. Int J Cancer 2008; 123:2073 - 2079
- Guo M, Ren J, House MG, Qi Y, Brock MV, Herman JG. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin Cancer Res 2006; 12:4515 - 4522
- Kuester D, Dar AA, Moskaluk CC, Krueger S, Meyer F, Hartig R, et al. Early involvement of death-associated protein kinase promoter hypermethylation in the carcinogenesis of Barrett's esophageal adenocarcinoma and its association with clinical progression. Neoplasia 2007; 9:236 - 245
- Kuroki T, Trapasso F, Yendamuri S, Matsuyama A, Alder H, Mori M, et al. Allele loss and promoter hypermethylation of VHL, RAR-beta, RASSF1A and FHIT tumor suppressor genes on chromosome 3p in esophageal squamous cell carcinoma. Cancer Res 2003; 63:3724 - 3728
- Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, et al. Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol 2002; 12:253 - 265
- Song MS, Song SJ, Ayad NG, et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol 2004; 6:129 - 137
- Kuroki T, Trapasso F, Yendamuri S, Matsuyama A, Alder H, Mori M, et al. Promoter hypermethylation of RASSF1A in esophageal squamous cell carcinoma. Clin Cancer Res 2003; 9:1441 - 1445
- Oka D, Yamashita S, Tomioka T, Nakanishi Y, Kato H, Kaminishi M, et al. The presence of aberrant DNA methylation in noncancerous esophageal mucosae in association with smoking history: a target for risk diagnosis and prevention of esophageal cancers. Cancer 2009; 115:3412 - 3426
- Baumann S, Keller G, Pühringer F, Napieralski R, Feith M, Langer R, et al. The prognostic impact of O6-Methylguanine-DNA Methyltransferase (MGMT) promotor hypermethylation in esophageal adenocarcinoma. Int J Cancer 2006; 119:264 - 268
- Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009; 5:1000602
- Ishii T, Murakami J, Notohara K, Cullings HM, Sasamoto H, Kambara T, et al. Oesophageal squamous cell carcinoma may develop within a background of accumulating DNA methylation in normal and dysplastic mucosa. Gut 2007; 56:13 - 19
- Girault I, Tozlu S, Lidereau R, Bièche I. Expression analysis of DNA methyltransferases 1, 3A and 3B in sporadic breast carcinomas. Clin Cancer Res 2003; 9:4415 - 4422
- Ellinger J, Albers P, Perabo FG, Müller SC, von Ruecker A, Bastian PJ. CpG island hypermethylation of cell-free circulating serum DNA in patients with testicular cancer. J Urol 2009; 182:324 - 329
- Munck-Wikland E, Kuylenstierna R, Wahren B, Lindholm J, Haglund S. Tumor markers carcinoembryonic antigen, CA 50 and CA 19-9 and squamous cell carcinoma of the esophagus. Pretreatment screening. Cancer 1988; 62:2281 - 2286
- Banki F, Yacoub WN, Hagen JA, Mason RJ, Ayazi S, DeMeester SR, et al. Plasma DNA is more reliable than carcinoembryonic antigen for diagnosis of recurrent esophageal cancer. J Am Coll Surg 2008; 207:30 - 35
- Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci USA 2000; 97:710 - 715